1. Introduction
Thermoplastic polymers are deployed in a multitude of applications ranging from single-use packaging to their use in the automotive and aerospace industries. Engineering polymers such as polyamides (PAs) have gained widespread use in the automotive sector due to their desirable physical properties, such as high toughness, good thermal stability, mechanical strength, and impact resistance [
1,
2]. Polyamide 6 (PA6) is particularly popular due to its ductility, strength, and hot-forming properties [
3]. Compared with PA6, which has a molecular backbone based on six carbon atoms, PA12 has longer methyl (CH
2) chains and a lower melting point [
4]. Its unique combination of properties, such as high chemical resistance, high pressure and impact resistance, and relatively low moisture absorption, make it an attractive material for various applications [
4]. However, it should be noted that polyamides are more prone to moisture absorption, which can cause degradation during processing [
3,
5]. Commodity polymers such as polyethylene (PE) are extensively used as a flexible packaging material due to adequate flexibility and strength coupled with low cost [
6]. Unlike polyamides, it is well-established that PE is a hydrophobic material with non-polar characteristics, thus making it a challenging polymer to wet or print on [
7]. In general, polymeric surfaces are often characterised by poor wettability [
8], which limits their potential in certain applications, such as joining dissimilar materials using adhesives.
To overcome this challenge and improve the versatility of polymeric materials in applications that require adhesive bonding, various surface modification techniques are commonly employed. Numerous studies have shown that plasma treatments are some of the most promising surface modification methods [
9,
10]. These techniques are capable of altering the physical and chemical properties of a material surface, thereby significantly enhancing the surface wettability and adhesion properties without influencing the bulk properties of the material [
11,
12,
13,
14]. Among the plasma treatments currently available, μPlasma surface modification is particularly interesting because, unlike traditional plasma treatments that require low-pressure conditions (typically 50–400 Pa) [
15], μPlasma modifications can be performed under atmospheric conditions, thus significantly reducing the processing costs [
16]. Given these advantages over conventional plasma treatment methods, μPlasma technology may enable future integration with robotic systems, thereby enabling precise, localised, and multi-axially adjustable μPlasma modification of geometrically complex industrial components.
Although it is well established that plasma surface treatment can change the surface morphology of a polymer and enhance surface wettability via the formation of new functional groups on the surface, it is not a permanent modification. Instead, the polymer surface is subject to post-treatment ageing phenomena. These ageing phenomena can be subdivided into degradative and non-degradative processes. Degradative processes may involve chain scission, crosslinking, and oxidation [
17,
18], whereas non-degradative processes may involve hydrophobic recovery that involves chain reorientation and diffusion back towards the bulk of the material. Previous studies have shown that post-treatment ageing is affected by temperature [
19], relative humidity [
20], and polymer crystallinity [
21].
In this study, μPlasma surface modification of HDPE, PA12, and PA6 is explored. These polymeric materials were selected in order to assess the effect of hydrophilicity on the post-treatment ageing process. This study extends previous work via a decoupling of the effects of crystallinity and hydrophilicity on the ageing process. It also develops the characterisation of the ageing process via the application of a stretched exponential function to analyse and characterise the ageing data.
2. Materials and Methods
2.1. Materials
The materials utilised in this study are listed in
Table 1. All were supplied in sheet form and were dried in a desiccator for 7 days prior to processing.
2.2. Sample Preparation and Crystallinity Control
The crystallinities of the polymers were varied via a controlled cool from the melt. This was carried out in a heated press (George E. Moore & Sons Ltd., Birmingham, UK).
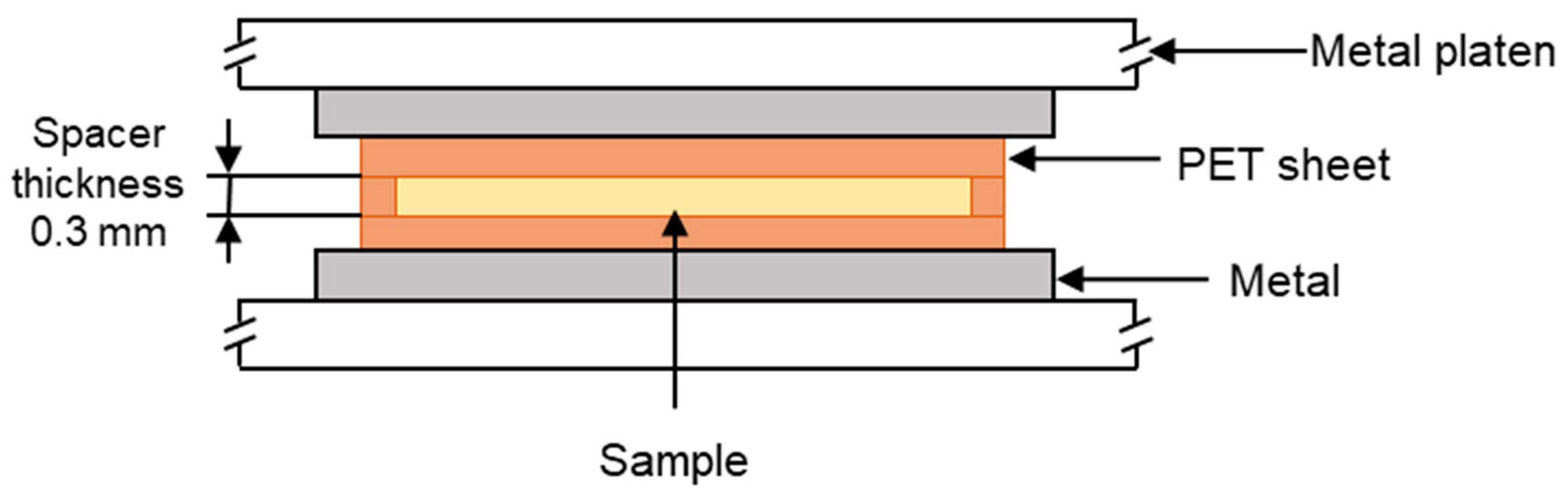
The polymer samples were placed into a polyethylene terephthalate (PET) mould that was composed of three sections. There was a PET spacer of thickness 0.3 mm, and on either side of this, there was a sheet of PET that functioned as a release film. This assembly was then placed in the preheated hot press, as shown in
Figure 1. The polymers were heated for 3 min to initiate melting. Then, a load of 10 tonnes was applied via the platens for a further 5 min, causing the polymer melt to flow and then fill the mould. The polymers were then recrystallised by cooling from the melt under different cooling regimes, which are listed in
Table 2. The melting temperatures of the ‘as received’ materials were determined using differential scanning calorimetry (DSC) as described in
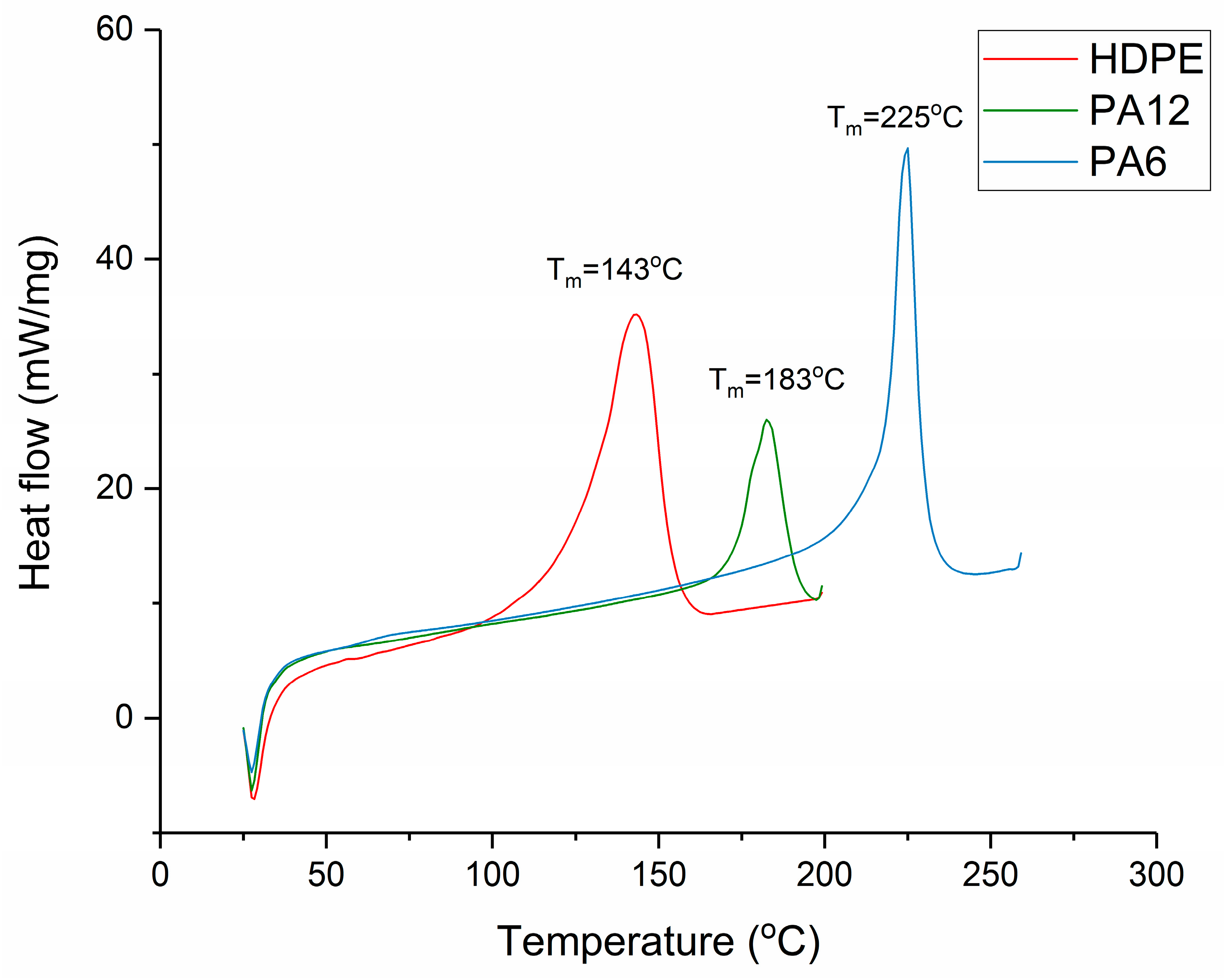
Section 2.3. The melting regions of each material are shown in
Figure 2, and this informed the selection of the hot-press temperatures, which were as follows: 160 °C for HDPE, 200 °C for PA12, and 240 °C for PA6. The heat of fusion and the degree of crystallinity for each recrystallised sample were then determined via DSC (as per
Section 2.3). Samples with comparable crystallinities were then selected for μPlasma treatment, and these are listed in
Table 3.
2.3. Measurement of Crystallinity Using Differential Scanning Calorimetry (DSC)
A differential scanning calorimeter (PerkinElmer DSC-7, PerkinElmer Inc., Waltham, MA, USA) was used for determining the melting point and the degree of crystallinity of the cooled polymers. Disc-shaped samples were placed in aluminium pans and encapsulated with lids (aluminium 40 μL flat DSC pans and lids, Thermal Instruments Ltd., Lancashire, UK). The sample weight was 5 ± 0.1 mg. The samples were heated at 50 K min
−1, and the melting point and heat of fusion were calculated. The resulting degrees of crystallinity were determined from the melting region according to a method described previously [
22]. It is displayed in
Figure 2 that the T
m values for HDPE, PA12, and PA6 were 143 °C, 183 °C, and 225 °C, respectively.
2.4. μPlasma Surface Modification
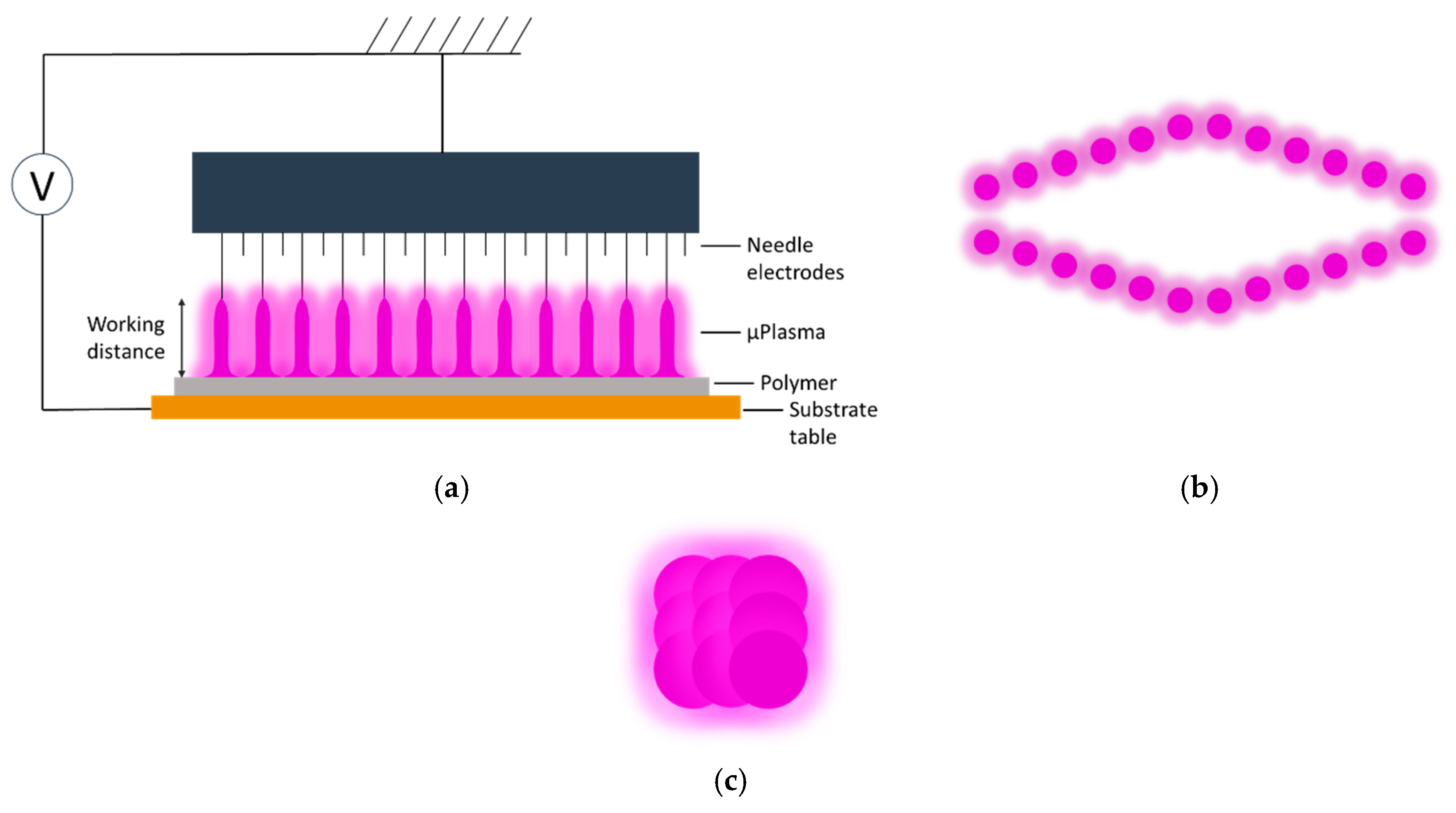
Both the plaque and film polymer surfaces were cleaned using ethanol before μPlasma modification. The μPlasma modification was carried out using a Roth & Rau Pixdro LP50 plasma inkjet printer (InnoPhysics, Eindhoven, The Netherlands) with an InnoPhysics POD24 print-head as shown in
Figure 3a,b. A printing rate of 20 mm/s, a working distance from the sample surface to the tips of the printing needles of 100 μm, and an accelerating voltage of 7 kV were chosen as the μPlasma modification parameters, which had previously been reported to induce the most significant enhancement in wettability [
20]. Additionally, in this study, the polymer surfaces were modified using a single treatment scan. The plasma spots were designed to overlap with each other (shown in
Figure 3c) to produce uniform treatment coverage.
2.5. Atomic Force Microscopy (AFM)
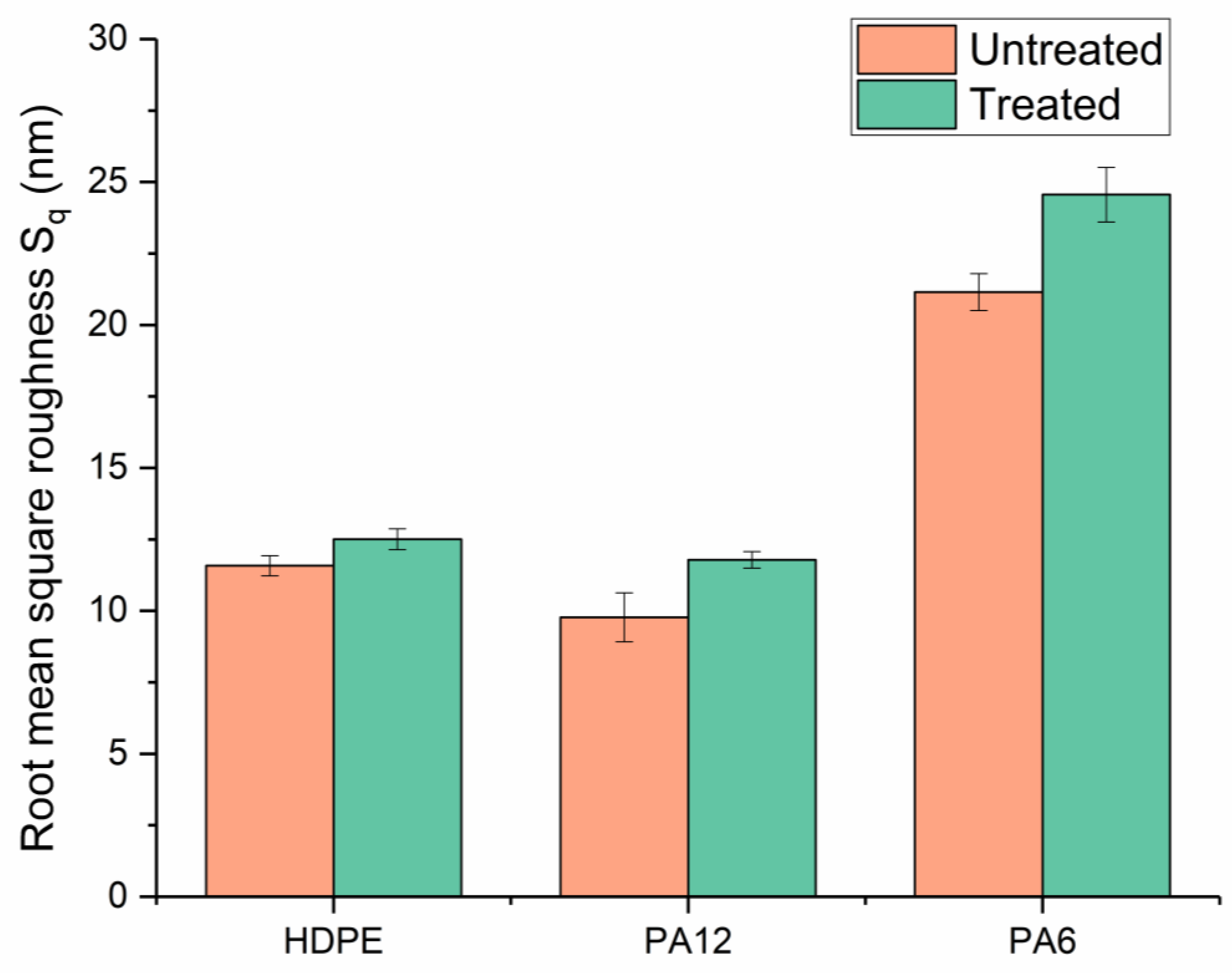
Surface morphologies were examined using an atomic force microscope (Bruker Multimode AFM, Billerica, MA, USA) operating with a silicon cantilever (Nanosensors PPP-NCHR, Neuchatel, Switzerland, tip radius < 10 nm, length 125 µm, resonant frequency 330 kHz) under a “tapping mode”. Before use, the cantilever was cleaned by rinsing it with ethanol and exposing it to a UV–ozone chamber for 10 min. Images were generated across an area of 16 µm × 16 µm, with a minimum of 3 different positions recorded for each type of surface. Three-dimensional height profiles, average roughness, and root-mean-square (RMS) roughness data were obtained from the AFM data using an open-source image processing tool (Gwyddion, SourceForge, San Diego, CA, USA). To limit the influence of macroscopic surface deviations (e.g., striations or bumps), three smaller areas (16 µm2) were chosen for each sample type to obtain comparable roughness measurements.
2.6. Measurement and Analysis of Contact Angles
The contact angles of the untreated and treated polymer surfaces were measured in accordance with the ISO 19403-2:2020 standard [
23] using an experimental apparatus under the sessile drop method. The apparatus was adjusted to allow for samples to be placed in line with the camera and light source to ensure a clear and flat view of the droplet, which, therefore, enabled comparable measurements between polymers. An optimised measuring technique from our previous work was utilised [
20]. Ionised water droplets with a volume of 6 μL were formed on sample surfaces using a calibrated pipette. Averages and standard deviations of contact-angle values were obtained using three repeat droplets for each sample type. Both left and right contact angles were measured for each droplet using the Ossila contact-angle measurement software (Version 4.1.4, Ossila Ltd., Sheffield, UK).
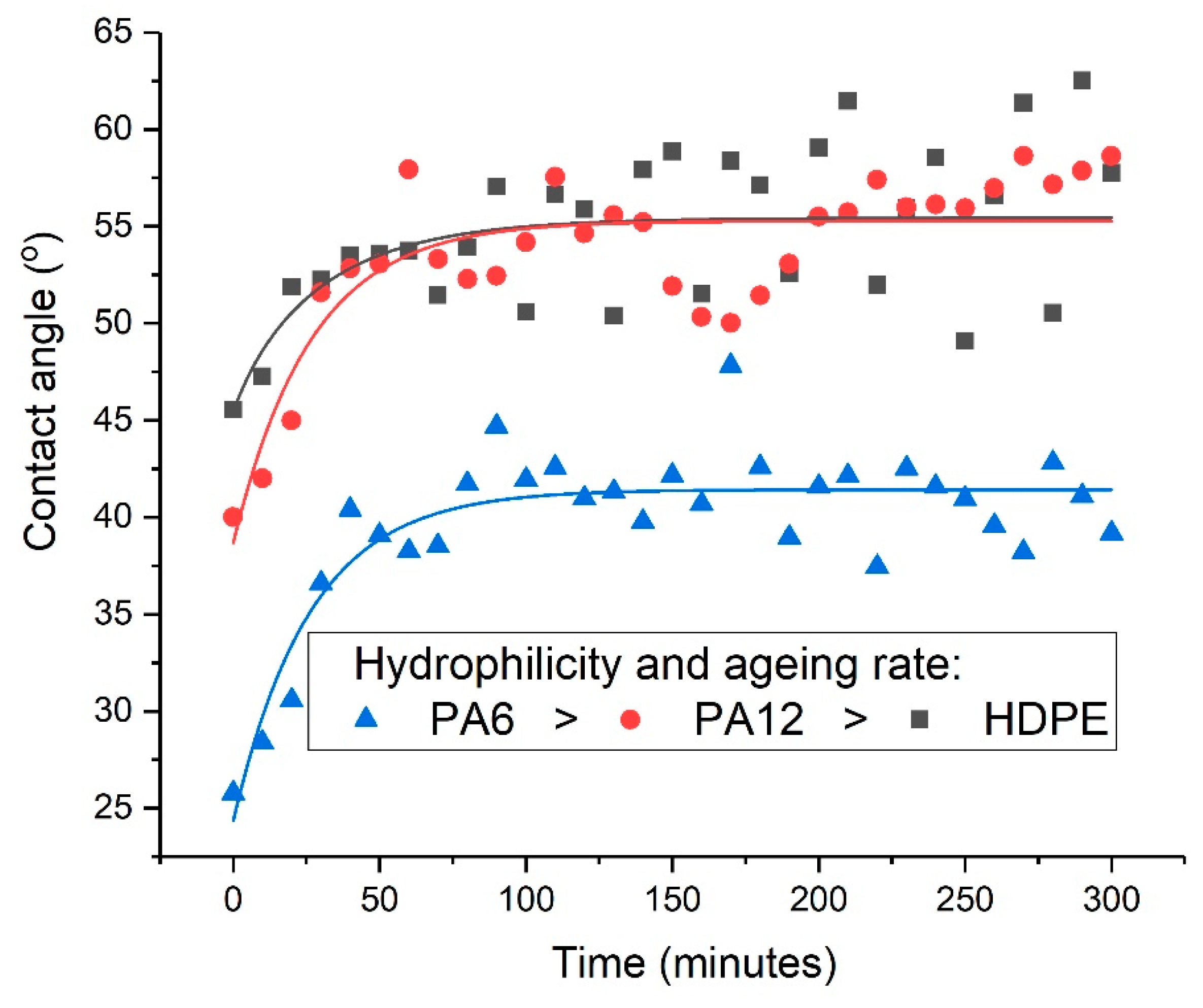
2.7. Phenomenological Modelling of the Post-Treatment Ageing Process
A stretched exponential function was used as the basis of a phenomenological model of the post-treatment ageing (or relaxation) process. The Kohlrausch–Williams–Watts (KWW) function has been applied to various relaxation phenomena in polymers, including dielectric relaxation [
24], and in a slightly modified form, to enthalpy relaxation (or physical ageing) [
25]. The basic stretched exponential function is shown in Equation (1), where the relaxation of the quantity
is given by
where
t is the time,
is the characteristic relaxation time constant, and
is the stretching exponent with a value between 0 and 1, which determines the shape of the ageing curve. To characterise the post-treatment ageing process, a modified KWW function was employed in which the change in contact angle (
) varied with time according to Equation (1).
The three unknown parameters,
,
(the contact angle when
approaches
), and
, were determined using non-linear least-squares fitting. The modification shown in Equation (2) is after that of Cowie and Ferguson [
25].
2.8. Fourier-Transform Infrared Spectroscopy (FTIR)
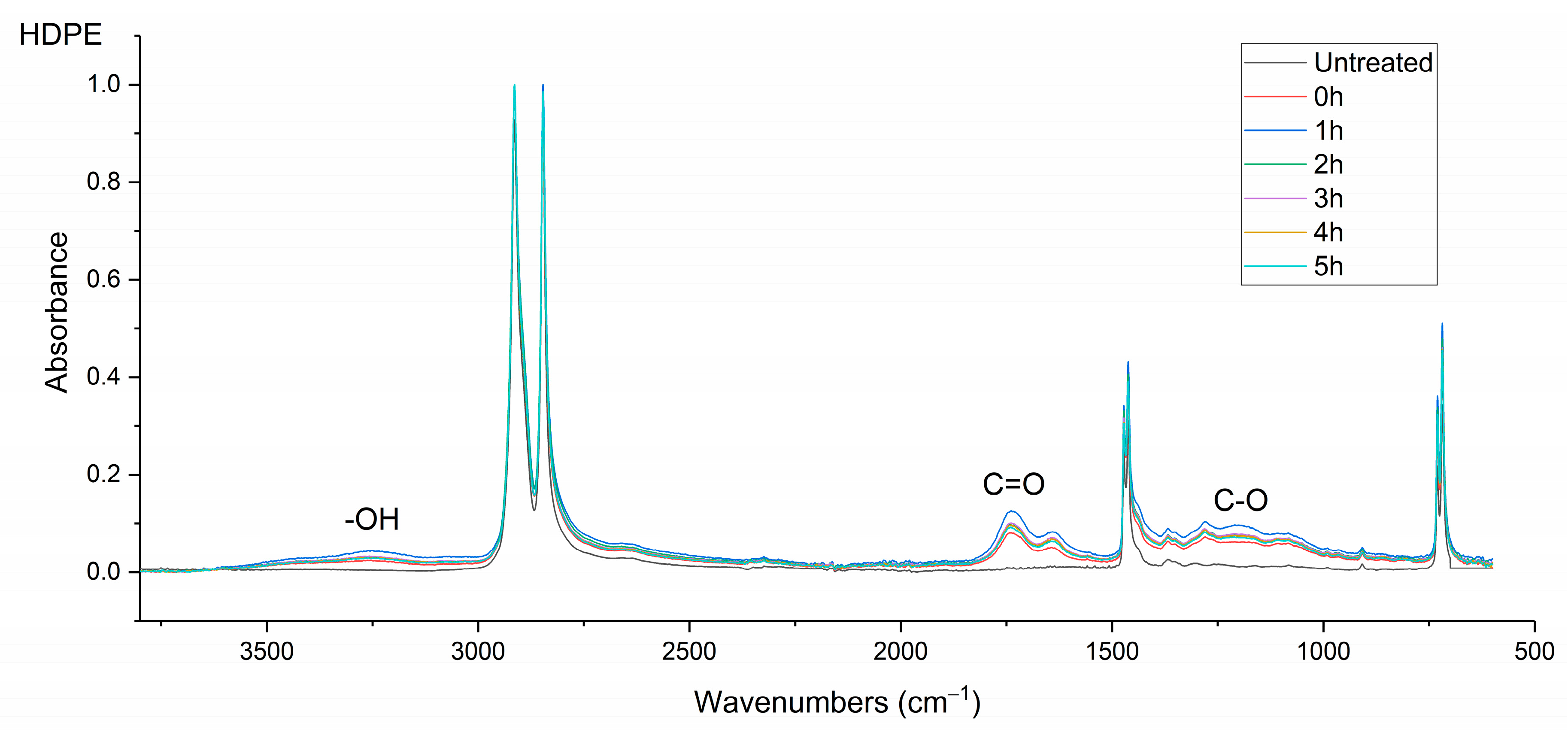
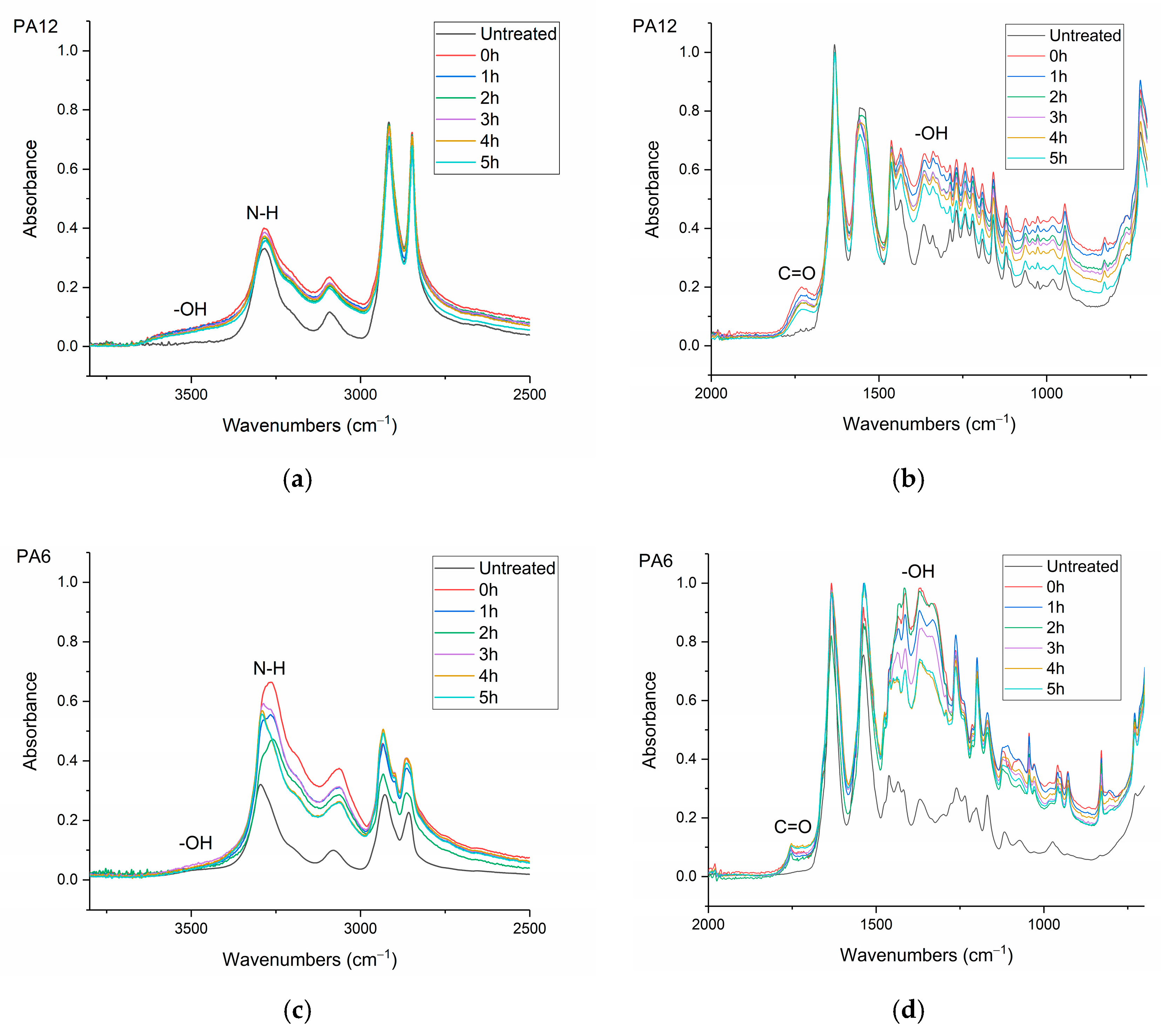
The infrared (IR) spectra were obtained using a Nicolet MAGNA 860 FTIR spectrometer (Thermo-Scientific, Waltham, MA, USA) fitted with an attenuated total reflectance (ATR) accessory (Golden Gate ATR, Specac, Orpington, UK). The samples were μPlasma treated 10 times and then transferred instantly to the ATR (treated surface in contact with the ATR crystal). The spectral range was between 500 and 4000 cm–1. In total, 100 scans were recorded at a resolution of 2 cm–1.
4. Conclusions
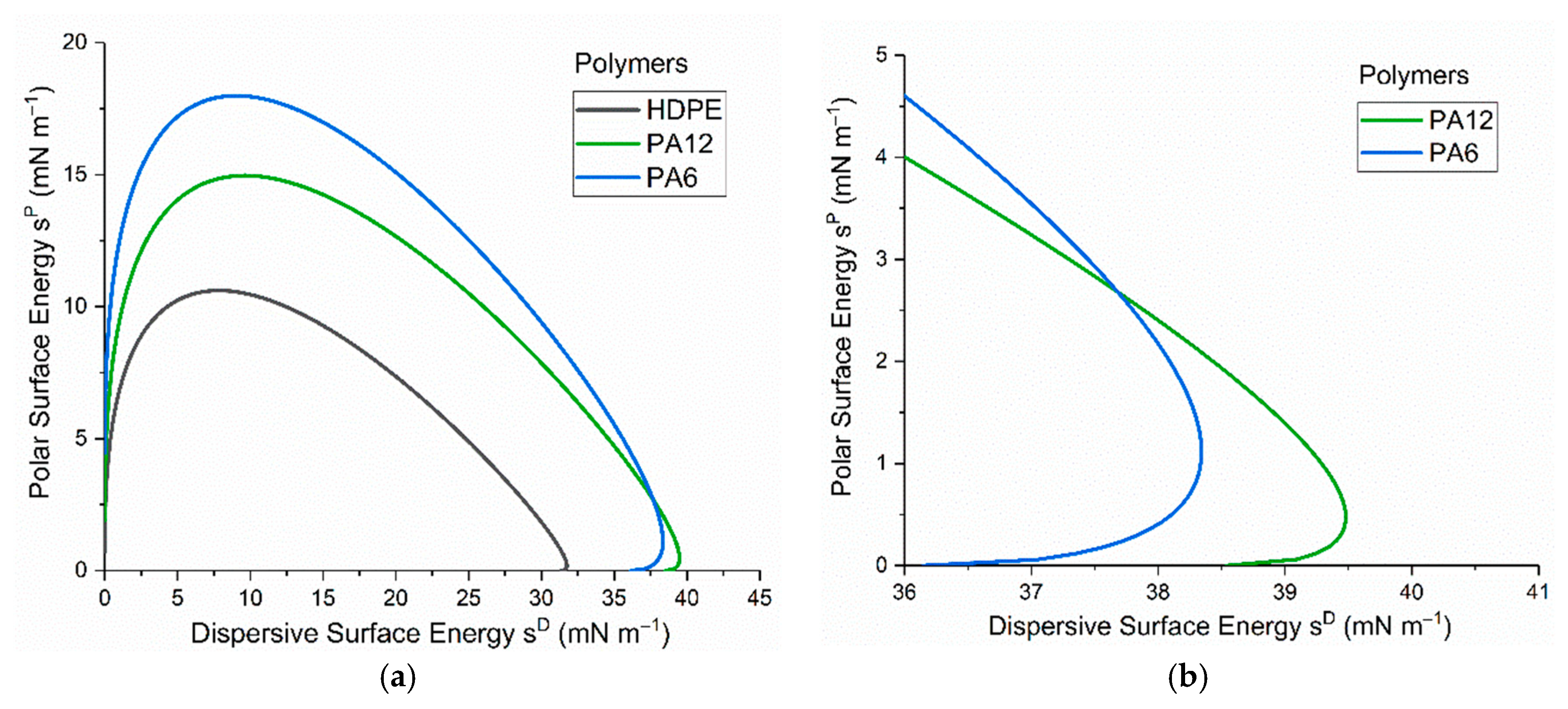
Surface contact-angle measurements, moisture absorption capability measurements, and calculations of wetting envelopes revealed that the hydrophilicity of the three polymers was in the order of PA6 > PA12 > HDPE, with HDPE being identified as hydrophobic. The wettability of the polymers was improved substantially through μPlasma modification, with increases in surface roughness and the introduction of more polar functional groups. The changes in chemical compositions of the polymer surfaces were believed to be the dominant factor for the improvement in wettability. Although the wettability enhancement of the three polymer surfaces following μPlasma modification was significant, it was subjected to an ageing phenomenon. The contact angle increased by 27.6% for HDPE, 50.1% for PA12, and 68.2% for PA6 following five-hour ageing, compared to their treated but unaged equivalents. However, the wettability was still partially remained. With the assistance of KWW modelling, it was found that the ageing rate was dependent on the degree of hydrophilicity of the polymers, i.e., the higher the hydrophilicity, the faster the ageing process. This finding was further confirmed by FTIR through the reduction in the polar functional groups, including hydroxyl (-OH), carbonyl (C=O), amide bands (N-H), and ether (C-O) groups.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}