
High-Entropy Diborides—Silicon Carbide Composites by Reactive and Non-Reactive Spark Plasma Sintering: A Comparative Study



 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
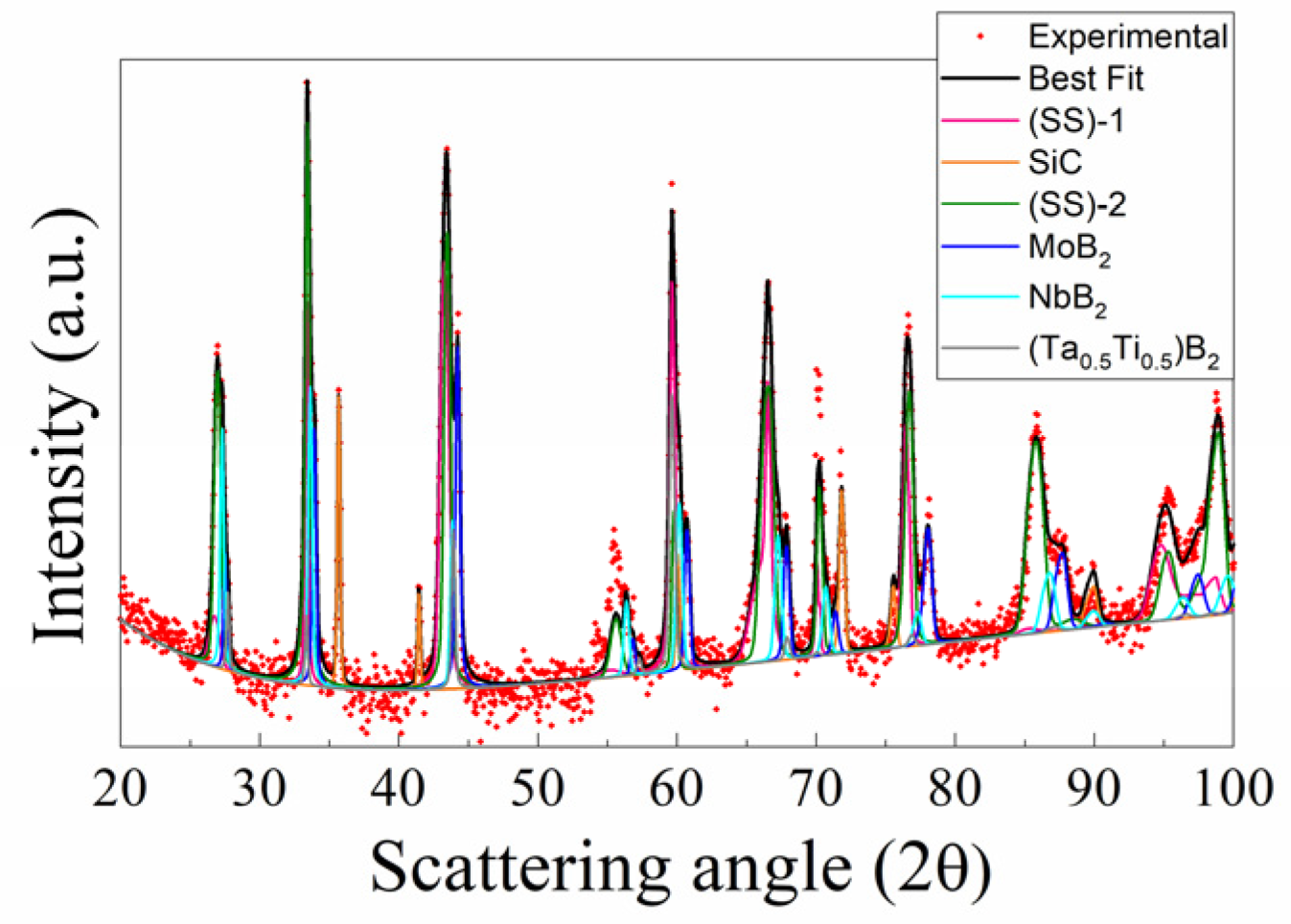
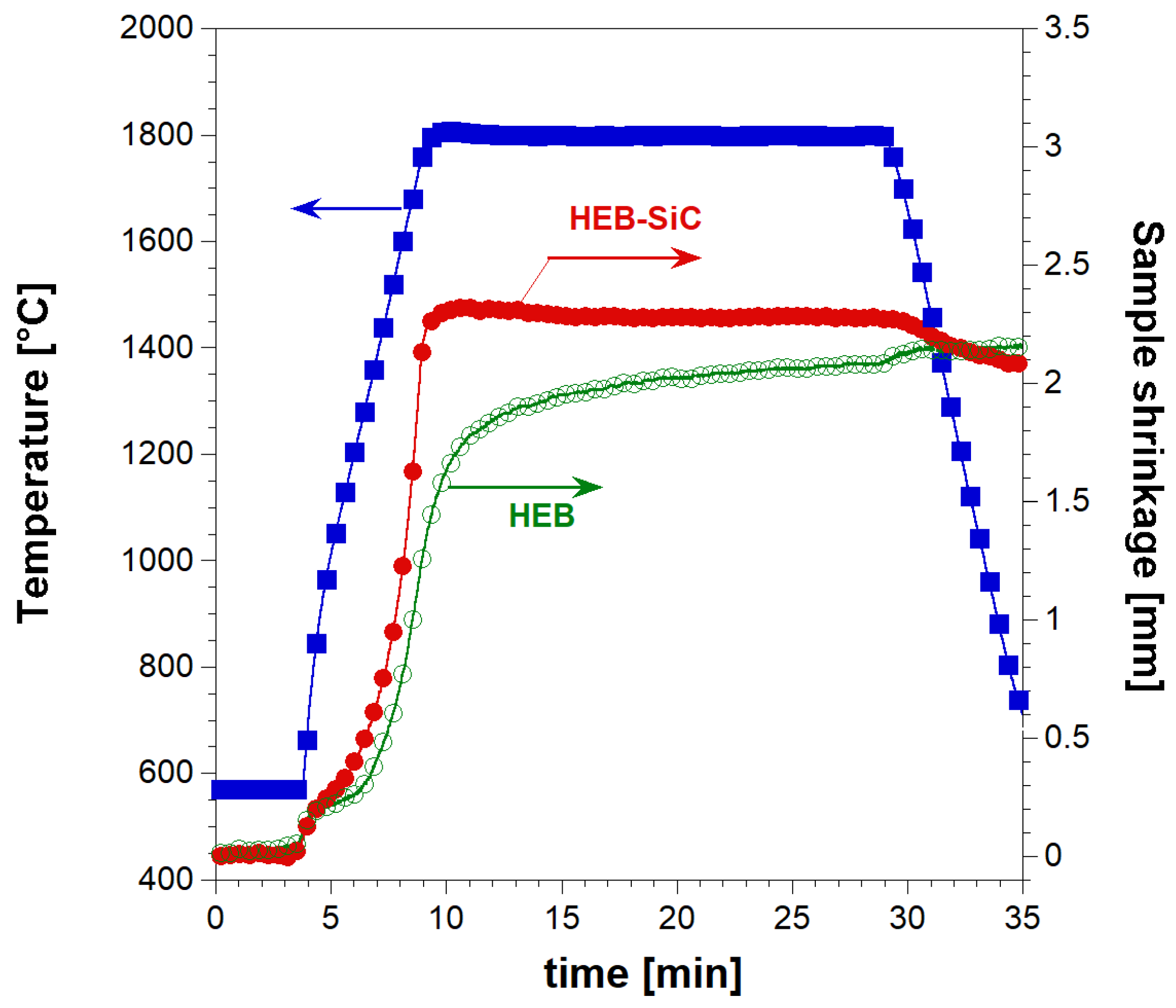
3.1. Reactive Spark Plasma Sintering Route
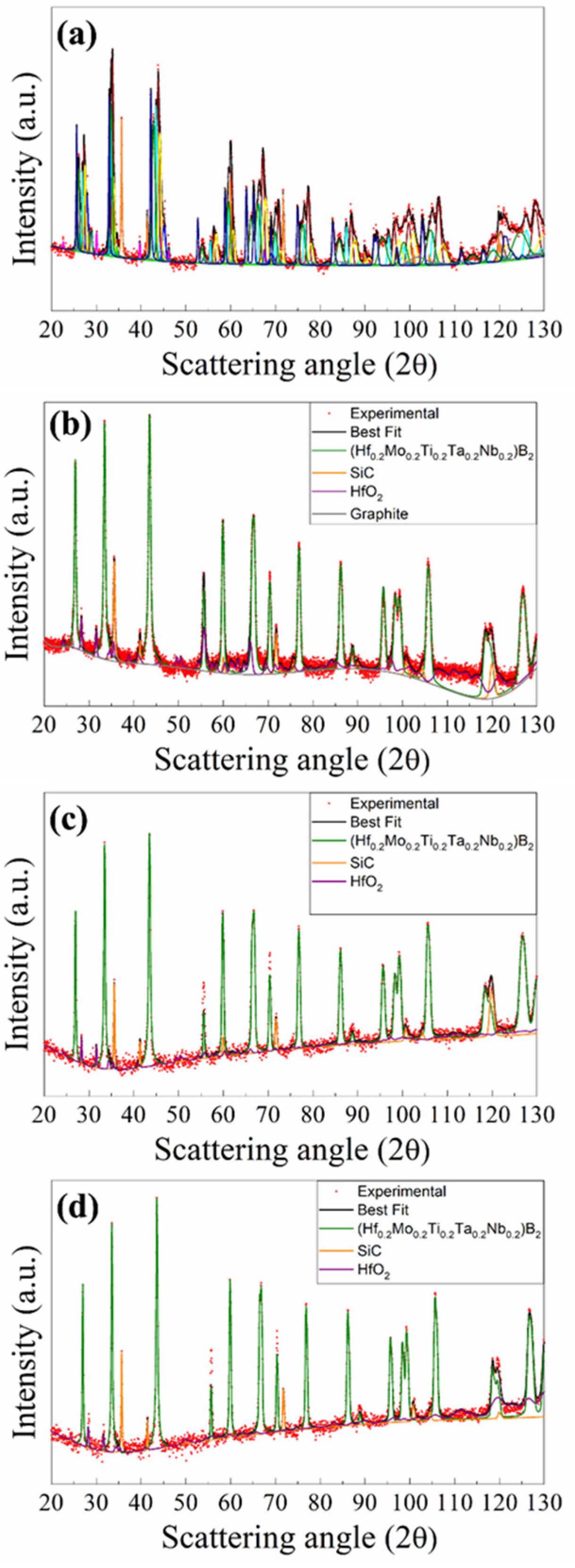
3.2. SHS–SPS Route
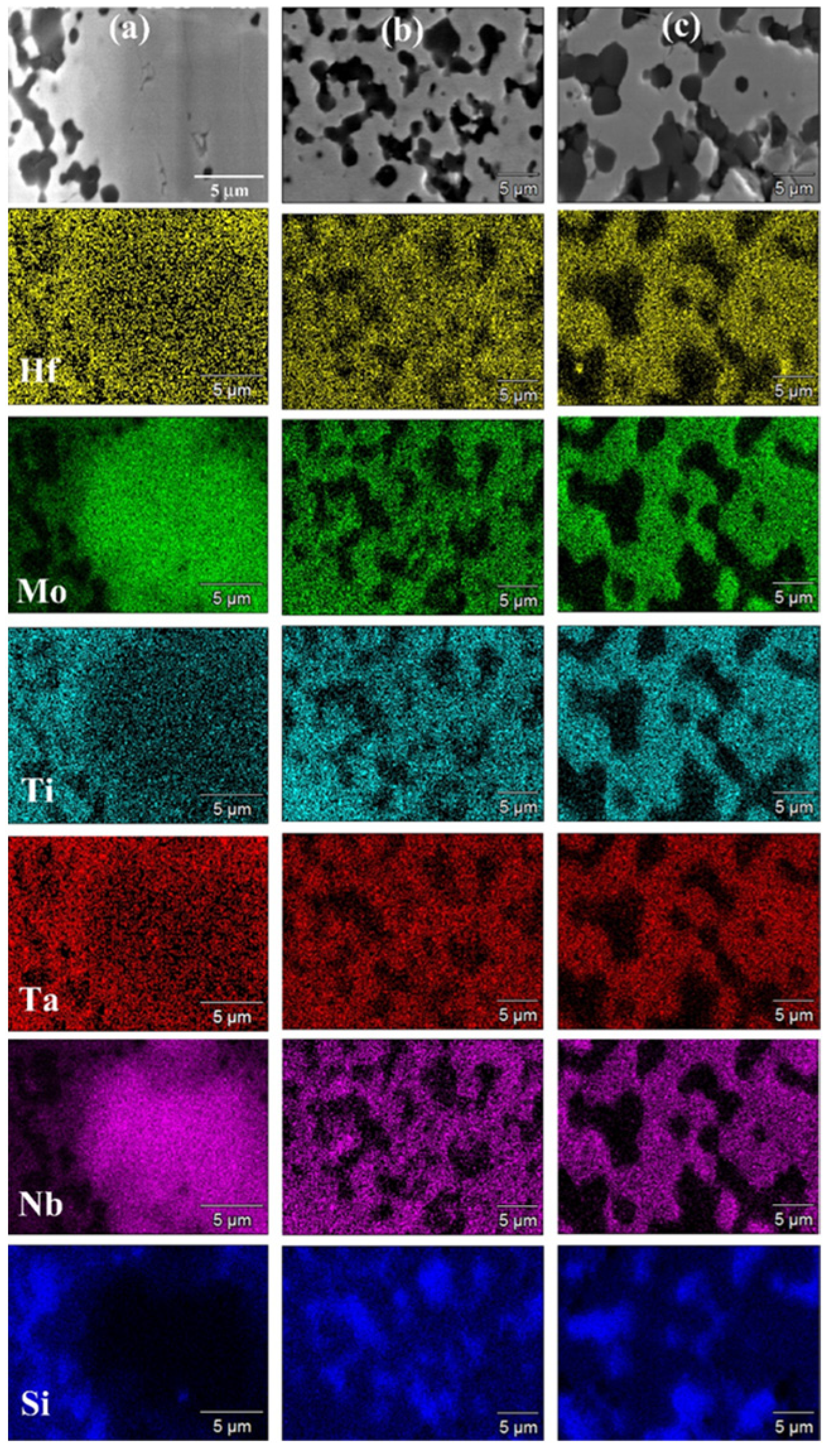
3.2.1. The (Hf0.2Mo0.2Ti0.2Ta0.2Nb0.2)B2–SiC System
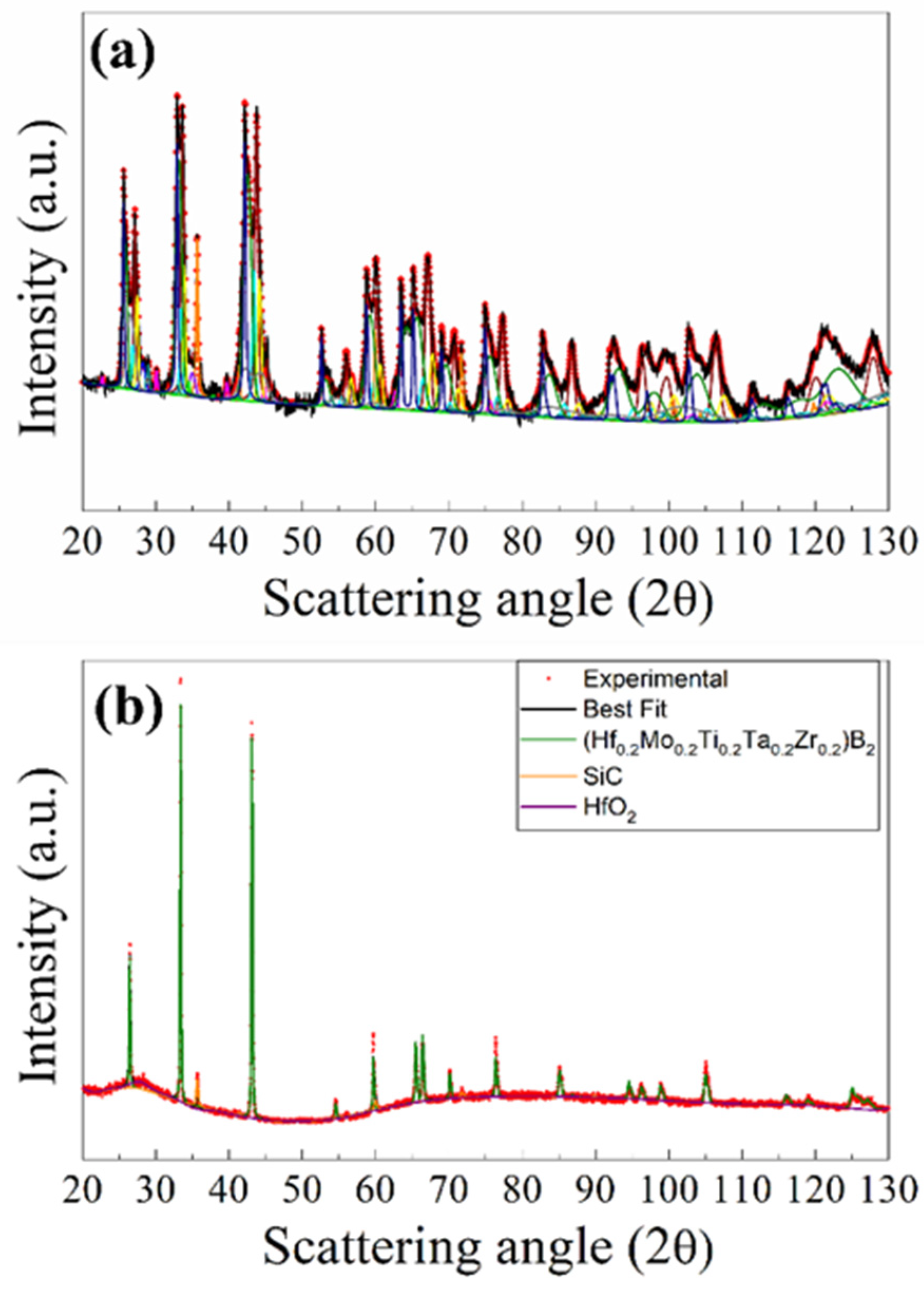
3.2.2. The (Hf0.2Mo0.2Ti0.2Ta0.2Zr0.2)B2–SiC System
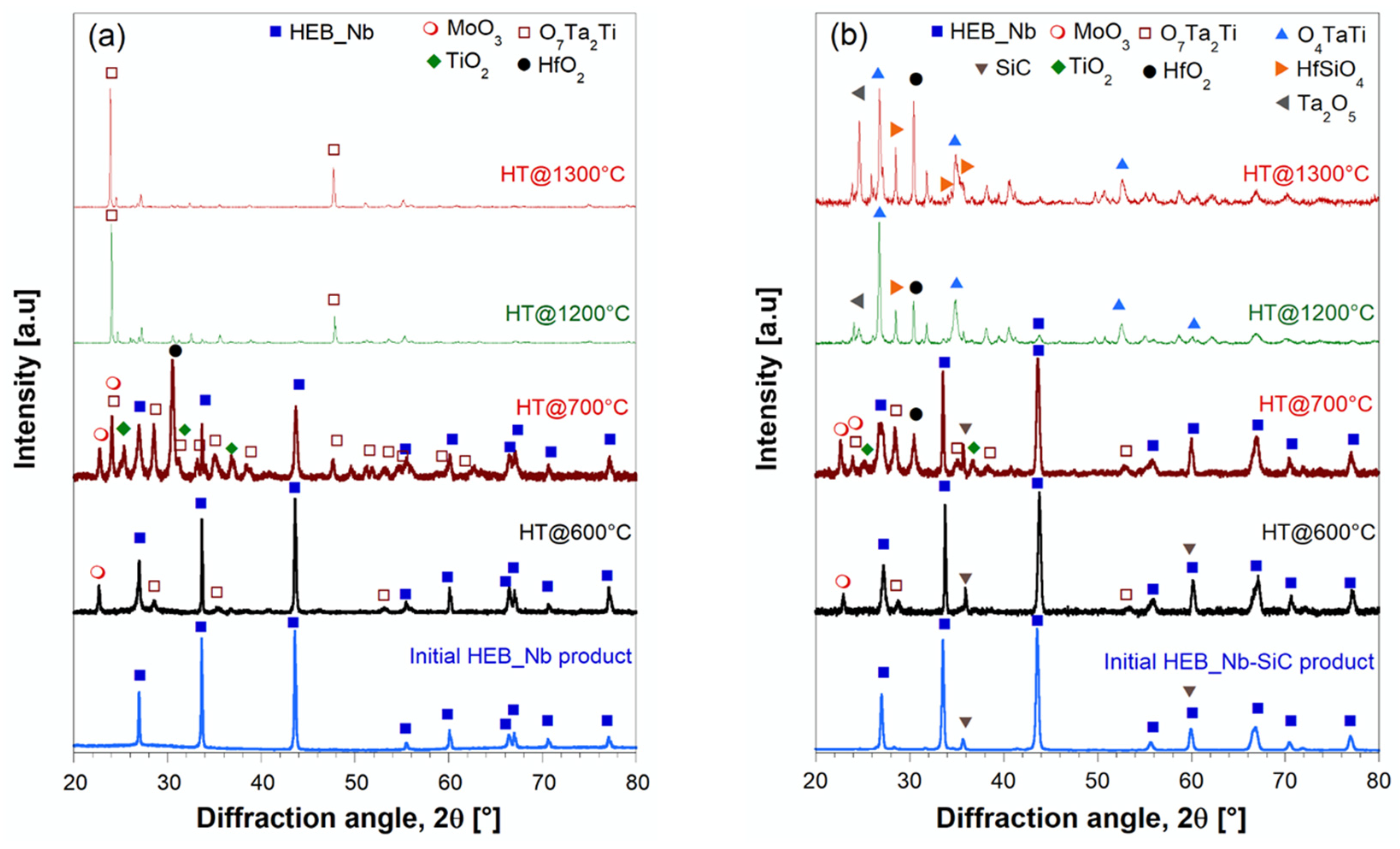
3.3. Oxidation Behavior
3.4. Mechanical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fahrenholtz, W.G.; Wuchina, E.J.; Lee, W.E.; Zhou, Y. Ultra-High Temperature Ceramics: Materials for Extreme Environment Applications, 1st ed.; Wiley-American Ceramic Society: Hoboken, NJ, USA, 2014; pp. 1–441. [Google Scholar]
- Fahrenholtz, W.G.; Hilmas, G.E.; Talmy, I.G.; Zaykoski, J.A. Refractory diborides of zirconium and hafnium. J. Am. Ceram. Soc. 2007, 90, 1347–1364. [Google Scholar] [CrossRef]
- Licheri, R.; Orrù, R.; Musa, C.; Cao, G. Efficient technologies for the fabrication of dense TaB2-based ultra-high-temperature ceramics. ACS Appl. Mater. Interfaces 2010, 2, 2206–2212. [Google Scholar] [CrossRef]
- Sebakhy, K.O.; Vitale, G.; Hassan, A.; Pereira-Almao, P. New Insights into the Kinetics of Structural Transformation and Hydrogenation Activity of Nano-crystalline Molybdenum Carbide. Catal. Lett. 2018, 148, 904–923. [Google Scholar] [CrossRef]
- Sciti, D.; Balbo, A.; Bellosi, A. Oxidation behaviour of a pressureless sintered HfB2-MoSi2 composite. J. Eur. Ceram. Soc. 2009, 29, 1809–1815. [Google Scholar] [CrossRef]
- Musa, C.; Licheri, R.; Orrù, R.; Cao, G. Synthesis, sintering, and oxidative behavior of HfB2-HfSi2 ceramics. Ind. Eng. Chem. Res. 2014, 53, 9101–9108. [Google Scholar] [CrossRef]
- Aguirre, T.G.; Lamm, B.W.; Cramer, C.L.; Mitchell, D.J. Zirconium-diboride silicon-carbide composites: A review. Ceram. Int. 2022, 48, 7344–7361. [Google Scholar] [CrossRef]
- Gild, J.; Zhang, Y.; Harrington, T.; Jiang, S.; Hu, T.; Quinn, M.C.; Mellor, W.M.; Zhou, N.; Vecchio, K.; Luo, J. High-Entropy Metal Diborides: A New Class of High-Entropy Materials and a New Type of Ultrahigh Temperature Ceramics. Sci. Rep. 2016, 6, 37946. [Google Scholar] [CrossRef]
- Mayrhofer, P.H.; Kirnbauer, A.; Ertelthaler, P.; Koller, C.M. High-entropy ceramic thin films; A case study on transition metal diborides. Scr. Mater. 2018, 149, 93–97. [Google Scholar] [CrossRef]
- Tallarita, G.; Licheri, R.; Garroni, S.; Barbarossa, S.; Orrù, R.; Cao, G. High-entropy transition metal diborides by reactive and non-reactive spark plasma sintering: A comparative investigation. J. Eur. Ceram. Soc. 2020, 40, 942–952. [Google Scholar] [CrossRef]
- Feng, L.; Fahrenholtz, W.G.; Brenner, D.W. High-Entropy Ultra-High-Temperature Borides and Carbides: A New Class of Materials for Extreme Environments. Annu. Rev. Mater. Res. 2021, 51, 165–185. [Google Scholar] [CrossRef]
- Wang, F.; Monteverde, F.; Cui, B. Will high-entropy carbides and borides be enabling materials for extreme environments? Int. J. Extrem. Manuf. 2023, 5, 022002. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, Z.B.; Sun, S.K.; Guo, W.M.; Chen, Q.S.; Qiu, J.X.; Plucknett, K.; Lin, H.T. Microstructure and mechanical properties of high-entropy borides derived from boro/carbothermal reduction. J. Eur. Ceram. Soc. 2019, 39, 3920–3924. [Google Scholar] [CrossRef]
- Murchie, A.C.; Watts, J.L.; Fahrenholtz, W.G.; Hilmas, G.E. Room-temperature mechanical properties of a high-entropy diboride. Int. J. Appl. Ceram. Technol. 2022, 19, 2293–2299. [Google Scholar] [CrossRef]
- Guo, R.; Li, Z.; Li, L.; Liu, Y.; Zheng, R.; Ma, C. Microstructures and oxidation mechanisms of (Zr0.2Hf0.2Ta0.2Nb0.2Ti0.2)B2 high-entropy ceramic. J. Eur. Ceram. Soc. 2022, 42, 2127–2134. [Google Scholar] [CrossRef]
- Shen, X.Q.; Liu, J.X.; Li, F.; Zhang, G.J. Preparation and characterization of diboride-based high entropy (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)B2–SiC particulate composites. Ceram. Int. 2019, 45, 24508–24514. [Google Scholar] [CrossRef]
- Liu, J.X.; Shen, X.Q.; Wu, Y.; Li, F.; Liang, Y.; Zhang, G.J. Mechanical properties of hot-pressed high-entropy diboride-based ceramics. J. Adv. Ceram. 2020, 9, 503–510. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, S.K.; Guo, W.M.; Xu, L.; Zhang, W.; Lin, H.T. Optimal preparation of high-entropy boride-silicon carbide ceramics. J. Adv. Ceram. 2021, 10, 173–180. [Google Scholar] [CrossRef]
- Gong, Y.; Yang, Z.; Wei, X.; Song, S.; Ma, S. Synthesis and electromagnetic wave absorbing properties of high-entropy metal diboride-silicon carbide composite powders. J. Mater. Sci. 2022, 57, 9218–9230. [Google Scholar] [CrossRef]
- Cheng, Y.Y.; Zhou, L.; Liu, J.X.; Tan, Y.F.; Zhang, G.J. Grain growth inhibition by sluggish diffusion and Zener pinning in high-entropy diboride ceramics. J. Am. Ceram. Soc. 2023, 106, 4997–5004. [Google Scholar] [CrossRef]
- Barbarossa, S.; Orrù, R.; Garroni, S.; Licheri, R.; Cao, G. Ultra High Temperature High-Entropy Borides: Effect of Graphite Addition on Oxides Removal and Densification Behaviour. Ceram. Int. 2021, 47, 6220–6231. [Google Scholar] [CrossRef]
- Barbarossa, S.; Orrù, R.; Cannillo, V.; Iacomini, A.; Garroni, S.; Murgia, M.; Cao, G. Fabrication and Characterization of Quinary High Entropy-Ultra-High Temperature Diborides. Ceramics 2021, 4, 108–120. [Google Scholar] [CrossRef]
- Musa, C.; Orrù, R.; Sciti, D.; Silvestroni, L.; Cao, G. Synthesis, consolidation and characterization of monolithic and SiC whiskers reinforced HfB2 ceramics. J. Eur. Ceram. Soc. 2013, 33, 603–614. [Google Scholar] [CrossRef]
- Cincotti, A.; Licheri, R.; Locci, A.M.; Orrù, R.; Cao, G. A review on combustion synthesis of novel materials: Recent experimental and modeling results. J. Chem. Technol. Biotechnol. 2003, 78, 122–127. [Google Scholar] [CrossRef]
- Matthews, F.L.; Rawlings, R. Composite Materials: Engineering and Science, 1st ed.; Woodhead Publishing: Sawston, UK, 1999; pp. 1–480. [Google Scholar]
- Shackelford, J.F.; Alexander, W. CRC Materials Science and Engineering Handbook, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2001; pp. 1–1980. [Google Scholar]
- Lutterotti, L.; Ceccato, R.; Dal Maschio, R.; Pagani, E. Quantitative analysis of silicate glass in ceramic materials by the Rietveld method. Mater. Sci. Forum 1998, 87, 278–281. [Google Scholar] [CrossRef]
- Ponton, C.B.; Rawlings, R.D. Vickers Indentation Fracture Toughness Test. Part 1: Review of Literature and Formulation of Standardised Indentation Toughness Equations. Mater. Sci. Technol. 1989, 5, 865–872. [Google Scholar] [CrossRef]
- Ponton, C.B.; Rawlings, R.D. Vickers Indentation Fracture Toughness Test. Part 2: Application and Critical Evaluation of Standardised Indentation Toughness Equations. Mater. Sci. Technol. 1989, 5, 961–976. [Google Scholar] [CrossRef]
- Licheri, R.; Orrù, R.; Locci, A.M.; Cao, G. Efficient synthesis/sintering routes to obtain fully dense ultra-high-temperature ceramics (UHTCs). Ind. Eng. Chem. Res. 2007, 46, 9087–9096. [Google Scholar] [CrossRef]
- Orrù, R.; Cao, G. Comparison of reactive and non-reactive spark plasma sintering routes for the fabrication of monolithic and composite Ultra High Temperature Ceramics (UHTC) materials. Materials 2013, 6, 1566–1583. [Google Scholar] [CrossRef]
- Licheri, R.; Musa, C.; Orrù, R.; Cao, G. Influence of the heating rate on the in situ synthesis and consolidation of ZrB2 by reactive Spark Plasma Sintering. J. Eur. Ceram. Soc. 2015, 35, 1129–1137. [Google Scholar] [CrossRef]
- Licheri, R.; Orrù, R.; Musa, C.; Locci, A.M.; Cao, G. Consolidation via spark plasma sintering of HfB2/SiC and HfB2/HfC/SiC composite powders obtained by self-propagating high-temperature synthesis. J. Alloys Compd. 2009, 478, 572–578. [Google Scholar] [CrossRef]
- Gild, J.; Wright, A.; Quiambao-Tomko, K.; Qin, M.; Tomko, J.A.; bin Hoque, M.S.; Braun, J.L.; Bloomfield, B.; Martinez, D.; Harrington, T.; et al. Thermal conductivity and hardness of three single-phase high-entropy metal diborides fabricated by borocarbothermal reduction and spark plasma sintering. Ceram. Int. 2020, 46, 6906–6913. [Google Scholar] [CrossRef]
- Crystallography Open Database. Available online: https://www.crystallography.net/cod/index.php (accessed on 15 December 2023).
- Licheri, R.; Orrù, R.; Musa, C.; Cao, G. Combination of SHS and SPS Techniques for fabrication of fully dense ZrB2-ZrC-SiC composites. Mater. Lett. 2008, 62, 432–435. [Google Scholar] [CrossRef]
- Grazulis, S.; Chateigner, D.; Downs, R.T.; Yokochi, A.T.; Quiros, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P.; Le Bail, A. Crystallography Open Database—An open-access collection of crystal structures. J. Appl. Crystallogr. 2009, 42, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Tallarita, G.; Licheri, R.; Garroni, S.; Orrù, R.; Cao, G. Novel processing route for the fabrication of bulk high-entropy metal diborides. Scr. Mater. 2019, 158, 100–104. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactant | Vendor (Code) | Particle Size (µm) | Purity (%) |
|---|---|---|---|
| Hf | Alfa Aesar, Karlsruhe, Germany (10201) | <44 | 99.6 |
| Mo | Alfa Aesar (10031) | <149 | ≥99 |
| Ta | Alfa Aesar (00337) | <44 | 99.8 |
| Ti | Aldrich, St. Louis, MI, USA (26.849-6) | <149 | 99.7 |
| Nb | Alfa Aesar (010275) | <44 | 99.8 |
| Zr | Thermo Scientific, Waltham, MA, USA (00847) | 2–3 | - |
| Si | Aldrich (21561-9) | <44 | 99% |
| B4C | Alfa Aesar (40504) | 1–7 | 99.4 |
| System ID | d10 (µm) | d50 (µm) | d90 (µm) | d[4,3] (µm) |
|---|---|---|---|---|
| HEB_Nb–SiC | 0.155 ± 0.025 | 0.825 ± 0.215 | 6.645 ± 1.055 | 2.185 ± 0.395 |
| HEB_Zr–SiC | 0.13 ± 0.01 | 0.61 ± 0.04 | 5.715 ± 0.435 | 1.88 ± 0.15 |
| System | Fabrication Method | Sintering Conditions (TD, td, P) | ρ (%) | HV (Load, N) (GPa) | KIC (MPa m1/2) | Reference |
|---|---|---|---|---|---|---|
| (Hf0.2Mo0.2Ti0.2Ta0.2Nb0.2)–0 vol.%SiC | SHS–SPS | 1950 °C/2 min/20 MPa | 97.4 ± 0.3 | 27.0 ± 1.3 (1) | 2.61 ± 0.17 | This work |
| (Hf0.2Mo0.2Ti0.2Ta0.2Nb0.2)–27.7 vol.%SiC | SHS–SPS | 1800 °C/20 min/20 MPa | 97.2 ± 1.4 | 26.0 ± 1.0 (1) | 7.35 ± 0.66 | This work |
| SHS–SPS | 1900 °C/5 min/20 MPa | 98.3 ± 1.3 | 27.0 ± 1.7 (1) | 6.23 ± 0.50 | This work | |
| SHS–SPS | 1900 °C/20 min/20 MPa | 98.3 ± 1.2 | 27.0 ± 1.7 (1) | 5.36 ± 0.37 | This work | |
| (Hf0.2Mo0.2Ti0.2Ta0.2Zr0.2)B2–0 vol.%SiC | SHS–SPS | 1950 °C/20 min/20 MPa | 96.5 ± 0.7 | 25.0 ± 1.6 (1) | 2.11 ± 0.15 | This work |
| (Hf0.2Mo0.2Ti0.2Ta0.2Zr0.2)B2–27.4 vol.%SiC | SHS–SPS | 1800 °C/20 min/20 MPa | 97.7 ± 0.6 | 27.0 ± 1.5 (1) | 4.11 ± 0.32 | This work |
| (Hf0.2Mo0.2Ti0.2Ta0.2Nb0.2)–20 vol.%SiC | BR–SPS | 2000 °C/10 min/30 MPa | 99.1 ± 0.1 | 26.2 ± 1.8 (1.96) | 4.41 ± 0.21 | [18] |
| BCR–SPS | 100.0 ± 0.5 | 28.1 ± 0.9 (1.96) | 4.25 ± 0.37 | |||
| (Hf0.2Mo0.2Ti0.2Zr0.2Nb0.2)–20 vol.%SiC | BR–SPS | 2000 °C/10 min/30 MPa | 100.0 ± 0.4 | 25.8 ± 1.2 (1.96) | 4.53 ± 0.66 | [18] |
| BCR–SPS | 98.6 ± 0.2 | 29.0 ± 1.3 (1.96) | 3.80 ± 0.33 | |||
| (Hf0.2Nb0.2Ti0.2Ta0.2Zr0.2)B2–10 vol.%SiC | BCR–SPS | 1800 °C/10 min/30 MPa | 99.47 | ~20.1 (49) | ~5.00 | [20] |
| (Hf0.2Nb0.2Ti0.2Ta0.2Zr0.2)B2–20 vol.%SiC | BCR–SPS | 1800 °C/10 min/30 MPa | 99.51 | ~20.7 (49) | ~5.20 | [20] |
| BCR–HP | 1800 °C/60 min/n.r. | >99 | 24.8 ± 1.2 (1.96) | 4.85 ± 0.33 | [17] | |
| BCR–SPS | 1600–1900 °C/10 min/30 MPa | >97 | ~23 (49) | ~4.7 | [16] | |
| (Hf0.2Nb0.2Ti0.2Ta0.2Zr0.2)B2–30 vol.%SiC | BCR–SPS | 1800 °C/10 min/30 MPa | 99.73 | ~21.1 (49) | ~5.20 | [20] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pakhomova, E.; Cao, G.; Orrù, R.; Garroni, S.; Ferro, P.; Licheri, R. High-Entropy Diborides—Silicon Carbide Composites by Reactive and Non-Reactive Spark Plasma Sintering: A Comparative Study. Materials 2024, 17, 718. https://doi.org/10.3390/ma17030718
Pakhomova E, Cao G, Orrù R, Garroni S, Ferro P, Licheri R. High-Entropy Diborides—Silicon Carbide Composites by Reactive and Non-Reactive Spark Plasma Sintering: A Comparative Study. Materials. 2024; 17(3):718. https://doi.org/10.3390/ma17030718
Chicago/Turabian StylePakhomova, Ekaterina, Giacomo Cao, Roberto Orrù, Sebastiano Garroni, Paolo Ferro, and Roberta Licheri. 2024. "High-Entropy Diborides—Silicon Carbide Composites by Reactive and Non-Reactive Spark Plasma Sintering: A Comparative Study" Materials 17, no. 3: 718. https://doi.org/10.3390/ma17030718
APA StylePakhomova, E., Cao, G., Orrù, R., Garroni, S., Ferro, P., & Licheri, R. (2024). High-Entropy Diborides—Silicon Carbide Composites by Reactive and Non-Reactive Spark Plasma Sintering: A Comparative Study. Materials, 17(3), 718. https://doi.org/10.3390/ma17030718