
Al(SO4)(OH)·5H2O Stemming from Complexation of Aluminum Sulfate with Water-Soluble Ternary Copolymer and further Stabilized by Silica Gel as Effective Admixtures for Enhanced Mortar Cementing
 , , and
, , and
Abstract
1. Introduction
2. Experimental
2.1. Starting Materials
2.2. Instruments for Measuring Setting Time of Cement Paste and Mechanical Strength of Mortar
2.3. Instruments for Microstructural and Morphological Characterizations
2.4. Synthesis of Admixtures
2.5. Determination of Monomer Conversion of Copolymer (A1)
2.6. Measurement of Setting Time of Cement Paste
2.7. Measurement of Compressive and Flexural Strengths of Cement Mortar
3. Results and Discussion
3.1. Characterization of Admixtures
3.2. Effects of Admixtures in Cementing
3.3. Experimental Insights into the Cementing Process Facilitated by Admixture
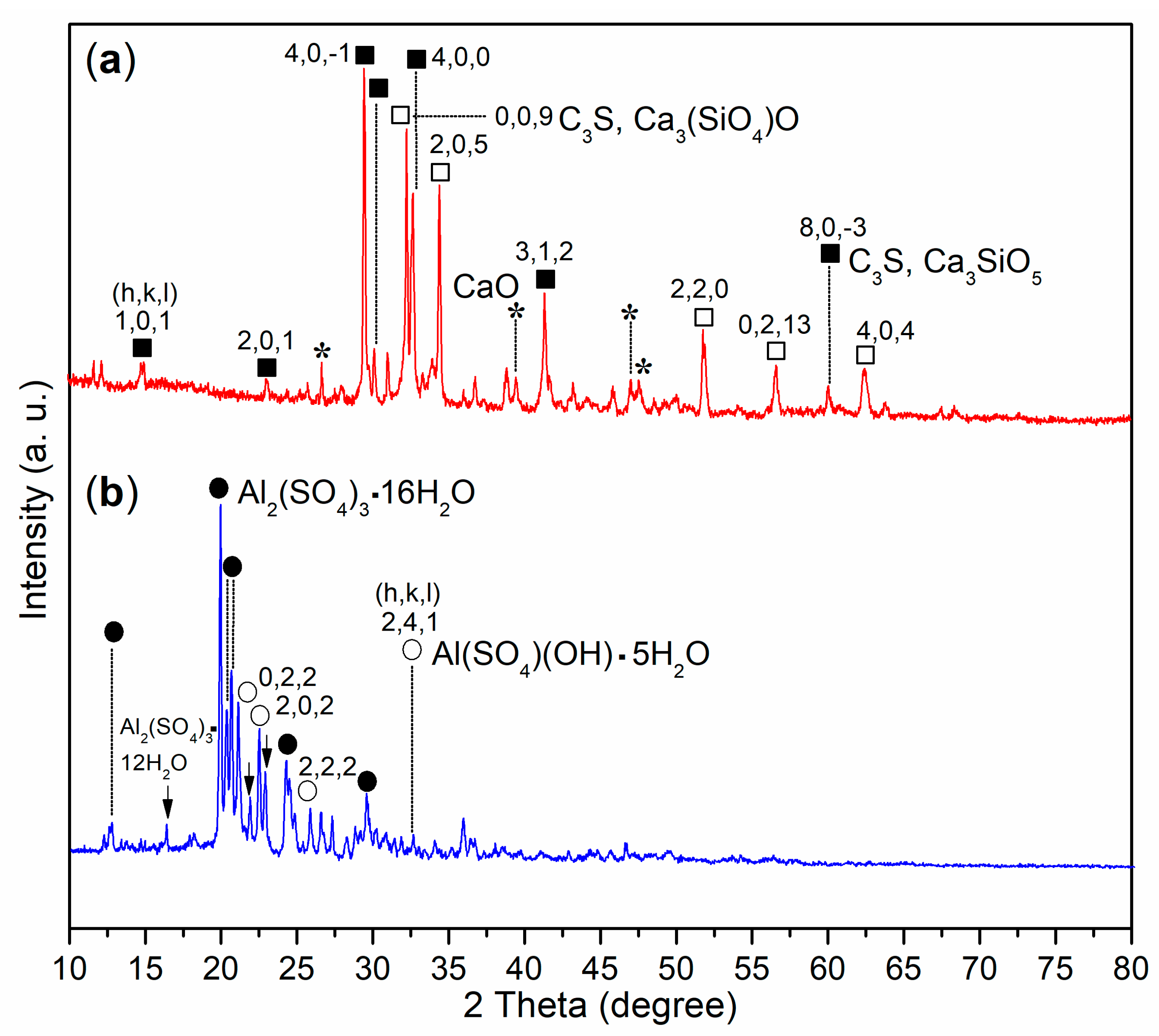
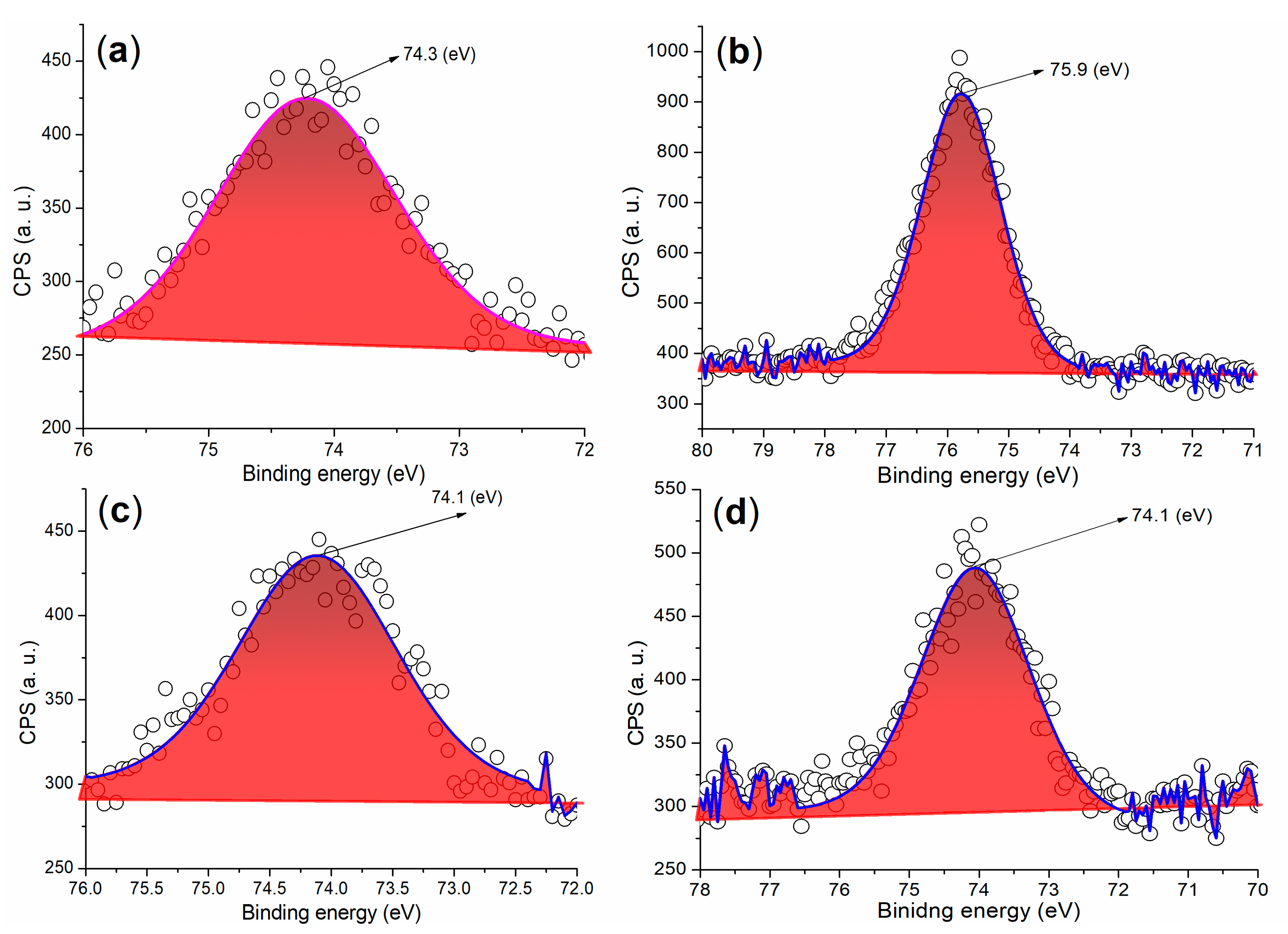
3.3.1. Elemental and Composition Analysis
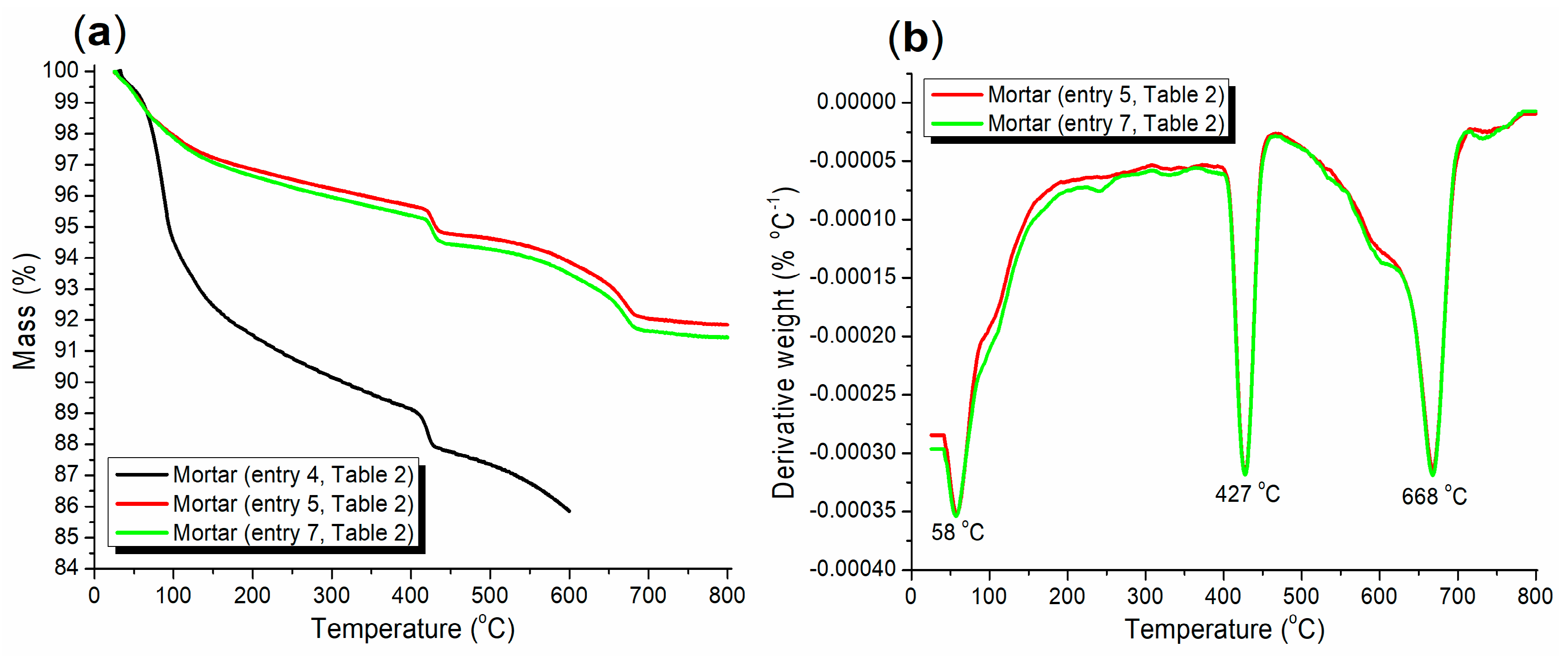
3.3.2. Morphological, Thermogravimetric, and Functional Group Analyses
3.4. Proposed Mechanism for Cement Hydration Facilitated by A2
4. Conclusions
- The admixture-blank experiment revealed significantly prolonged initial and final setting times of the cement paste, along with reduced compressive and flexural strengths of the mortar. The addition of pure aluminum sulfate and the synthesized copolymer as admixtures notably shortened the setting times and enhanced the mechanical strengths. However, these improvements still fell short of meeting the requirements specified by the Chinese standard GB/T 35159-2017.
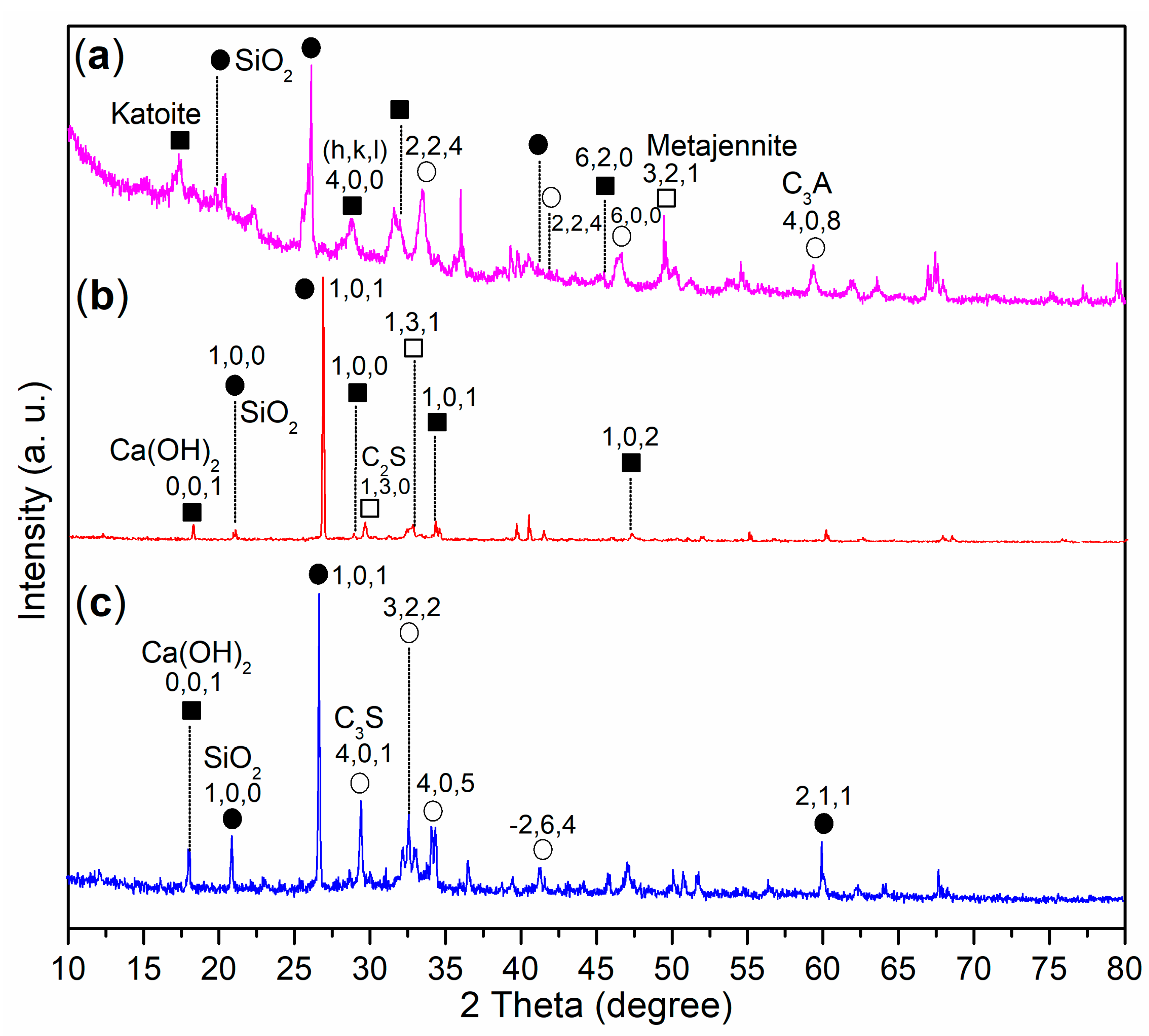
- The introduction of a complex admixture, combining copolymer and aluminum sulfate, not only reduced the setting times of cement paste but also enhanced the mechanical strengths of mortar compared to the use of aluminum sulfate alone. Characterization results revealed that this complex admixture led to the formation of katoite and metajennite in the mortar after 24 h, alongside C3A. These phases contributed to improved setting times and mechanical strength development. In contrast, the mortar treated with pure aluminum sulfate contained a significant amount of unreacted C3S, highlighting the superior activity of the complex admixture in accelerating cement setting.
- The further addition of silica gel to the complex admixture would shorten the setting times of the paste even more, slightly reduce compressive strength, but enhance flexural strength compared to the initial complex admixture. The presence of residual C2S in the mortar treated with the silica gel-incorporated complex admixture suggests that silicon components may fill the micropores and mesopores of the mortar. This process accelerated cement setting and improved flexural strength, although it slightly decreased compressive strength.
- Increasing the dosage of the silica gel-incorporated complex admixture negatively affected the setting times of the paste, while the compressive strength remained largely unchanged; however, the flexural strength decreased. This indicated that the cement hydration performance is highly sensitive to the amount of silicon components used.
- Conversely, under the same dosage of admixture, the complex formulation containing a higher amount of silica gel prolonged the setting times, decreased compressive strength, but significantly improved flexural strength compared to the formulation with less silica gel.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yuan, P.; Zhang, B.; Yang, Y.; Jiang, T.; Li, J.; Qiu, J.; He, H. Application of polymer cement repair mortar in underground engineering: A review. Case Stud. Constr. Mat. 2023, 19, e02555. [Google Scholar] [CrossRef]
- Phair, J.W. Green chemistry for sustainable cement production and use. Green Chem. 2006, 8, 763–780. [Google Scholar] [CrossRef]
- Han, J.; Wang, K.; Wang, Y.; Shi, J. Study of aluminum sulfate and anhydrite on cement hydration process. Mater. Struct. 2016, 49, 1105–1114. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, J.; Sun, S.; Zhong, K.; Shao, Q.; Xu, H.; Huang, H.; Wei, J. Dispersion, fluidity retention and retardation effect of polyacrylate-based ether superplasticizer nanomicelles in Portland cement. Constr. Build. Mater. 2021, 290, 123149. [Google Scholar] [CrossRef]
- Aslania, F.; Zhang, Y.; Manning, D.; Valdez, L.C.; Manning, N. Additive and alternative materials to cement for well plugging and abandonment: A state-of-the-art review. J. Petrol. Sci. Eng. 2022, 215, 110728. [Google Scholar] [CrossRef]
- Marchon, D.; Juilland, P.; Gallucci, E.; Frunz, L.; Flatt, R.J. Molecular and submolecular scale effects of comb−copolymers on tri−calcium silicate reactivity: Toward molecular design. J. Am. Ceram. Soc. 2017, 100, 817–841. [Google Scholar] [CrossRef]
- Cheung, J.; Jeknavorian, A.; Roberts, L.; Silva, D. Impact of admixtures on the hydration kinetics of Portland cement. Cem. Concr. Res. 2011, 41, 1289–1309. [Google Scholar] [CrossRef]
- Yang, H.; Plank, J.; Sun, Z. Investigation on the optimal chemical structure of methacrylate ester based polycarboxylate superplasticizers to be used as cement grinding aid under laboratory conditions: Effect of anionicity, side chain length and dosage on grinding efficiency, mortar workability and strength development. Constr. Build. Mater. 2019, 224, 1018–1025. [Google Scholar]
- Davoodia, S.; Al-Shargabi, M.; Wood, D.A.; Rukavishnikov, V.S. Recent advances in polymers as additives for wellbore cementing applications: A review. Fuel 2024, 357, 129692. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, J.Y.; Suh, H.W.; Cho, B.Y.; Park, W.J.; Bae, S.C. Mechanical degradation and thermal decomposition of ethylene-vinyl acetate (EVA) polymer-modified cement mortar (PCM) exposed to high-temperature. Sustainability 2019, 11, 500. [Google Scholar] [CrossRef]
- Mercante, I.; Alejandrino, C.; Ojeda, J.P.; Chini, J.; Maroto, C.; Fajardo, N. Mortar and concrete composites with recycled plastic: A review. Sci. Technol. Mater. 2018, 30, 69–79. [Google Scholar] [CrossRef]
- Brancher, L.R.; Nunes, M.F.D.O.; Grisa, A.M.C.; Pagnussat, D.T.; Zeni, M. Acoustic behavior of subfloor lightweight mortars containing micronized poly (Ethylene Vinyl Acetate) (EVA). Materials 2016, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Snoecka, D.; Goethals, W.; Hovind, J.; Trtik, P.; Van Mullem, T.; Van den Heede, P.; De Belie, N. Internal curing of cement pastes by means of superabsorbent polymers visualized by neutron tomography. Cem. Concr. Res. 2021, 147, 106528. [Google Scholar] [CrossRef]
- Mechtcherine, V.; Secrieru, E.; Schröfl, C. Effect of superabsorbent polymers (SAPs) on rheological properties of fresh cement-based mortars—Development of yield stress and plastic viscosity over time. Cem. Concr. Res. 2015, 67, 52–65. [Google Scholar] [CrossRef]
- Snoeck, D.; Steuperaert, S.; Van Tittelboom, K.; Dubruel, P.; De Belie, N. Visualization of water penetration in cementitious materials with superabsorbent polymers by means of neutron radiography. Cem. Concr. Res. 2012, 42, 1113–1121. [Google Scholar] [CrossRef]
- Mechtcherine, V.; Schröfl, C.; Wyrzykowski, M.; Gorges, M.; Cusson, D.; Margeson, J.; De Belie, N.; Snoeck, D.; Ichimiya, K.; Igarashi, S.-I.; et al. Effect of superabsorbent polymers (SAP) on the freeze-thaw resistance of concrete: Results of a RILEM interlaboratory test. Mater. Struct. 2017, 50, 1–19. [Google Scholar] [CrossRef]
- Shao, L.; Feng, P.; Liu, Q.; Chen, C.; Cai, Y.; Xu, G. A review on performance improvement and multi-functionalization of cement composites using capsules. Constr. Build. Mater. 2023, 409, 133977. [Google Scholar] [CrossRef]
- Xue, C.; Li, W.; Li, J.; Tam, V.W.Y.; Ye, G. A review study on encapsulation-based self-healing for cementitious materials. Struct. Concr. 2019, 20, 198–212. [Google Scholar] [CrossRef]
- Du, W.; Yu, J.; Gu, Y.; Li, Y.; Han, X.; Liu, Q. Preparation and application of microcapsules containing toluene-di-isocyanate for self-healing of concrete. Constr. Build. Mater. 2019, 202, 762–769. [Google Scholar] [CrossRef]
- Lv, L.; Yang, Z.; Chen, G.; Zhu, G.; Han, N.; Schlangen, E.; Xing, F. Synthesis and characterization of a new polymeric microcapsule and feasibility investigation in self-healing cementitious materials. Constr. Build. Mater. 2016, 105, 487–495. [Google Scholar] [CrossRef]
- Brooks, J.J.; Megat Johari, M.A.; Mazloom, M. Effect of admixtures on the setting times of high-strength concrete. Cem. Concr. Comp. 2000, 22, 293–301. [Google Scholar] [CrossRef]
- Rakhimova, N. Montmorillonite clays in Portland clinker-reduced, non-clinker cements, and cement composites: A review. Constr. Build. Mater. 2024, 411, 134678. [Google Scholar] [CrossRef]
- Hwang, C.L.; Shen, D.H. The effects of blast-furnace slag and fly ash on the hydration of Portland cement. Cem. Concr. Res. 1991, 21, 410–425. [Google Scholar] [CrossRef]
- Zhang, M.H.; Lastra, R.; Malhotra, V.M. Rice-husk ash paste and concrete: Some aspects of hydration and the microstructure of the interfacial zone between the aggregate and paste. Cem. Concr. Res. 1996, 26, 963–977. [Google Scholar] [CrossRef]
- Thomas, M.; Hooton, R.D.; Rogers, C.; Fournier, B. 50 years old and still going strong. Concr. Int. 2012, 34, 35. [Google Scholar]
- Rao, G.A. Investigations on the performance of silica fume-incorporated cement paste sand mortars. Cem. Concr. Res. 2003, 33, 1765–1770. [Google Scholar] [CrossRef]
- Hou, P.; Wang, X.; Zhou, X.; Cheng, X.; Shah, S.P. Regulations on the hydration, morphology, and sulfate-attack resistivity of C3A with micro/nano-silica particles. Constr. Build. Mater. 2022, 324, 126388. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, C.; Ma, Y.; Xiao, Y.; Liu, Y. Accelerators for shotcrete–Chemical composition and their effects on hydration, microstructure and properties of cement-based materials. Constr. Build. Mater. 2021, 281, 122557. [Google Scholar] [CrossRef]
- Chen, L.; Nakamura, K.; Hama, T. Review on stabilization/solidification methods and mechanism of heavy metals based on OPC-based binders. J. Environ. Manag. 2023, 332, 117362. [Google Scholar] [CrossRef]
- Rincon, J.; Camarillo, R.; Martín, A. Solubility of aluminum sulfate in near-critical and supercritical water. J. Chem. Eng. Data 2012, 57, 2084–2094. [Google Scholar] [CrossRef]
- GB/T 17671-2021; Test method of cement mortar strength (ISO method). China National Standardization Administration Committee: Beijing, China, 2021.
- GB/T 10535-2014; Water treatment chemicals—Hydrolyzed polymaleic anhydride. China National Standardization Administration Committee: Beijing, China, 2014.
- JC 477-2005; Flash setting admixtures for shotcrete. China National Standardization Administration Committee: Beijing, China, 2005.
- Bednarza, S.; Wesołowska-Piętaka, A.; Konefał, R.; Świergosz, T. Persulfate initiated free-radical polymerization of itaconic acid: Kinetics, end-groups and side products. Eur. Polym. J. 2018, 106, 63–71. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.M.; Zhang, L.; Gerritsen, G.; Abbenhuis, H.C.L.; van Santen, R.A.; Li, C. Enantioselective epoxidation epoxidation of β-methylstyrene catalyzed by immobilized Mn(salen) catalysts in different mesoporous silica supports. J. Catal. 2008, 256, 226–236. [Google Scholar] [CrossRef]
- GB/T 35159-2017; Flash setting admixtures for shotcrete. China National Standardization Administration Committee: Beijing, China, 2017.
- GB/T 34231-2017; Coal. Determination of Loss on Ignition for Coal Combustion Residues. National Standardization Administration of China: Beijing, China, 2017.
- Shivani; Sharma, N.; Kumar, M.; Kumar, M. Low resistance ohmic contact of multi-metallic Mo/Al/Au stack with ultra-wide bandgap Ga2O3 thin film with post-annealing and its in-depth interface studies for next-generation high-power devices. Surf. Interfaces 2024, 46, 103937. [Google Scholar] [CrossRef]
- van den Brand, J.; Snijders, P.C.; Sloof, W.G.; Terryn, H.; de Wit, J.H.W. Acid-base characterization of aluminum oxide surfaces with XPS. J. Phys. Chem. B 2004, 108, 6017–6024. [Google Scholar] [CrossRef]
- Iatsunskyi, I.; Gottardi, G.; Micheli, V.; Canteri, R.; Coy, E.; Bechelany, M. Atomic layer deposition of palladium coated TiO2/Si nanopillars: ToF-SIMS, AES and XPS characterization study. Appl. Surf. Sci. 2021, 542, 148603. [Google Scholar] [CrossRef]
- Abbass, A.M.; Elrahman, M.A.; Sikora, P.; Strzałkowski, J.; Stephan, D.; Abdel-Gawwad, H.A. From dolomite waste to katoite-based binder: Synthesis, performance and characterization. J. Build. Eng. 2023, 75, 106971. [Google Scholar] [CrossRef]
- Zunino, F.; Scrivener, K. The influence of sulfate addition on hydration kinetics and C-S-H morphology of C3S and C3S/C3A systems. Cem. Concr. Res. 2022, 160, 106930. [Google Scholar] [CrossRef]
- Yang, Y.; Lai, Y.; Wu, K.; Wang, W.; Fang, J.; Gao, Y.; Yang, Z. Effect of Cr2O3 on the clinkerization and carbonation properties of γ-C2S. J. Build. Eng. 2024, 90, 109395. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | m (g) b | V (mL) c | X (mg g−1) d | α (%) e |
|---|---|---|---|---|
| blank | - | 20.66 | - | - |
| A1 | 0.50 | 16.52 | 66.21 | 97.16 |
| Entry a | Admixture (Dosage) b | Setting Time (min, Cement Paste) c | Compressive Strength (MPa, Mortar) d | Flexural Strength (MPa, Mortar) d | |||||
|---|---|---|---|---|---|---|---|---|---|
| Initial (IST) | Final (FST) | 6 h | 24 h | 28 d | 6 h | 24 h | 28 d | ||
| 1 | blank | 29.10 ± 0.71 e | 40.07 ± 0.85 | 0.7 ± 0.18 | 4.9 ± 0.11 | 22.3 ± 0.44 | 0.4 ± 0.02 | 2.8 ± 0.16 | 10.0 ± 0.11 |
| 2 | AS (7%) | 18.81 ± 0.92 | 36.59 ± 1.85 | 1.1 ± 0.19 | 6.1 ± 0.12 | 24.7 ± 0.51 | 0.8 ± 0.10 | 2.9 ± 0.11 | 11.6 ± 0.09 |
| 3 | A1 (7%) | 26.53 ± 0.16 | 35.69 ± 0.17 | 0.9 ± 0.02 | 6.1 ± 0.10 | 23.6 ± 0.31 | 0.4 ± 0.01 | 2.9 ± 0.25 | 11.8 ± 0.27 |
| 4 | A2 (7%) | 4.65 ± 0.10 | 8.33 ± 0.31 | 1.6 ± 0.03 | 10.4 ± 0.20 | 26.9 ± 0.17 | 0.8 ± 0.02 | 3.6 ± 0.15 | 12.9 ± 0.11 |
| 5 | A3a (7%) | 1.00 ± 0.02 | 1.95 ± 0.04 | 1.3 ± 0.03 | 10.0 ± 0.13 | 23.9 ± 0.05 | 1.3 ± 0.05 | 4.9 ± 0.32 | 13.9 ± 0.08 |
| 6 | A3a (8%) | 1.36 ± 0.11 | 2.19 ± 0.02 | 1.2 ± 0.01 | 9.9 ± 0.20 | 24.0 ± 0.23 | 1.6 ± 0.07 | 5.8 ± 0.26 | 12.7 ± 0.31 |
| 7 | A3b (7%) | 2.21 ± 0.17 | 3.07 ± 0.10 | 1.1 ± 0.05 | 7.9 ± 0.16 | 23.2 ± 0.49 | 1.5 ± 0.06 | 6.3 ± 0.20 | 16.9 ± 0.08 |
| Entry a | R28 (%) b |
|---|---|
| 5 | 107 |
| 7 | 104 |
| Composition | CaO | SiO2 | Al2O3 | Fe2O3 | SO3 | MgO | K2O | Na2O | Ignition loss b |
|---|---|---|---|---|---|---|---|---|---|
| Content (wt.%) | 61.06 | 18.02 | 6.01 | 3.76 | 4.55 | 1.69 | 1.25 | 0.33 | 3.33 |
| Entry | C (1s) | O (1s) | S (2p) | Si (2p) | K (2p) | Ca (2p) or N (1s) | Al (2p) |
|---|---|---|---|---|---|---|---|
| Cement a | 284.80 (34.92) b | 530.80 (32.27) | 168.80 (2.84) | 100.80 (7.08) | 292.80 (8.79) | 346.80 (11.74) | 73.80 (2.35) |
| A2 | 284.80 (59.72) | 532.80 (28.26) | 169.80 (7.31) | - c | - | 400.80 (1.27) d | 74.80 (3.44) |
| 4 (24 h) e | 284.80 (30.49) | 530.80 (38.53) | 167.80 (1.18) | 101.80 (8.67) | 292.80 (7.68) | 346.80 (11.70) | 73.80 (1.55) |
| 5 (24 h) f | 284.80 (33.02) | 531.80 (35.57) | 168.80 (3.11) | 101.80 (6.67) | 292.80 (8.32) | 346.80 (10.68) | 74.80 (2.07) |
| 7 (24 h) g | 284.80 (33.13) | 531.80 (37.05) | 168.80 (3.04) | 101.80 (6.28) | 293.0 (8.10) | 346.80 (12.15) | 74.30 (0.25) |
| Entry b | Si (wt.%) | Al (wt.%) | S (wt.%) | Mg (wt.%) | Na (wt.%) |
|---|---|---|---|---|---|
| 5 | 24.47 | 1.41 | 0.69 | 0.25 | 0.16 |
| 7 | 24.24 | 1.74 | 0.80 | 0.32 | 0.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Z.; Bibi, Z.; Chaudhary, S.; Jia, Q.; Li, X.; Sun, Y. Al(SO4)(OH)·5H2O Stemming from Complexation of Aluminum Sulfate with Water-Soluble Ternary Copolymer and further Stabilized by Silica Gel as Effective Admixtures for Enhanced Mortar Cementing. Materials 2024, 17, 4762. https://doi.org/10.3390/ma17194762
Song Z, Bibi Z, Chaudhary S, Jia Q, Li X, Sun Y. Al(SO4)(OH)·5H2O Stemming from Complexation of Aluminum Sulfate with Water-Soluble Ternary Copolymer and further Stabilized by Silica Gel as Effective Admixtures for Enhanced Mortar Cementing. Materials. 2024; 17(19):4762. https://doi.org/10.3390/ma17194762
Chicago/Turabian StyleSong, Zhiyuan, Zainab Bibi, Sidra Chaudhary, Qinxiang Jia, Xiaoyong Li, and Yang Sun. 2024. "Al(SO4)(OH)·5H2O Stemming from Complexation of Aluminum Sulfate with Water-Soluble Ternary Copolymer and further Stabilized by Silica Gel as Effective Admixtures for Enhanced Mortar Cementing" Materials 17, no. 19: 4762. https://doi.org/10.3390/ma17194762
APA StyleSong, Z., Bibi, Z., Chaudhary, S., Jia, Q., Li, X., & Sun, Y. (2024). Al(SO4)(OH)·5H2O Stemming from Complexation of Aluminum Sulfate with Water-Soluble Ternary Copolymer and further Stabilized by Silica Gel as Effective Admixtures for Enhanced Mortar Cementing. Materials, 17(19), 4762. https://doi.org/10.3390/ma17194762