Synthesis, Characterization, and Biological Performances of Magnesium-Substituted Dicalcium Phosphate Anhydrous


Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Mg-DCPA Powders
2.2. Preparation of Extracts
2.3. Cytotoxicity Test
2.4. Osteo/Odontogenic Differentiation Behavior
2.4.1. Alkaline Phosphatase (ALP) Staining
2.4.2. Alizarin Red S (ARS) Staining
2.5. Statistical Analysis
3. Results and Discussion
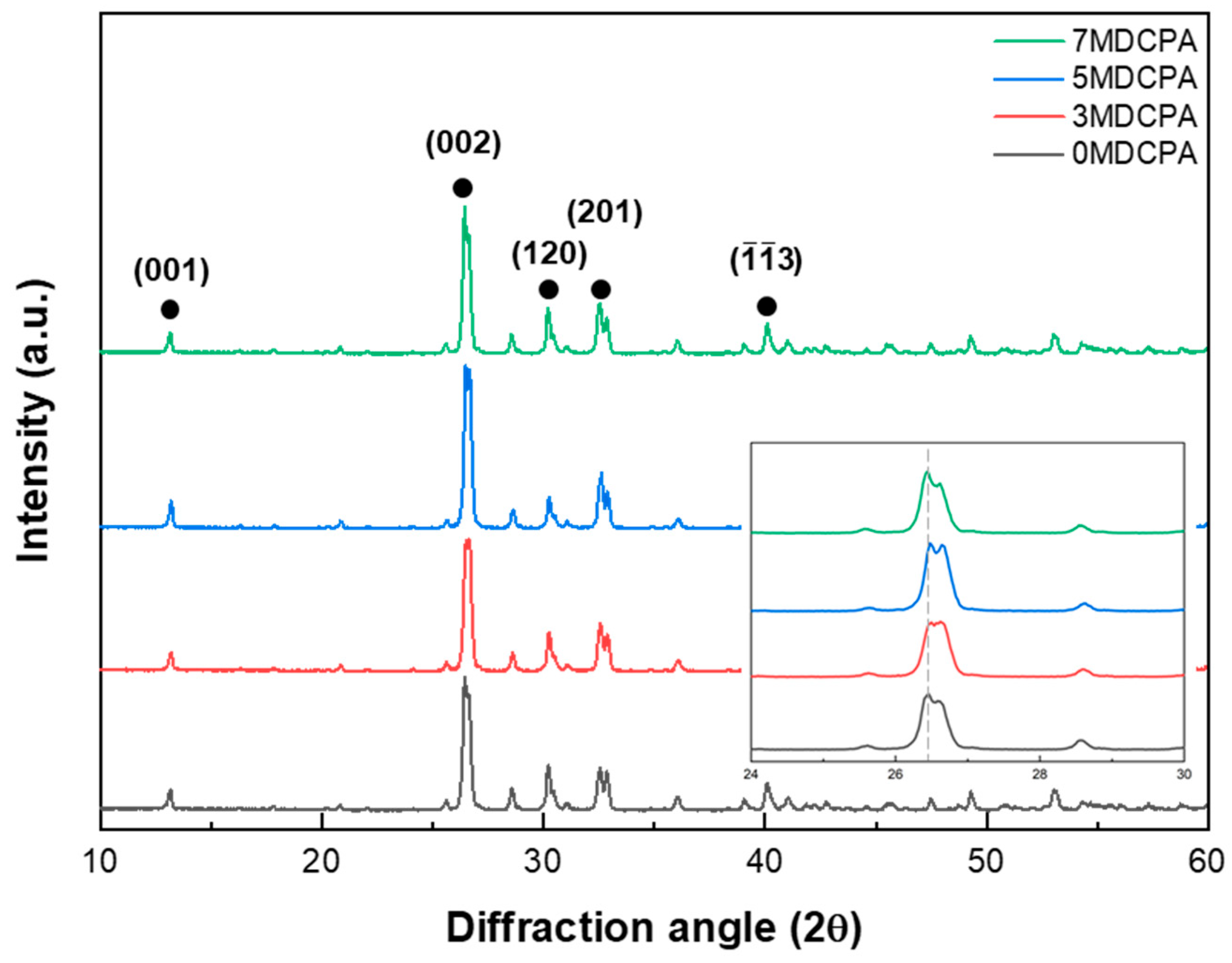
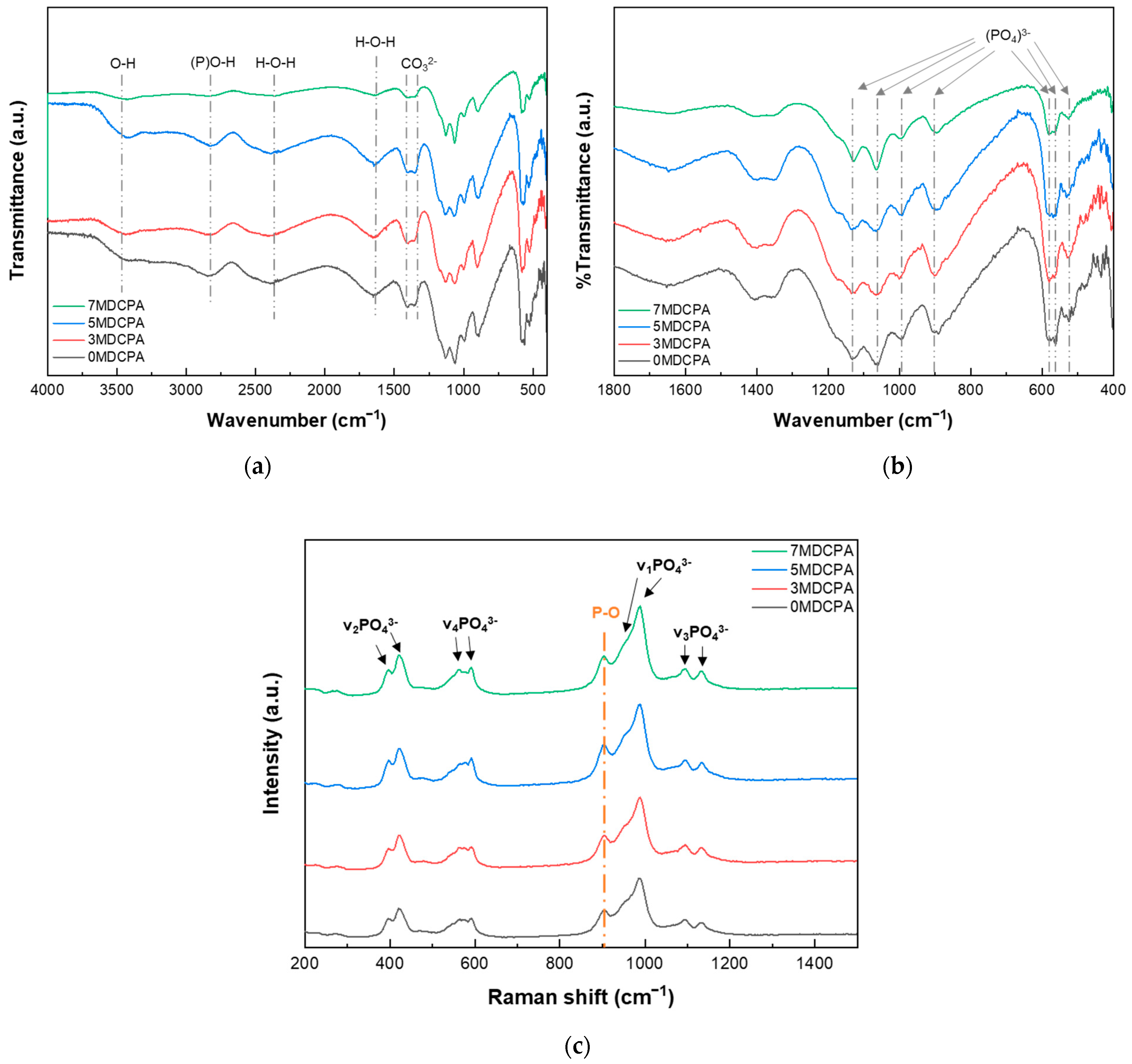
3.1. Characterization of the Synthesized Powder
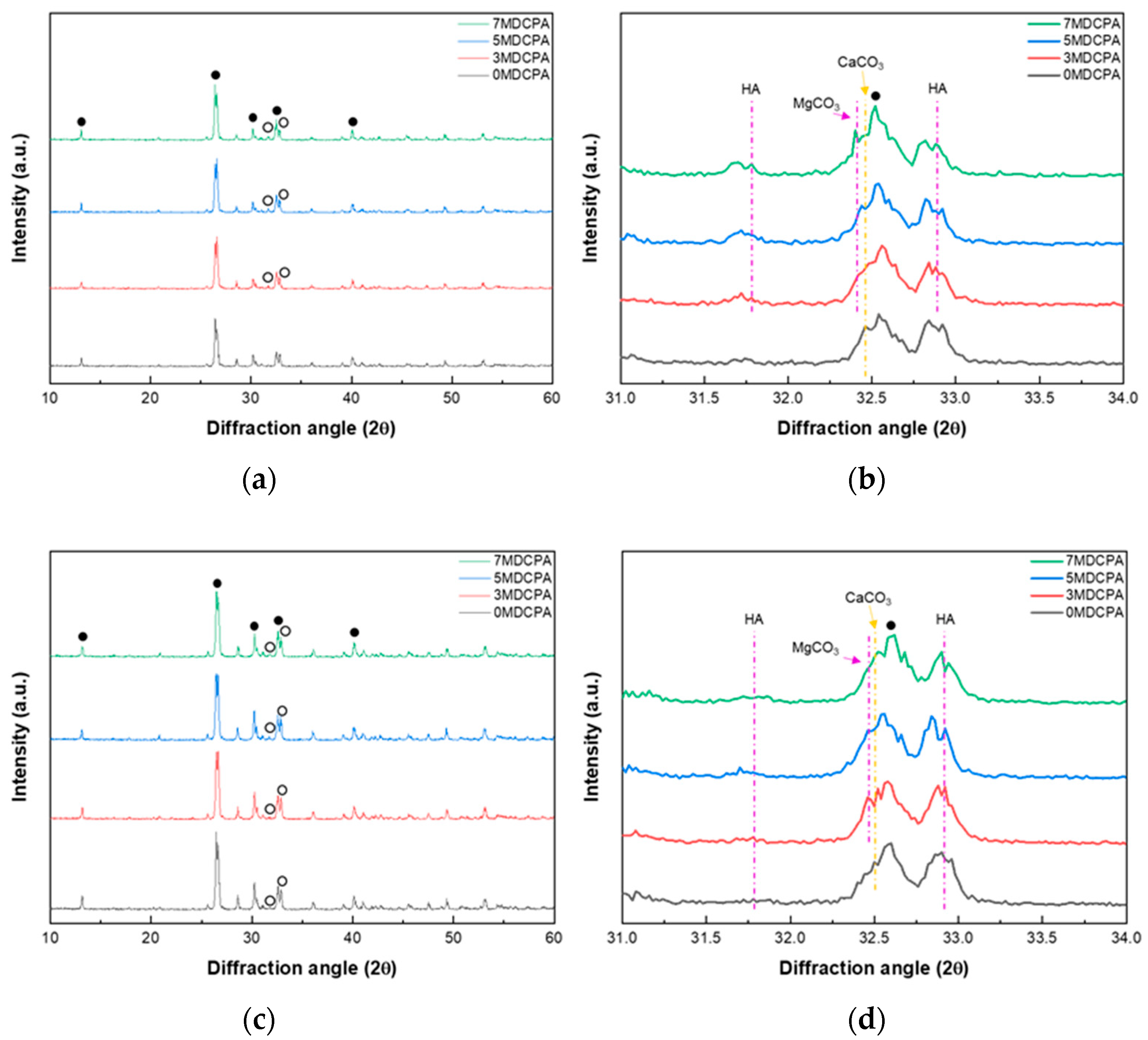
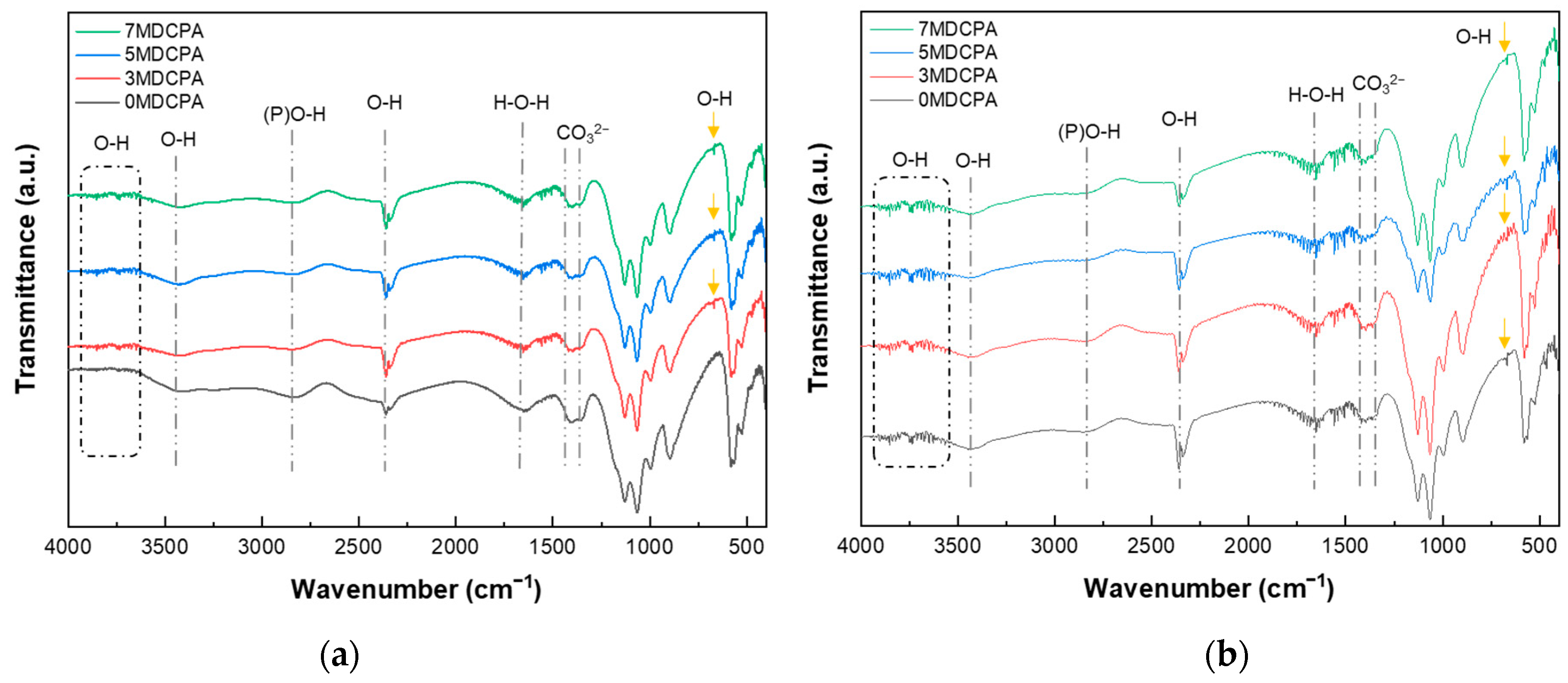
3.2. In Vitro Mineralization Behavior
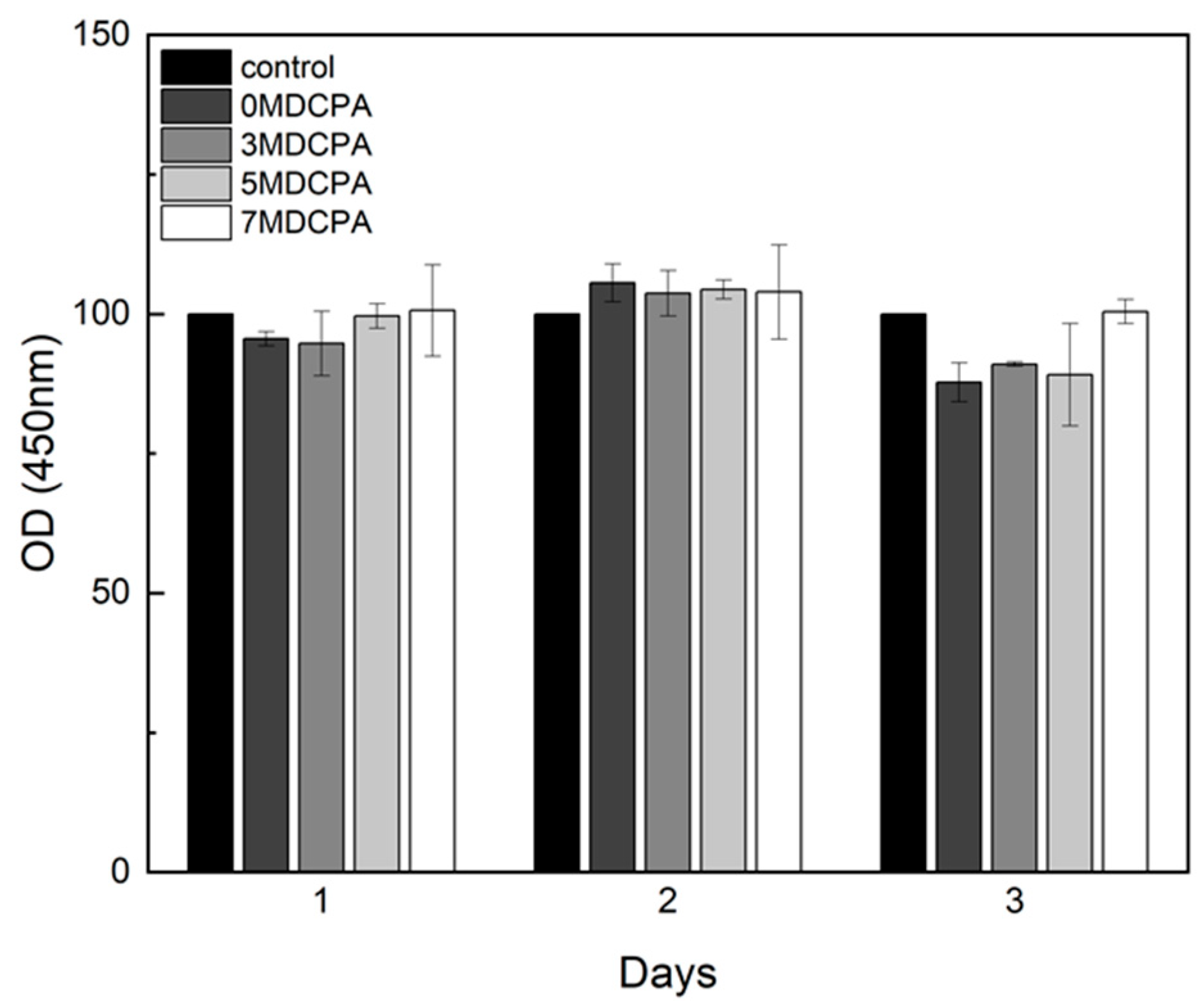
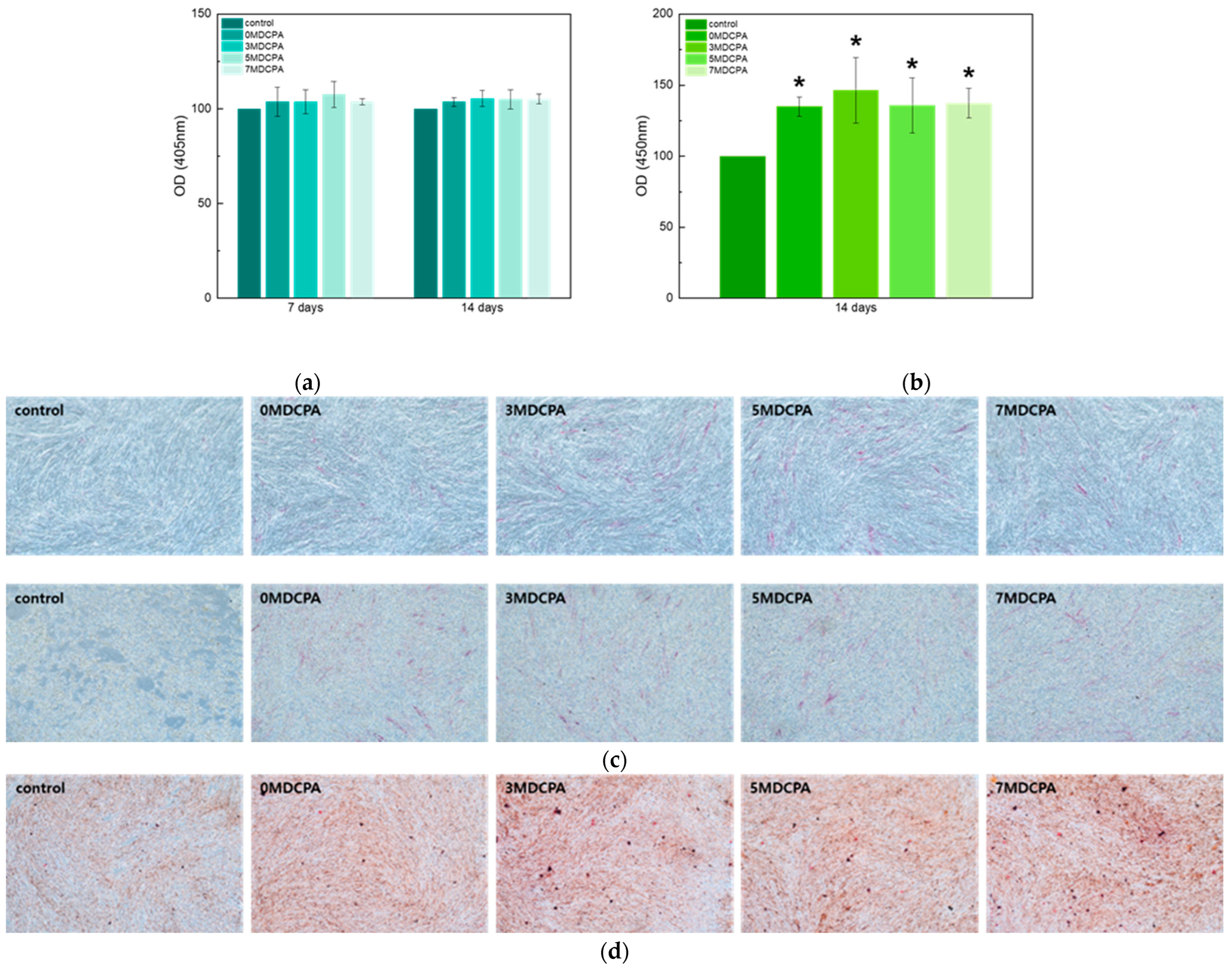
3.3. Cytotoxicity and Osteo/Odontogenic Differentiation Behavior
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenwald, A.S.; Boden, S.D.; Barrack, R.L.; Bostrom, M.P.; Goldberg, V.M.; Yaszemski, M.J.; Heim, C.S. The evolving role of bone-graft substitutes. In Proceedings of the American Academy of Orthopaedic Surgeons 77th Annual Meeting, New Orleans, LA, USA, 9–13 March 2010. [Google Scholar]
- Driessens, F. The Mineral in Bone, Dentin and Tooth Enamel. Bull. Soc. Chim. Belg. 1980, 89, 663–689. [Google Scholar] [CrossRef]
- Cengiz, B.; Gokce, Y.; Yildiz, N.; Aktas, Z.; Calimli, A. Synthesis and characterization of hydroxyapatite nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2008, 322, 29–33. [Google Scholar] [CrossRef]
- Dumitrescu, C.R.; Neacsu, I.A.; Surdu, V.A.; Nicoara, A.I.; Iordache, F.; Trusca, R.; Ciocan, L.T.; Ficai, A.; Andronescu, E. Nano-hydroxyapatite vs. Xenografts: Synthesis, characterization, and in vitro behavior. Nanomaterials 2021, 11, 2289. [Google Scholar] [CrossRef] [PubMed]
- Harun, W.; Asri, R.; Alias, J.; Zulkifli, F.; Kadirgama, K.; Ghani, S.; Shariffuddin, J. A comprehensive review of hydroxyapatite-based coatings adhesion on metallic biomaterials. Ceram. Int. 2018, 44, 1250–1268. [Google Scholar] [CrossRef]
- Szcześ, A.; Hołysz, L.; Chibowski, E. Synthesis of hydroxyapatite for biomedical applications. Adv. Colloid Interface Sci. 2017, 249, 321–330. [Google Scholar] [CrossRef]
- Bohner, M.; Gbureck, U.; Barralet, J. Technological issues for the development of more efficient calcium phosphate bone cements: A critical assessment. Biomaterials 2005, 26, 6423–6429. [Google Scholar] [CrossRef]
- Tonino, A.J.; van der Wal, B.C.H.; Heyligers, I.C.; Grimm, B. Bone remodeling and hydroxyapatite resorption in coated primary hip prostheses. Clin. Orthop. Relat. Res. 2009, 467, 478–484. [Google Scholar] [CrossRef]
- Tamimi, F.; Sheikh, Z.; Barralet, J. Dicalcium phosphate cements: Brushite and monetite. Acta Biomater. 2012, 8, 474–487. [Google Scholar] [CrossRef]
- Wang, C.-H.; Mutalik, C.; Yougbaré, S.; Teng, N.-C.; Kuo, T.-R. Calcium Phosphate nanoclusters for the repair of tooth enamel erosion. Nanomaterials 2022, 12, 1997. [Google Scholar] [CrossRef]
- Suzuki, O. Octacalcium phosphate (OCP)-based bone substitute materials. Jpn. Dent. Sci. Rev. 2013, 49, 58–71. [Google Scholar] [CrossRef]
- Torres, J.; Tamimi, I.; Cabrejos-Azama, J.; Tresguerres, I.; Alkhraisat, M.; López-Cabarcos, E.; Hernández, G.; Tamimi, F. Monetite granules versus particulate autologous bone in bone regeneration. Ann. Anat.-Anat. Anz. 2015, 200, 126–133. [Google Scholar]
- Idowu, B.; Cama, G.; Deb, S.; Di Silvio, L. In vitro osteoinductive potential of porous monetite for bone tissue engineering. J. Tissue Eng. 2014, 5, 2041731414536572. [Google Scholar] [PubMed]
- Wang, X.; Zhang, H.; Yu, X.; Mo, X.; Gao, J.; Hu, Y.; Min, J.; Ding, Q.; Fan, Y.; Jiang, W. Effects of water on cold-sintered highly dense dicalcium phosphate anhydrous bioceramic using its hydrate. J. Am. Ceram. Soc. 2024, 107, 4631–4640. [Google Scholar]
- Cerroni, L.; Filocamo, R.; Fabbri, M.; Piconi, C.; Caropreso, S.; Condò, S. Growth of osteoblast-like cells on porous hydroxyapatite ceramics: An in vitro study. Biomol. Eng. 2002, 19, 119–124. [Google Scholar] [PubMed]
- Hench, L.L.; Wilson, J. Surface-active biomaterials. Science 1984, 226, 630–636. [Google Scholar]
- Arcos, D.; Vallet-Regí, M. Substituted hydroxyapatite coatings of bone implants. J. Mater. Chem. B 2020, 8, 1781–1800. [Google Scholar]
- Adawy, A.; Diaz, R. Probing the structure, cytocompatibility, and antimicrobial efficacy of silver-, strontium-, and zinc-doped monetite. ACS Appl. Bio Mater. 2022, 5, 1648–1657. [Google Scholar]
- Suchanek, W.L.; Byrappa, K.; Shuk, P.; Riman, R.E.; Janas, V.F.; TenHuisen, K.S. Preparation of magnesium-substituted hydroxyapatite powders by the mechanochemical–hydrothermal method. Biomaterials 2004, 25, 4647–4657. [Google Scholar]
- Wolf, F.I.; Cittadini, A. Chemistry and biochemistry of magnesium. Mol. Asp. Med. 2003, 24, 3–9. [Google Scholar] [CrossRef]
- Nabiyouni, M.; Ren, Y.; Bhaduri, S.B. Magnesium substitution in the structure of orthopedic nanoparticles: A comparison between amorphous magnesium phosphates, calcium magnesium phosphates, and hydroxyapatites. Mater. Sci. Eng. C 2015, 52, 11–17. [Google Scholar]
- Bommala, V.K.; Krishna, M.G.; Rao, C.T. Magnesium matrix composites for biomedical applications: A review. J. Magnes. Alloys 2019, 7, 72–79. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Lu, T.; He, F.; Ye, J. Preparation and properties of porous calcium phosphate ceramic microspheres modified with magnesium phosphate surface coating for bone defect repair. Ceram. Int. 2024, 50, 7514–7527. [Google Scholar] [CrossRef]
- Kannan, S.; Goetz-Neunhoeffer, F.; Neubauer, J.; Pina, S.; Ferreira, J.M.F. Synthesis and structural characterization of strontium-and magnesium-co-substituted β-tricalcium phosphate. Acta Biomater. 2010, 6, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-W.; Lee, H.-S.; Kim, D.-H.; Jin, H.-H.; Hwang, K.-H.; Lee, J.K.; Park, H.-C.; Yoon, S.-Y. In situ synthesis of magnesium-substituted biphasic calcium phosphate and in vitro biodegradation. Mater. Res. Bull. 2012, 47, 2506–2512. [Google Scholar] [CrossRef]
- Devi, K.B.; Lalzawmliana, V.; Saidivya, M.; Kumar, V.; Roy, M.; Nandi, S.K. Magnesium phosphate bioceramics for bone tissue engineering. Chem. Rec. 2022, 22, e202200136. [Google Scholar] [CrossRef]
- Yoo, K.-H.; Kim, Y.-I.; Yoon, S.-Y. Physicochemical and biological properties of mg-doped calcium silicate endodontic cement. Materials 2021, 14, 1843. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef]
- ISO 10993–12: 2012; Biological Evaluation of Medical Devices—Part 12: Specimen Preparation and Reference Materials. International Organization for Standardization: Geneva, Switzerland, 2012.
- Yoo, K.-H.; Kim, Y.; Kim, Y.-I.; Bae, M.-K.; Yoon, S.-Y. Lithium doped biphasic calcium phosphate: Structural analysis and osteo/odontogenic potential in vitro. Front. Bioeng. Biotechnol. 2022, 10, 993126. [Google Scholar] [CrossRef]
- Eshtiagh-Hosseini, H.; Houssaindokht, M.R.; Chahkandhi, M.; Youssefi, A. Preparation of anhydrous dicalcium phosphate, DCPA, through sol–gel process, identification and phase transformation evaluation. J. Non-Cryst. Solids 2008, 354, 3854–3857. [Google Scholar] [CrossRef]
- Yoshida, K.; Hyuga, H.; Kondo, N.; Kita, H.; Sasaki, M.; Mitamura, M.; Hashimoto, K.; Toda, Y. Substitution model of monovalent (Li, Na, and K), divalent (Mg), and trivalent (Al) metal ions for β-tricalcium phosphate. J. Am. Ceram. Soc. 2006, 89, 688–690. [Google Scholar] [CrossRef]
- Catti, M.; Ferraris, G.; Filhol, A. Hydrogen bonding in the crystalline state. CaHPO4 (monetite), P1¯ or P1? A novel neutron diffraction study. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1977, 33, 1223–1229. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, L.; Gbureck, U.; Bhaduri, S.B.; Sikder, P. Monetite, an important calcium phosphate compound–Its synthesis, properties and applications in orthopedics. Acta Biomater. 2021, 127, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.W.; Cruickshank, D.W.J. The crystal structures of two calcium orthophosphates: CaHPO4 and Ca (H2PO4)2·H2O. Z. Krist.-Cryst. Mater. 1961, 116, 101–125. [Google Scholar] [CrossRef]
- Denne, W.A.; Jones, D.W. Neutron diffraction investigation of the hydrogen positions in the crystal structure of monetite, CaHPO4. J. Cryst. Mol. Struct. 1971, 1, 347–354. [Google Scholar] [CrossRef]
- Dickens, B.; Bowen, J.; Brown, W. A refinement of the crystal structure of CaHPO4 (synthetic monetite). Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 797–806. [Google Scholar] [CrossRef]
- Catti, M.; Ferraris, G.; Mason, S.A. Low-temperature ordering of hydrogen atoms in CaHPO4 (monetite): X-ray and neutron diffraction study at 145 K. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1980, 36, 254–259. [Google Scholar] [CrossRef]
- Rasskazova, L.A.; Zhuk, I.V.; Korotchenko, N.M.; Brichkov, A.S.; Chen, Y.-W.; Paukshtis, E.A.; Ivanov, V.K.; Kurzina, I.A.; Kozik, V.V. Synthesis of magnesium-and silicon-modified hydroxyapatites by microwave-assisted method. Sci. Rep. 2019, 9, 14836. [Google Scholar] [CrossRef]
- Tortet, L.; Gavarri, J.R.; Nihoul, G.; Dianoux, A. Study of protonic mobility in CaHPO4 2H2O (brushite) and CaHPO4 (monetite) by infrared spectroscopy and neutron scattering. J. Solid State Chem. 1997, 132, 6–16. [Google Scholar] [CrossRef]
- Petrov, I.; Šoptrajanov, B.; Fuson, N.; Lawson, J.R. Infra-red investigation of dicalcium phosphates. Spectrochim. Acta Part A Mol. Spectrosc. 1967, 23, 2637–2646. [Google Scholar] [CrossRef]
- Jillavenkatesa, A.; Condrate Sr, R.A. The infrared and Raman spectra of β-and α-tricalcium phosphate (Ca3 (PO4)2). Spectrosc. Lett. 1998, 31, 1619–1634. [Google Scholar] [CrossRef]
- Xu, J.; Butler, I.S.; Gilson, D.F. FT-Raman and high-pressure infrared spectroscopic studies of dicalcium phosphate dihydrate (CaHPO4·2H2O) and anhydrous dicalcium phosphate (CaHPO4). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1999, 55, 2801–2809. [Google Scholar] [CrossRef]
- Boanini, E.; Pagani, S.; Tschon, M.; Rubini, K.; Fini, M.; Bigi, A. Monetite vs. Brushite: Different influences on bone cell response modulated by strontium functionalization. J. Funct. Biomater. 2022, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Mozafari, M.; Banijamali, S.; Baino, F.; Kargozar, S.; Hill, R.G. Calcium carbonate: Adored and ignored in bioactivity assessment. Acta Biomater. 2019, 91, 35–47. [Google Scholar] [PubMed]
- Meejoo, S.; Maneeprakorn, W.; Winotai, P. Phase and thermal stability of nanocrystalline hydroxyapatite prepared via microwave heating. Thermochim. Acta 2006, 447, 115–120. [Google Scholar]
- Destainville, A.; Champion, E.; Bernache-Assollant, D.; Laborde, E. Synthesis, characterization and thermal behavior of apatitic tricalcium phosphate. Mater. Chem. Phys. 2003, 80, 269–277. [Google Scholar]
- Yang, J.; Fatima, K.; Zhou, X.; He, C. Meticulously engineered three-dimensional-printed scaffold with microarchitecture and controlled peptide release for enhanced bone regeneration. Biomater. Transl. 2024, 5, 69–83. [Google Scholar]
- Wei, J.; Baptista-Hon, D.T.; Wang, Z.; Li, G.; Herrler, T.; Dai, C.; Liu, K.; Yu, B.; Chen, X.; Yang, M.; et al. Bioengineered human tissue regeneration and repair using endogenous stem cells. Cell Rep. Med. 2023, 4, 101156. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples ID | Mg (mol%) |
|---|---|
| 0MDCPA | 0 |
| 3MDCPA | 3 |
| 5MDCPA | 5 |
| 7MDCPA | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Bae, J.-S.; Kim, Y.-I.; Yoo, K.-H.; Yoon, S.-Y. Synthesis, Characterization, and Biological Performances of Magnesium-Substituted Dicalcium Phosphate Anhydrous. Materials 2024, 17, 4605. https://doi.org/10.3390/ma17184605
Lee J, Bae J-S, Kim Y-I, Yoo K-H, Yoon S-Y. Synthesis, Characterization, and Biological Performances of Magnesium-Substituted Dicalcium Phosphate Anhydrous. Materials. 2024; 17(18):4605. https://doi.org/10.3390/ma17184605
Chicago/Turabian StyleLee, Jiyu, Jong-Seong Bae, Yong-Il Kim, Kyung-Hyeon Yoo, and Seog-Young Yoon. 2024. "Synthesis, Characterization, and Biological Performances of Magnesium-Substituted Dicalcium Phosphate Anhydrous" Materials 17, no. 18: 4605. https://doi.org/10.3390/ma17184605