
Structural Stability and Disorder Level of Moderately Reduced Paper-like Graphene Oxide Investigated with Micro-Raman Analysis

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
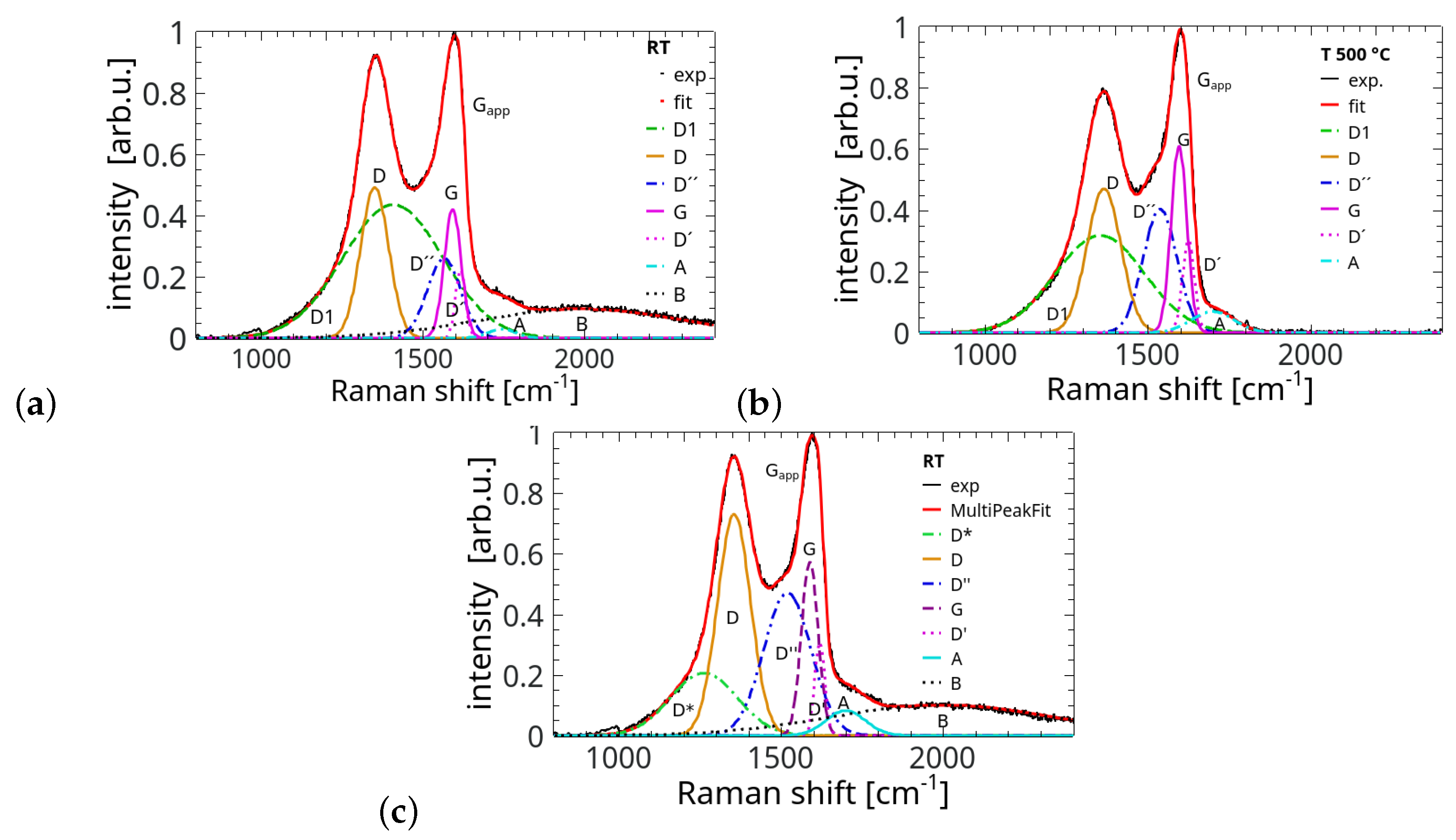
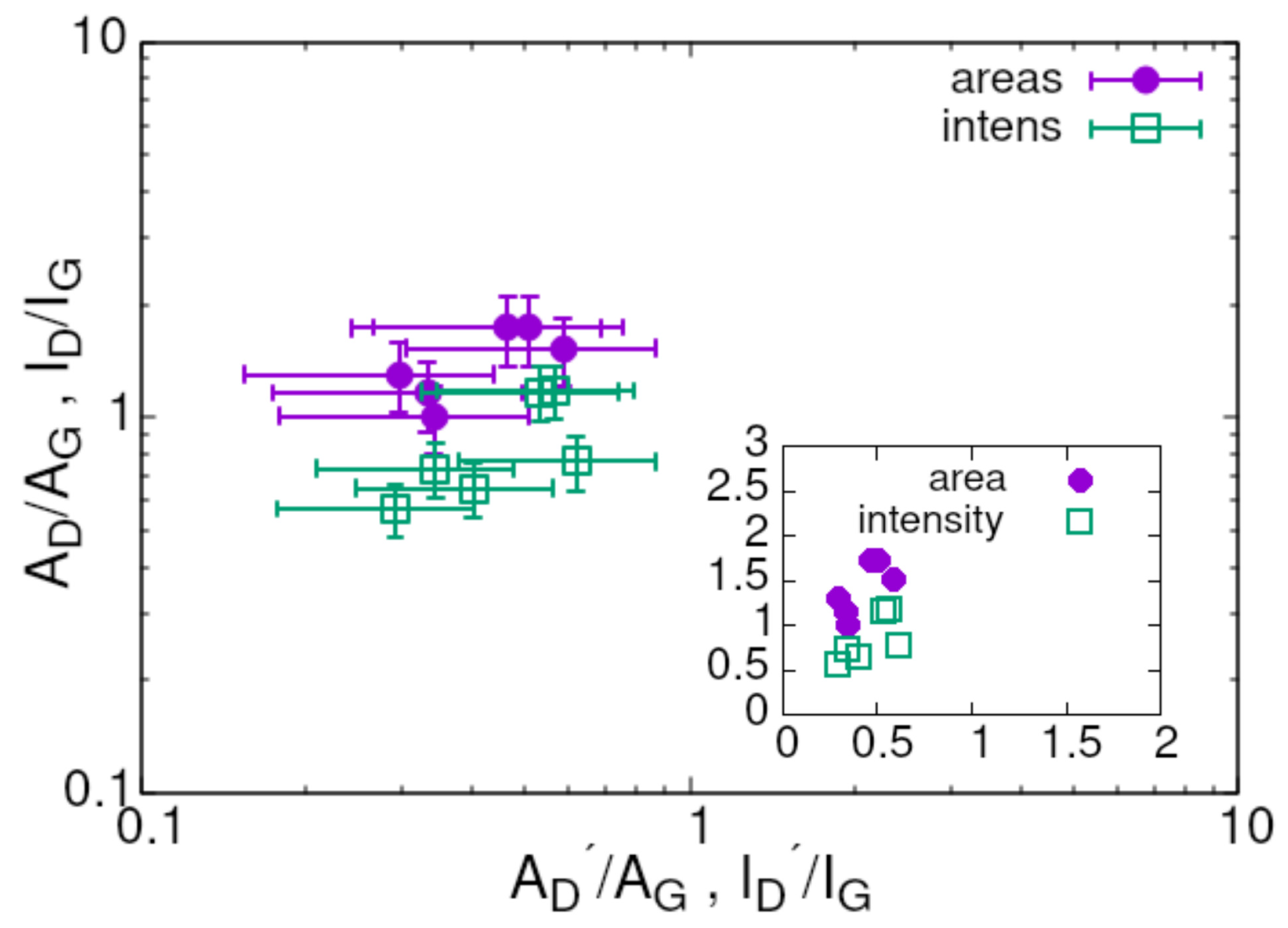
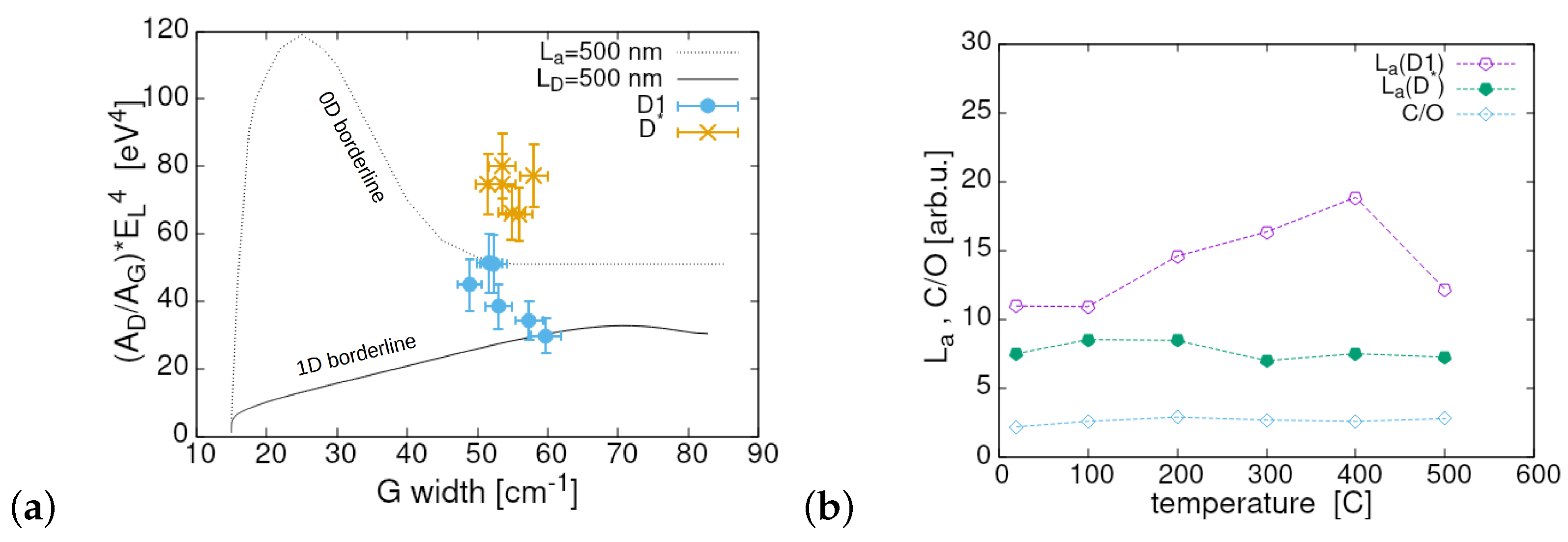
3. Results
4. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GO | graphene oxide |
| slGO | single-layer graphene oxide |
| flGO | few-layers graphene oxide |
References
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Eigler, S. Graphene Oxide: Fundamentals and Applications; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Dideikin, A.T.; Vul’, A.Y. Graphene Oxide and Derivatives: The Place in Graphene Family. Front. Phys. 2019, 6, 149. [Google Scholar] [CrossRef]
- Robinson, J.T.F.; Perkins, K.; Snow, E.S.; Wei, Z.; Sheehan, P.E. Reduced Graphene Oxide Molecular Sensors. Nano Lett. 2008, 8, 3137–3140. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Luo, B.; Zhi, L. Application of Graphene and Graphene-Based Materials in Clean Energy-Related Devices. Int. J. Energy Res. 2009, 33, 1161–1170. [Google Scholar] [CrossRef]
- Yu, D.; Dai, L. Self-Assembled Graphene/Carbon Nanotube Hybrid Films for Supercapacitors. J. Phys. Chem. Lett 2010, 1, 467–470. [Google Scholar] [CrossRef]
- Cassagneau, T.; Fendler, J.H. High Density Rechargeable Lithium-Ion Batteries Self-Asembled from Graphite Oxide Nanoplatelets and Polyelectrolytes. Adv. Mater. 1998, 10, 877–881. [Google Scholar] [CrossRef]
- Nair, R.R.; Wu, H.A.; Jayaram, P.N.; Grigorieva, I.V.; Geim, A.K. Unimpeded Permeation of Water Through Helium-Leak-Tight Graphene-Based Membranes. Science 2012, 335, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Acik, M.; Mattevi, C.; Gong, C.; Lee, G.; Cho, K.; Chhowalla, M.; Chabal, Y.J. The Role of Intercalated Water in Multilayered Graphene Oxide. ACS Nano 2010, 4, 5861–5868. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Hofmann, M.; Dresselhaus, G.; Jorio, A.; Dresselhaus, M.S. Raman spectroscopy of graphene and carbon nanotubes. Adv. Phys. 2011, 60, 413–550. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Basko, D.M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235–246. [Google Scholar] [CrossRef]
- Eckmann, A.; Felten, A.; Mishchenko, A.; Britnell, L.; Krupke, R.; Novoselov, K.S.; Casiraghi, C. Probing the Nature of Defects in Graphene by Raman Spectroscopy. Nano Lett. 2012, 12, 3925–3930. [Google Scholar] [CrossRef]
- Bagri, A.; Mattevi, C.; Acik, M.; Chabal, Y.J.; Chhowalla, M.; Shenoy, V.B. Structural evolution during reduction of chemically derived graphene oxide. Nat. Chem. 2010, 2, 581–587. [Google Scholar] [CrossRef]
- Larciprete, R.; Fabris, S.; Sun, T.; Lacovig, P.; Baraldi, A.; Lizzit, S. Dual Path Mechanism in the Thermal Reduction of Graphene Oxide. J. Amer. Chem. Soc. 2011, 133, 17315–17321. [Google Scholar] [CrossRef] [PubMed]
- Boschi, A.; Kovtun, A.; Liscio, F.; Xia, Z.; Kim, K.H.; Avila, S.L.; De Simone, S.; Mussi, V.; Barone, C.; Pagano, S.; et al. Mesoscopic 3D Charge Transport in Solution-Processed Graphene-Based Thin Films: A Multiscale Analysis. Small 2023, 19, 2303238. [Google Scholar] [CrossRef] [PubMed]
- Kaniyoor, A.; Ramaprabhu, S. A Raman spectroscopic investigation of graphite oxide derived graphene. AIP Adv. 2012, 2, 032183–032188. [Google Scholar] [CrossRef]
- Kim, S.; Zhou, S.; Hu, Y.; Acik, M.; Chabal, Y.J.; Berger, C.; de Heer, W.; Bongiorno, A.; Riedo, E. Room-temperature metastability of multilayer graphene oxide films. Nat. Mater. 2012, 11, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Fok, T.; Janulewicz, K.A.; Wachulak, P.; Bartnik, A.; Nasiłowska, B.; Kostecki, J.; Budner, B.; Fiedorowicz, H.; Djas, M.; Kuźmiuk, P.; et al. Electronic structure of multi-layered graphene oxide membrane moderately reduced in vacuum. J. Phys. Chem. Solids 2022, 164, 110623. [Google Scholar] [CrossRef]
- Kumar, P.V.; Bardhan, N.M.; Tongay, S.; Wu, J.; Belcher, A.M.; Grossman, J.C. Scalable enhancement of graphene oxide properties by thermally driven phase transformation. Nat: Chem. 2013, 6, 151–158. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X.; Bocquet, M.L. Structure and chemistry of graphene oxide in liquid water from first principle. Nat. Commun. 2020, 11, 1566. [Google Scholar] [CrossRef] [PubMed]
- Botas, C.; Botas, C.; Álvarez, P.; Blanco, C.; Santamaría, R.; Granda, M.; Gutiérrez, M.D.; Rodríguez-Reinoso, F.; Menéndez, R. Critical temperatures in the synthesis of graphene-like materials by thermal exfoliation–reduction of graphite oxide. Carbon 2013, 52, 476–485. [Google Scholar] [CrossRef]
- Lesiak, B.; Trykowski, G.; Tóth, J.; Biniak, S.; Kövér, L.; Rangam, N.; Stobinski, L.; Malolepszy, A. Chemical and structural properties of reduced graphene oxide - dependence on the reducing agent. J. Mater. Sci. 2021, 56, 3738–3754. [Google Scholar] [CrossRef]
- Singh, S.B.; Dastgheib, S.A. Physicochemical transformation of graphene oxide during heat treatment at 110–200 °C. Carbon Trends 2023, 10, 100251. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Qu, Y.; Engdahl, A.; Vajda, V.; McLoughlin, N. Ultrastructural heterogenity of carbonaceous material in ancient cherts: Investigating biosignature origin and preservation. Astrobiology 2015, 15, 825–842. [Google Scholar] [CrossRef] [PubMed]
- Claramunt, S.; Varea, A.; López-Díaz, D.; Velázquez, M.M.; Cornet, A.; Cirera, A. The Importance of Interbands on the Interpretation of the Raman Spectrum of Graphene Oxide. J. Phys. Chem. C 2015, 119, 10123–10129. [Google Scholar] [CrossRef]
- Díez-Betriu, X.; Álvarez García, S.; Botas, C.; Álvarez, P.; Sánchez-Marcos, J.; Prieto, C.; Menéndez, R.; de Andrés, A. Raman spectroscopy for the study of reduction mechanisms and optimization of conductivity in graphene oxide thin films. J. Mater. Chem. C 2013, 1, 6905–6912. [Google Scholar] [CrossRef]
- Cuesta, A.; Dhamelincourt, P.; Laureyns, J.; Martínez-Alonso, A.; Tascón, J.M.D. Raman microprobe studies on carbon materials. Carbon 1994, 12, 1523–1532. [Google Scholar] [CrossRef]
- King, A.A.K.; Davies, B.R.; Noorbehesht, N.; Newman, P.; Church, T.L.; Harris, A.T.; Razal, J.M.; Minett, A.I. A New Raman Metric for the Characterisation of Graphene Oxide and its Derivatives. Sci. Rep. 2016, 6, 19491. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C.; Robertson, J. Raman spectroscopy of amorphous, nanostructured, diamond-like carbon, and nanodiamond. Phil. Trans. R. Soc. Lond. A 2004, 362, 2477–2512. [Google Scholar] [CrossRef]
- Tamor, M.A.; Vassell, W.C. Raman “fingerprinting” of amorphous carbon films. J. Appl. Phys. 1994, 76, 3823–3830. [Google Scholar] [CrossRef]
- Merlen, A.; Buijnsters, J.G.; Pardanaud, C. A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for characterizing Defective Aromatic Carbon Solids: From Graphene to Amorphous Carbons. Coatings 2017, 7, 153. [Google Scholar] [CrossRef]
- Pardanaud, C.; Martin, C.; Roubin, P. Multiwavelength Raman spectroscopy analysis of a large sampling of disordered carbons extracted from the Tore Supra tokamak. Vib. Spectrosc. 2014, 70, 187–192. [Google Scholar] [CrossRef]
- Cançado, L.G.; Jorio, A.; Ferreira, E.M.; Stavale, F.; Achete, C.A.; Capaz, R.B.; Moutinho, M.V.D.O.; Lombardo, A.; Kulmala, T.S.; Ferrari, A.C. Quantifying Defects in Graphene via Raman Spectroscopy at Different Excitation Energies. Nano Lett. 2011, 11, 3190–3196. [Google Scholar] [CrossRef] [PubMed]
- Cançado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhães-Paniago, R.; Pimenta, M.A. General equation for the determination of the crystallite size L a of nanographite by Raman spectroscopy. Appl. Phys. Lett. 2006, 88, 163106–163109. [Google Scholar] [CrossRef]
- Jorio, A. Raman Spectroscopy in Graphene-Based Systems: Prototypes for Nanoscience and Nanometrology. ISRN Nanotechnol. 2012, 2012, 234216. [Google Scholar] [CrossRef]
- Cançado, L.G.; Kai Huang, A.; da Silva, M.G.; Ferreira, E.H.M.; Hof, F.; Kampioti, K.; Huang, K.; Penicaud, A.; Achete, C.A.; Capez, R.B.; et al. Disentangling contributions of point and line defects in the Raman spectra of graphene-related materials. 2D Mater. 2017, 4, 025039. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef]
- Shimodaira, N.; Masui, A. Raman spectroscopic investigation of activated carbon materials. J. Appl. Phys. 2002, 92, 902. [Google Scholar] [CrossRef]
- Mallet-Ladeira, P.; Puech, P.; Toulouse, C.; Cazayous, M.; Ratel-Ramond, N.; Weisbecker, P.; Vignoles, G.L.; Monthioux, M. A Raman study to obtain crystallite size of carbon materials: A better alternative to the Tuinstra–Koenig law. Carbon 2014, 80, 629–639. [Google Scholar] [CrossRef]
- Buchsteiner, A.; Lerf, A.; Pieper, J. Water Dynamics in Graphite Oxide Investigated with Neutron Scattering. J. Phys. Chem. B 2006, 110, 22328–22338. [Google Scholar] [CrossRef]
- Cross, P.C.; Burnham, J.; Leighton, P.A. The Raman Spectrum and the Structure of Water. J. Amer. Chem. Soc. 1937, 59, 1134–1147. [Google Scholar] [CrossRef]
- Senior, W.A.; Thompson, W.K. Assignment of the Infra-red and Raman Bands of Liquid Water. Nat. 1965, 205, 170. [Google Scholar] [CrossRef]
- Brooker, M.H.; Hancock, G.; Rice, B.C.; Shapter, J. Raman Frequency and Intensity Studies of Liquid H2O, H218O and D2O. J. Raman Spectrosc. 1989, 20, 683–694. [Google Scholar] [CrossRef]
- Medders, G.F.; Paesani, F. Infrared and Raman Spectroscopy of Liquid Water through “First- Principles” Many-Body Molecular Dynamics. J. Chem. Theory Comput. 2015, 11, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Fantini, C.; Cruz, E.; Jorio, A.; Terrones, M.; Terrones, H.; Van Lier, G.; Charlier, J.C.; Dresselhaus, M.S.; Saito, R.; Kim, Y.A.; et al. Resonance Raman study of linear carbon chains formed by the heat treatment of double-wall carbon nanotubes. Phys. Rev. B 2006, 73, 193408. [Google Scholar] [CrossRef]
- Nishide, D.; Dohi, H.; Wakabayashi, T.; Nishibori, E.; Aoyagi, S.; Ishida, M.; Kikuchi, S.; Kitaura, R.; Sugai, T.; Sakata, M.; et al. Single-wall carbon nanotubes encaging linear chain C10H2 polyyne molecules inside. Chem. Phys. Lett. 2006, 428, 356–360. [Google Scholar] [CrossRef]
- Lui, C.H.; Malard, L.M.; Kim, S.H.; Lantz, G.; Laverge, F.E.; Saito, R.; Heinz, T.F. Observation of Layer-Breathing Mode Vibrations in Few-Layer Graphene through Combination Raman Scattering. Nano Lett. 2012, 12, 5539–5544. [Google Scholar] [CrossRef]
- Ramos, S.L.L.M.; Pimenta, M.A.; Champi, A. Multiple-excitation study of the double-resonance Raman bands in rhombohedral graphite. Carbon 2021, 179, 683–691. [Google Scholar] [CrossRef]
- Bouhafs, C.; Pezzini, S.; Geisenhof, F.R.; Mishra, N.; Mišeikis, V.; Niu, Y.; Struzzi, C.; Weitz, R.T.; Zakharov, A.A.; Forti, S.; et al. Synthesis of large-area rhombohedral few-layer graphene by chemical vapor deposition on copper. Carbon 2021, 177, 282–290. [Google Scholar] [CrossRef]
- Colthup, N.B.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: Cambridge, MA, USA, 1990. [Google Scholar]
- Lucchese, M.M.; Stavale, F.; Ferreira, E.M.; Vilani, C.; Moutinho, M.V.D.O.; Capaz, R.B.; Achete, C.A.; Jorio, A. Quantifying ion-induced defects and Raman relaxation length in graphene. Carbon 2010, 48, 1592–1597. [Google Scholar] [CrossRef]
- Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Cançado, L.G.; Jorio, A.; Saito, R. Studying disorder in graphite-based systems by Raman spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Cançado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Speziali, N.L.; Jorio, A.; Pimenta, M.A. Measuring the degree of stacking order in graphite by Raman spectroscopy. Carbon 2008, 46, 272–275. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Production Method | T [°C] | / | Ref. | |||
|---|---|---|---|---|---|---|
| modif. Hummers/sediment. | RT | 1355/102 | 1589/52 | 1.2 (1.3) 1 | 2.2 | [18] |
| 500 | 1364/112 | 1592/49 | 0.77 (1.35) | 2.8 | [18] | |
| modif. Hummers/TPD | RT | 1364 | 1583 | 0.9 | 2.3 | [21] |
| 127 | 1364 | 1583/broad. 2 | 1.2 | 5.3 | [21] | |
| modif. Hummers | RT | 1337/119 | 1594/82 | 0.99 | [22] | |
| commerc. carbon powder | RT | 1343–1348 | 1590–1598 | 0.93 | [23] | |
| 140–200 | 1343–1348 | 1590–1598 | 1 | [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janulewicz, K.A.; Fok, T.; Bartosewicz, B.; Bartnik, A.; Fiedorowicz, H.; Wachulak, P. Structural Stability and Disorder Level of Moderately Reduced Paper-like Graphene Oxide Investigated with Micro-Raman Analysis. Materials 2024, 17, 877. https://doi.org/10.3390/ma17040877
Janulewicz KA, Fok T, Bartosewicz B, Bartnik A, Fiedorowicz H, Wachulak P. Structural Stability and Disorder Level of Moderately Reduced Paper-like Graphene Oxide Investigated with Micro-Raman Analysis. Materials. 2024; 17(4):877. https://doi.org/10.3390/ma17040877
Chicago/Turabian StyleJanulewicz, Karol Adam, Tomasz Fok, Bartosz Bartosewicz, Andrzej Bartnik, Henryk Fiedorowicz, and Przemysław Wachulak. 2024. "Structural Stability and Disorder Level of Moderately Reduced Paper-like Graphene Oxide Investigated with Micro-Raman Analysis" Materials 17, no. 4: 877. https://doi.org/10.3390/ma17040877
APA StyleJanulewicz, K. A., Fok, T., Bartosewicz, B., Bartnik, A., Fiedorowicz, H., & Wachulak, P. (2024). Structural Stability and Disorder Level of Moderately Reduced Paper-like Graphene Oxide Investigated with Micro-Raman Analysis. Materials, 17(4), 877. https://doi.org/10.3390/ma17040877