
Unraveling Broadband Near-Infrared Luminescence in Cr3+-Doped Ca3Y2Ge3O12 Garnets: Insights from First-Principles Analysis

 , ,
, , 
Abstract
:1. Introduction
2. Method of Calculations
3. Results and Discussion
3.1. Ground States of Both Neat and Cr3+-Doped Ca3Y2Ge3O12
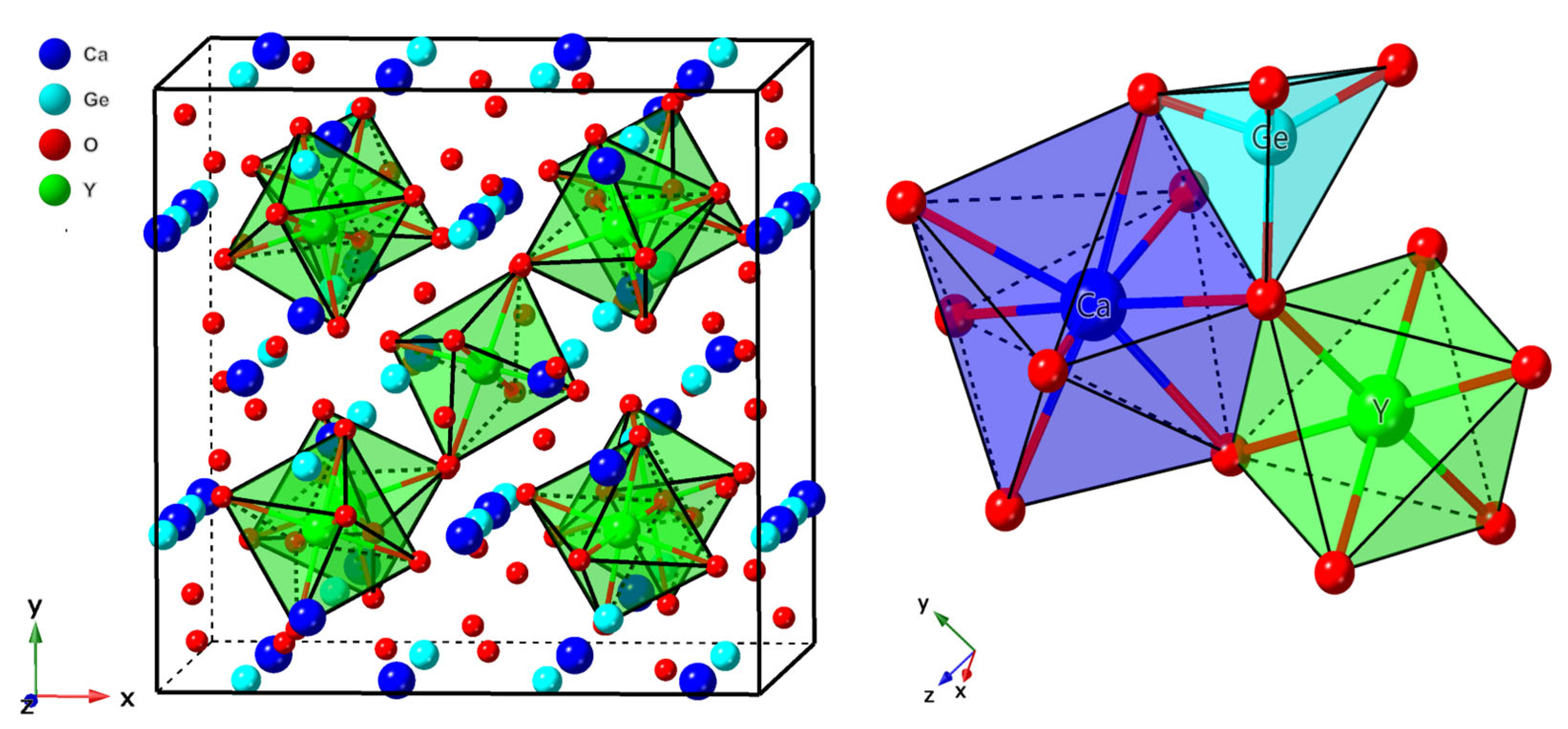
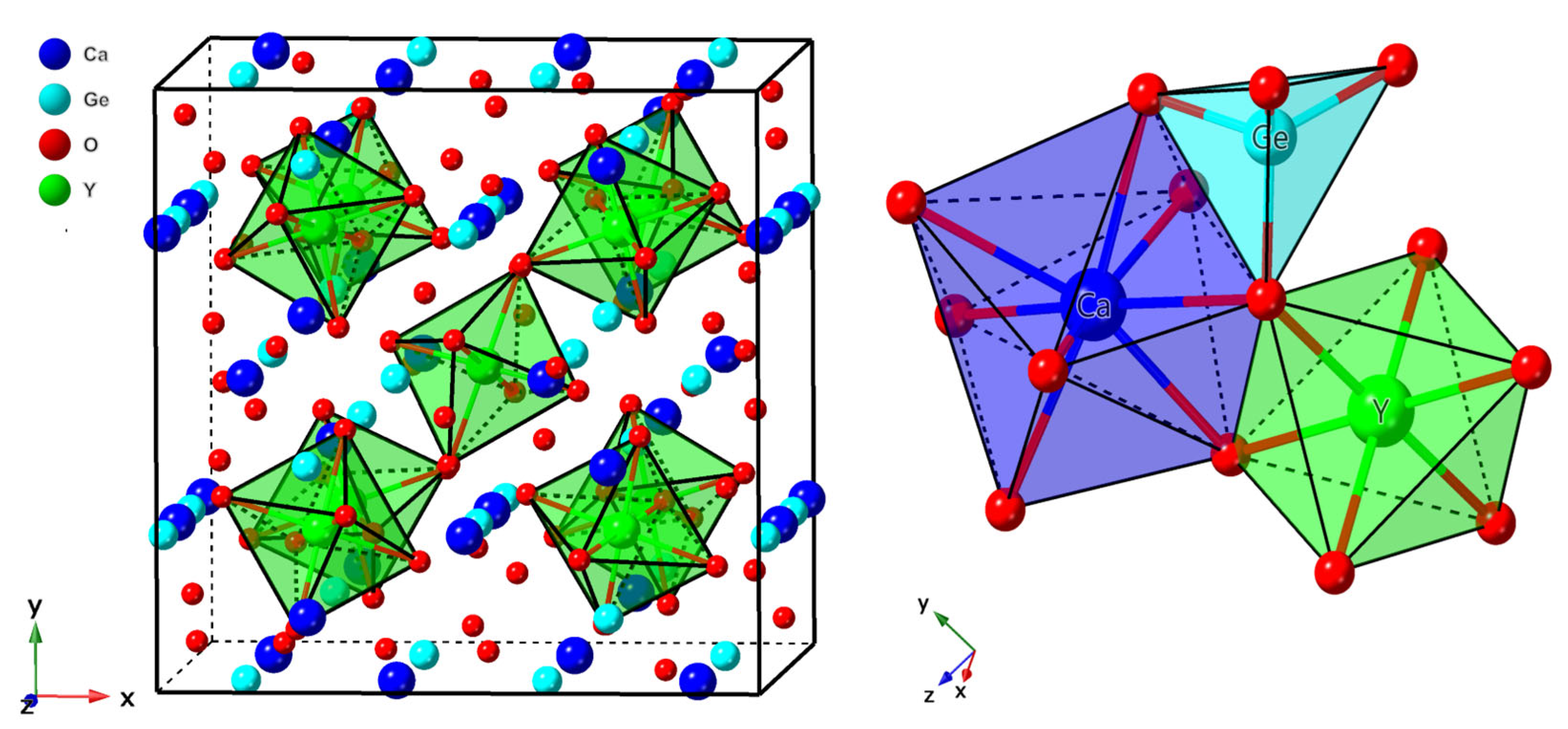
3.1.1. Structural Properties and Defect Site Occupancy
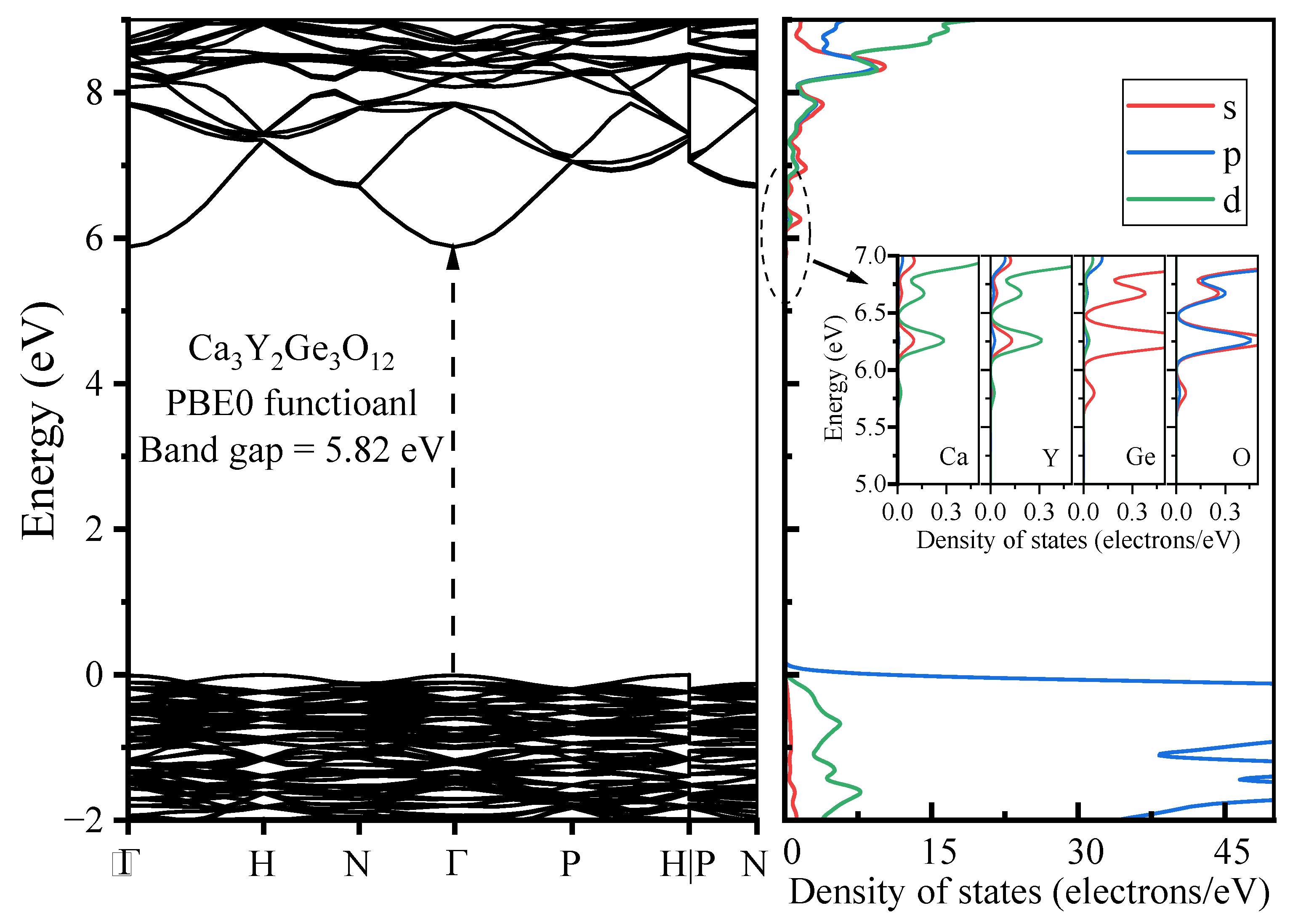
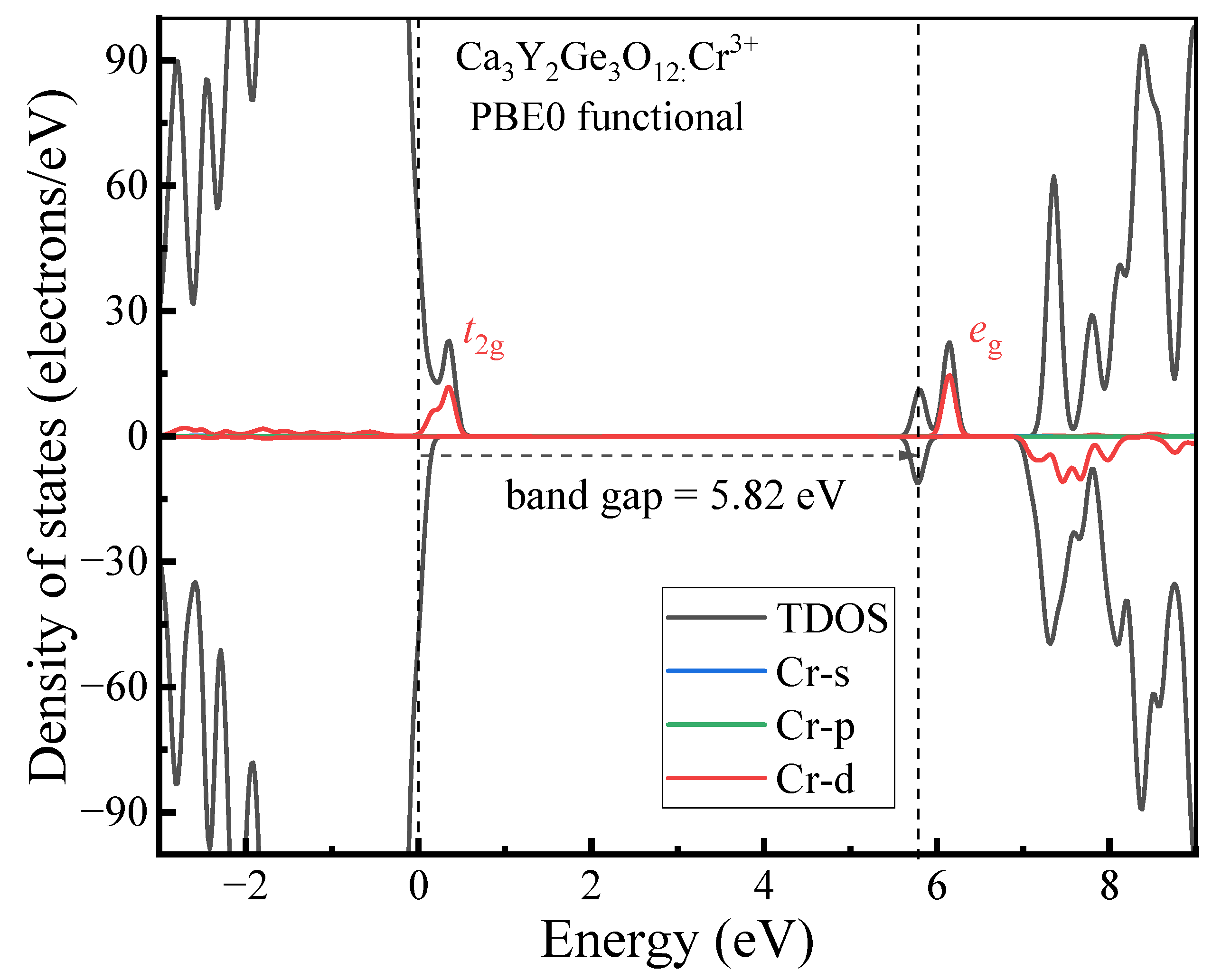
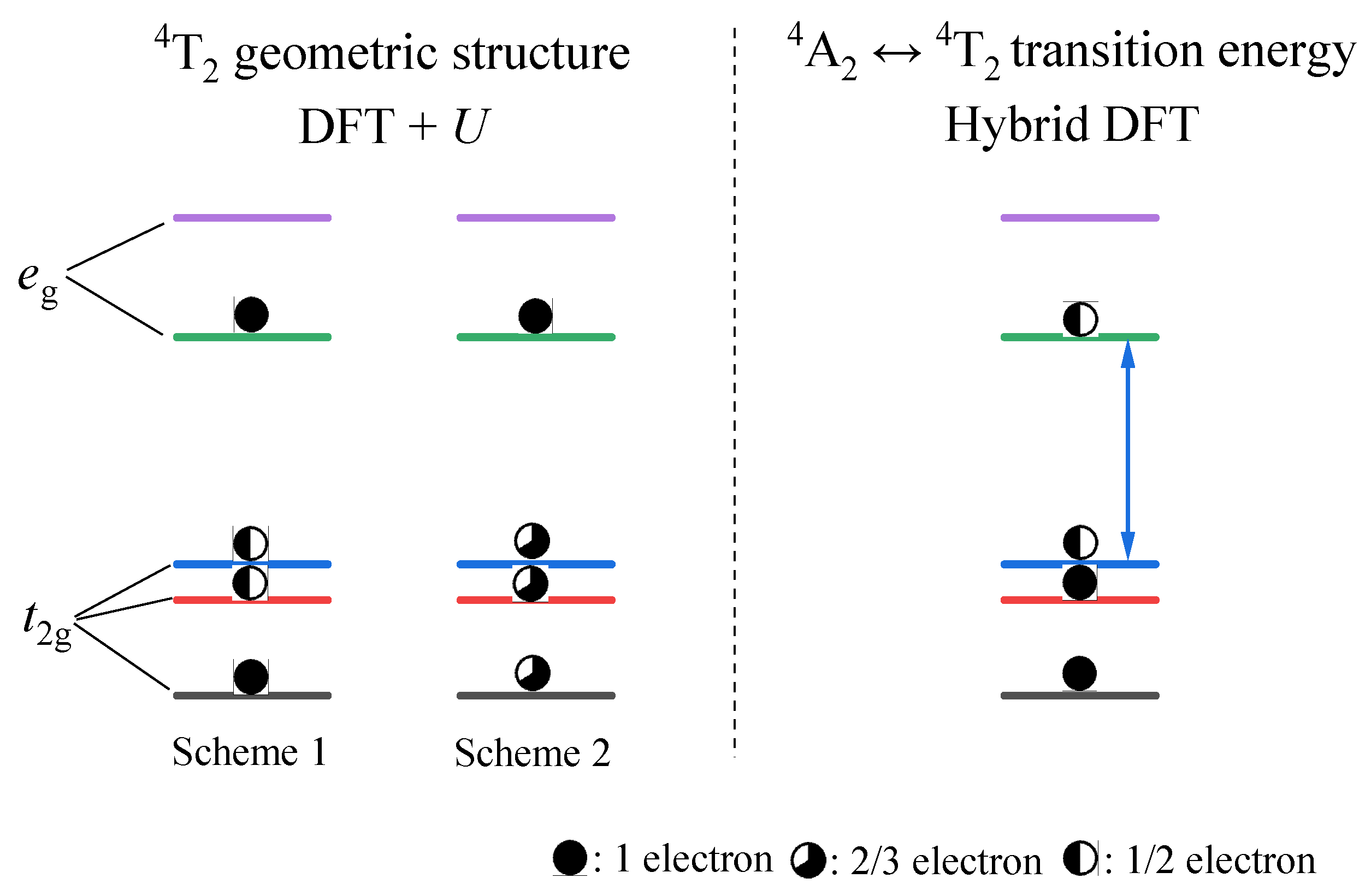
3.1.2. Electronic Properties
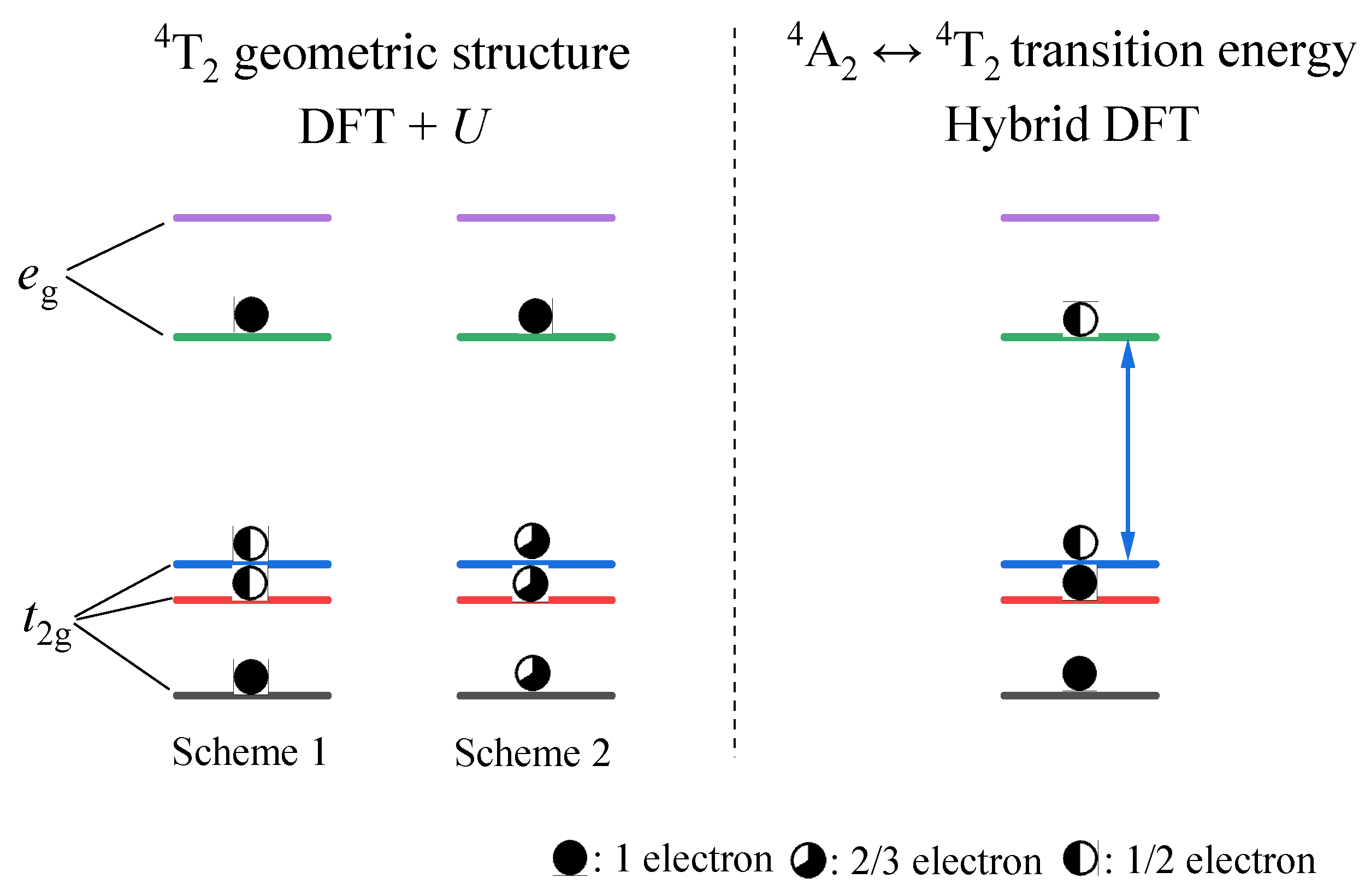
3.2. Excited States 2E and 4T2 of Cr3+-Doped Ca3Y2Ge3O12
3.2.1. Structural Properties
3.2.2. Optical Properties and Luminescence Mechanism
3.2.3. Thermal Stability
4. Conclusions
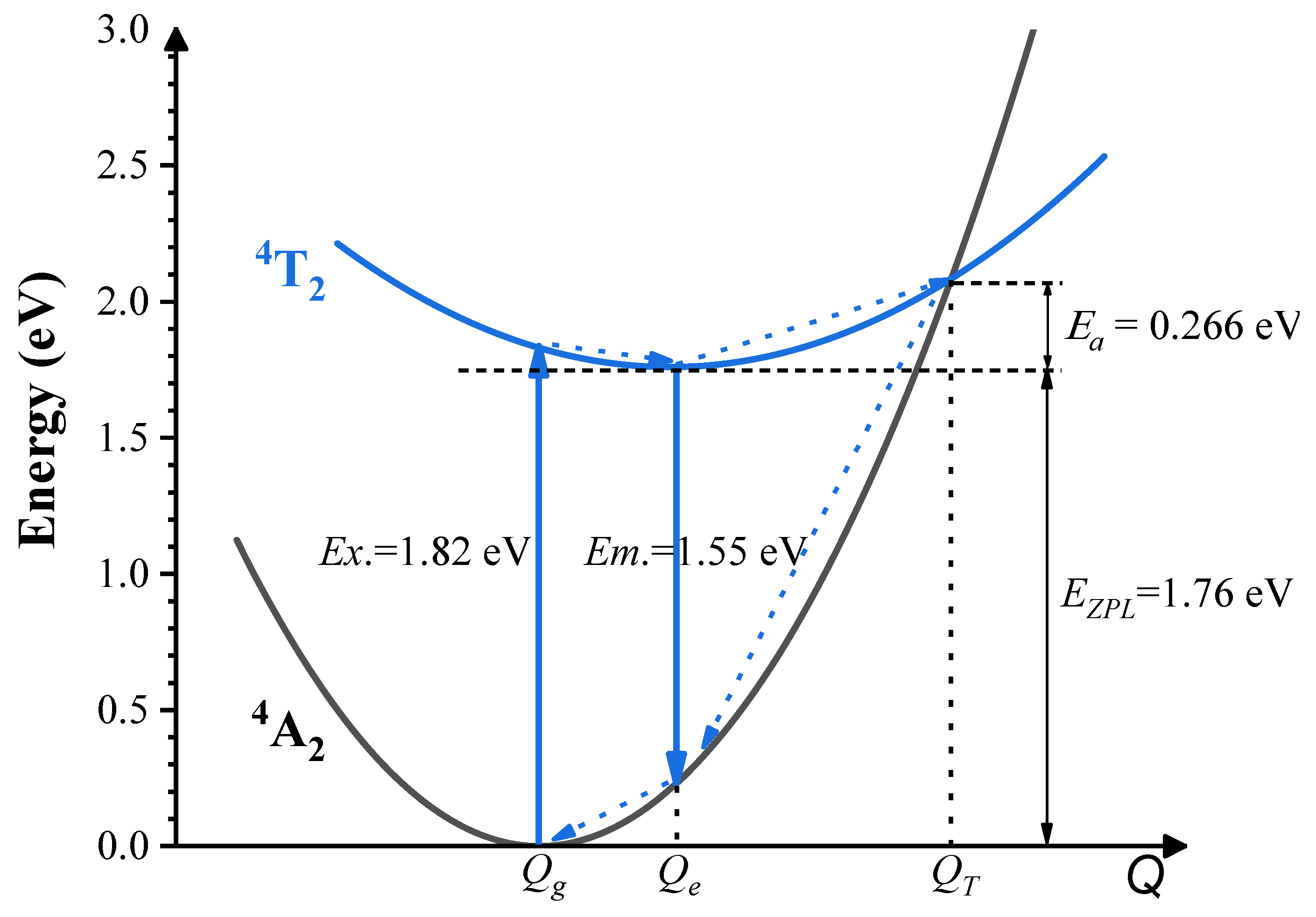
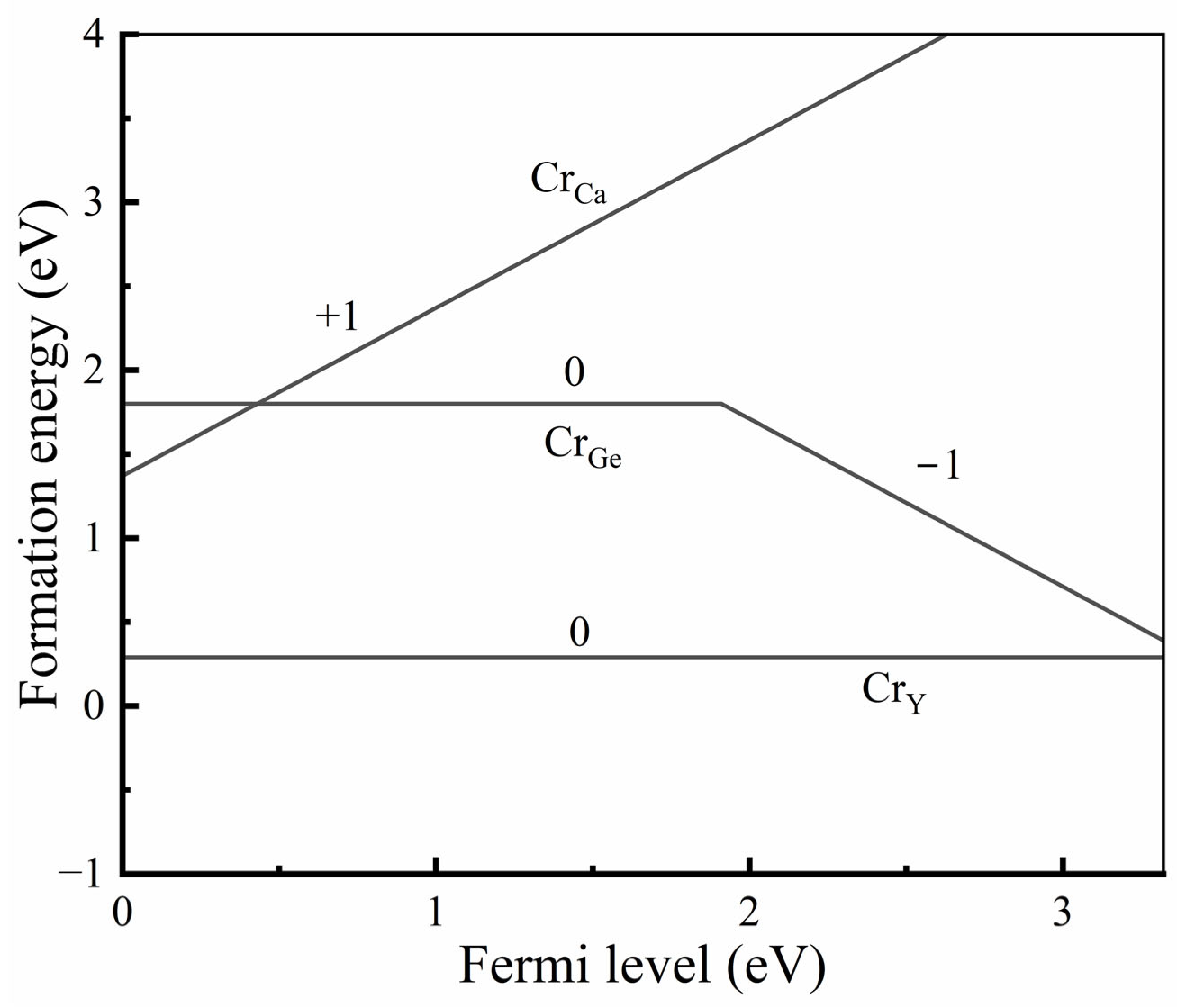
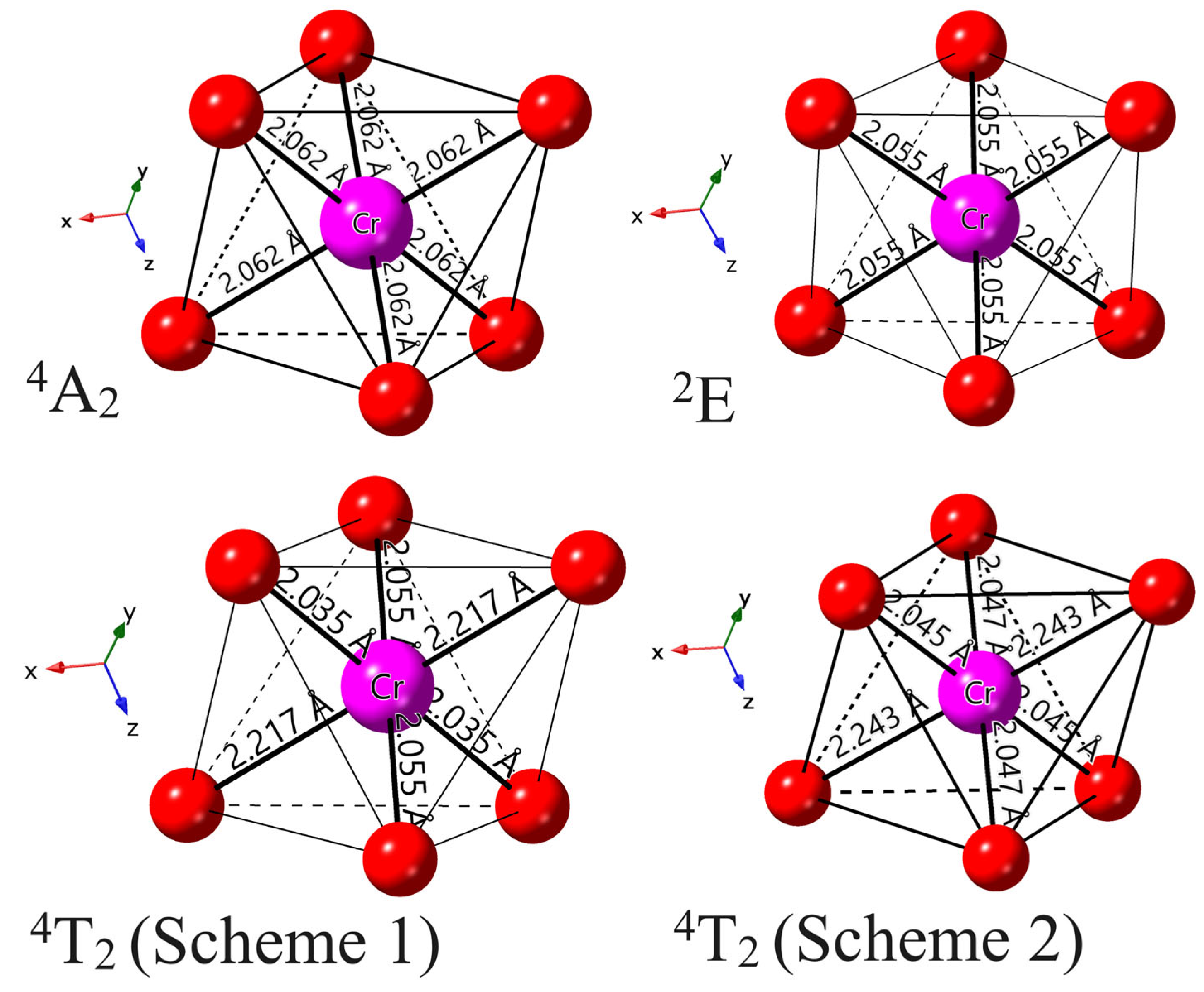
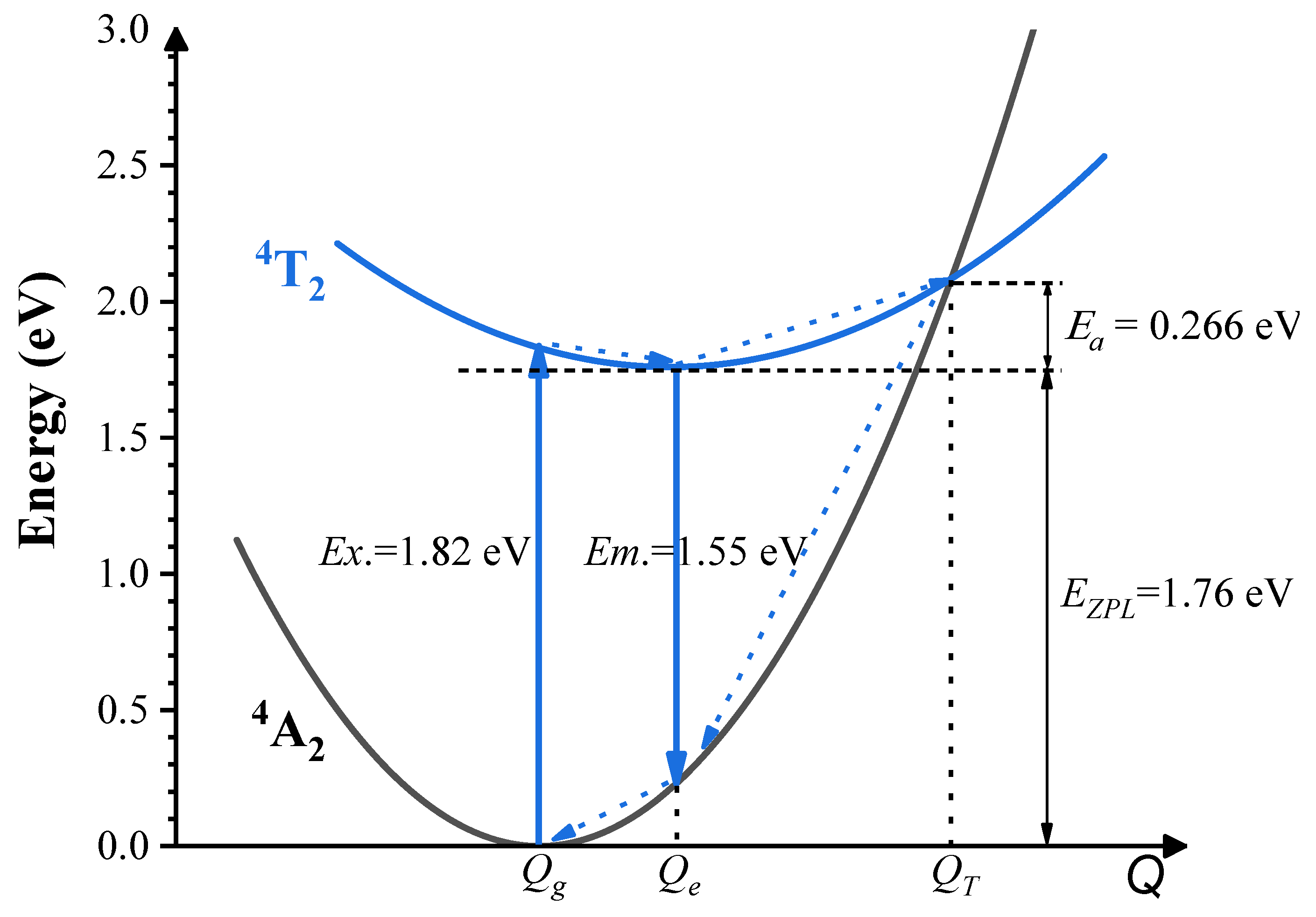
- The results from both the structural analysis and the defect formation energy calculations indicate a tendency for Cr3+ dopants to preferentially occupy Y3+ sites rather than Ca2+ and Ge4+ sites. Comparing the optimized geometric structure of the ground state 4A2 of Cr3+ ions, the excited state 4T2 exhibits a significant Jahn–Teller distortion, characterized by a notable axial expansion and a slight equatorial compression in the [CrO6]9− complex. In contrast, the excited state 2E primarily retains the initial ground-state structure, undergoing a negligible change.
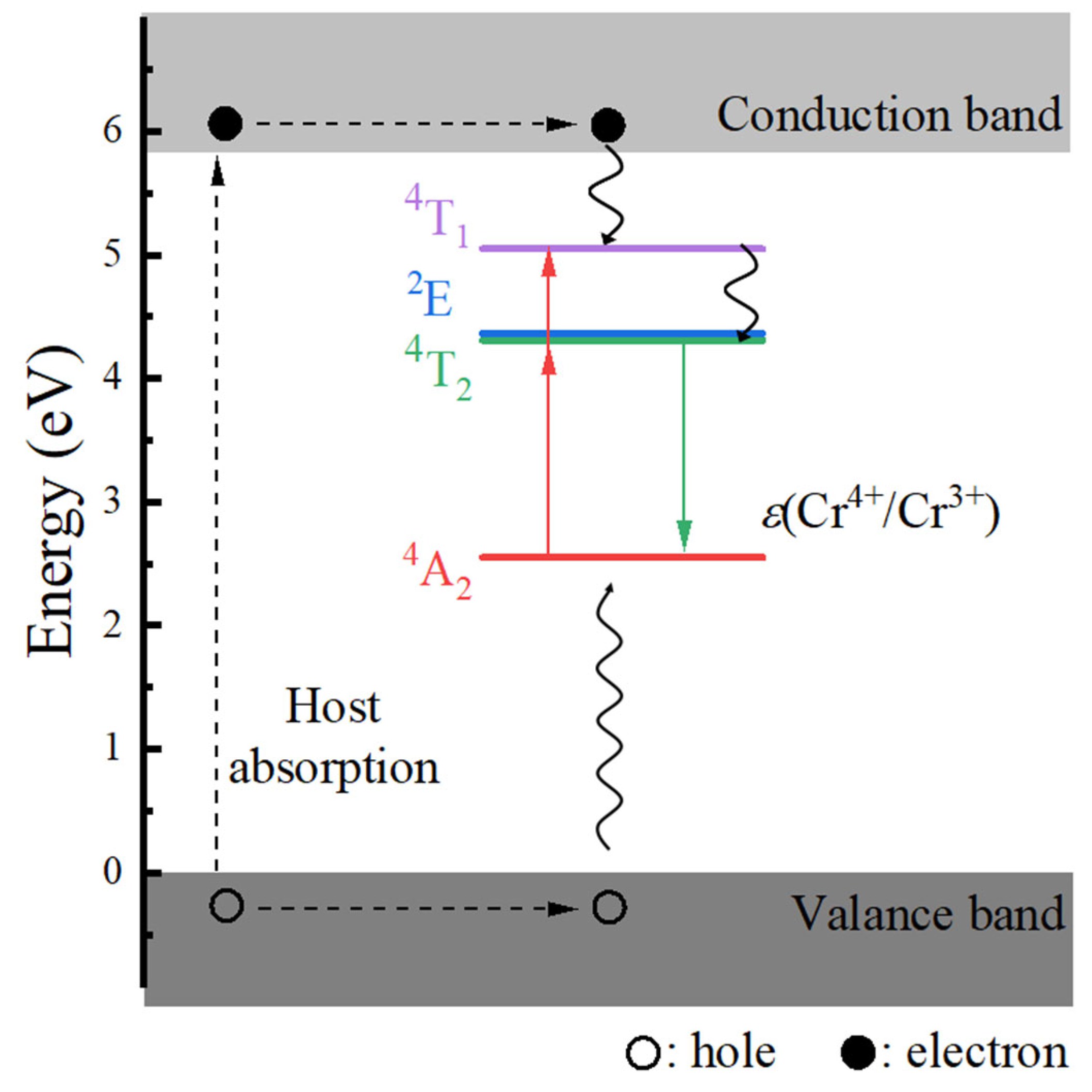
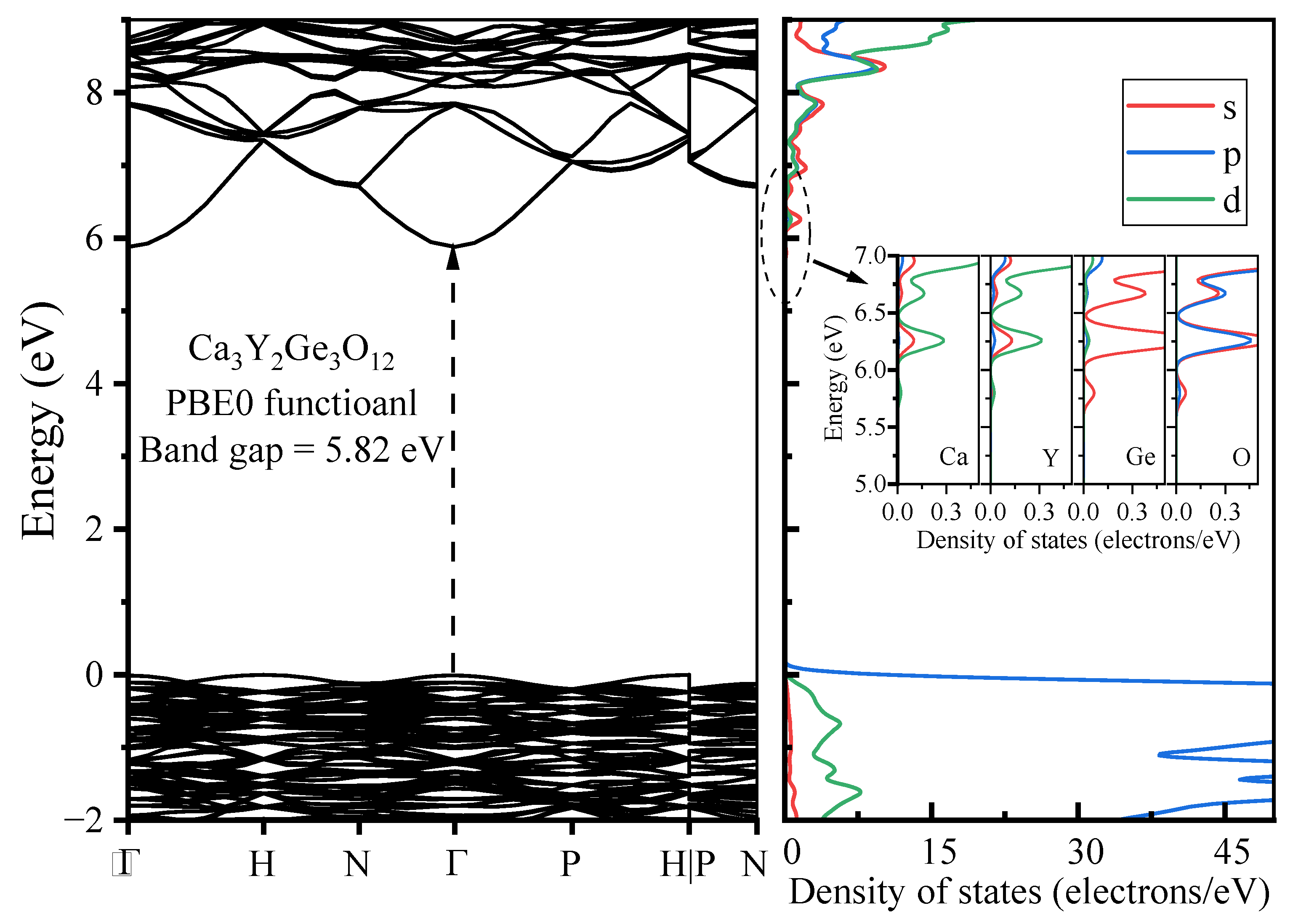
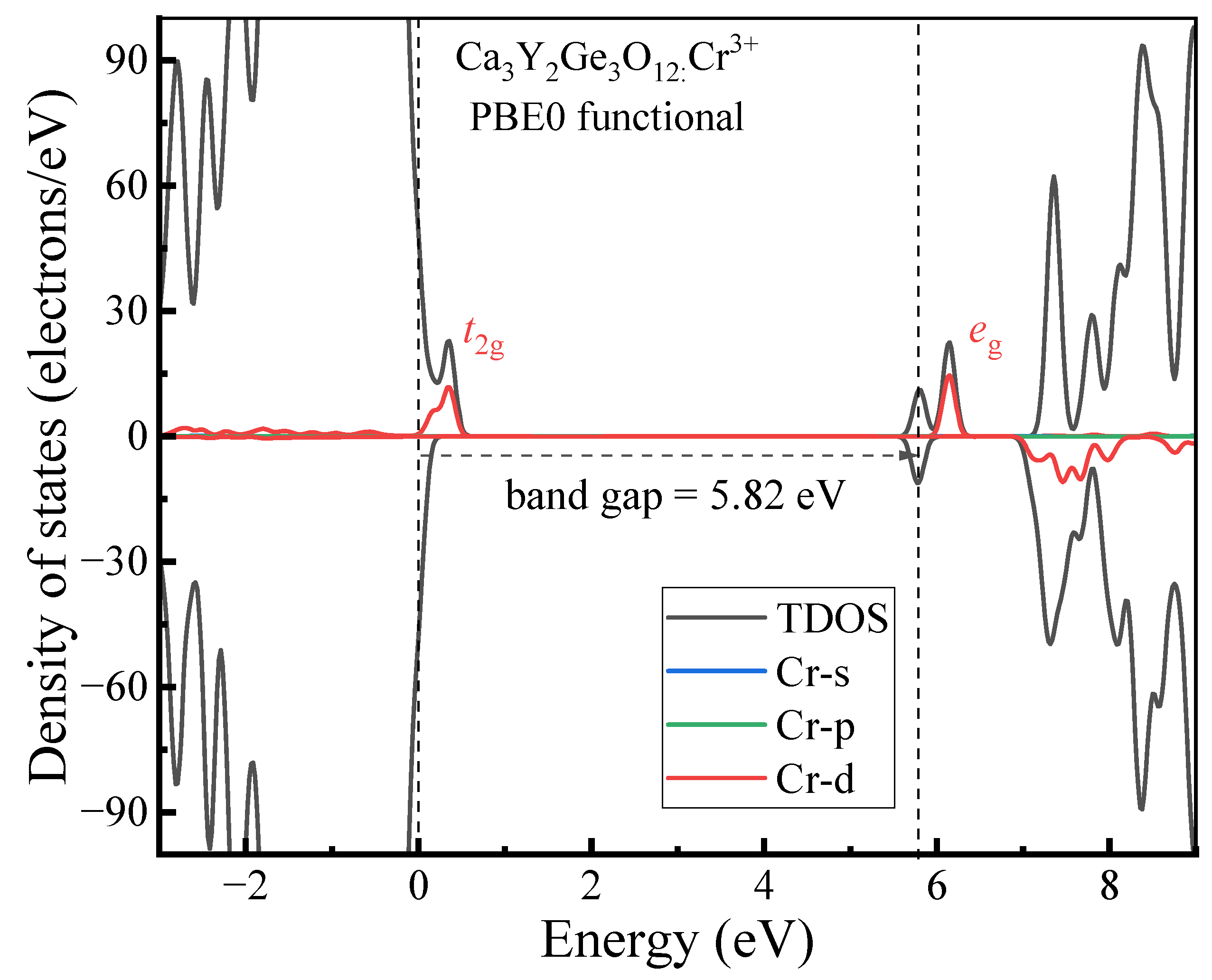
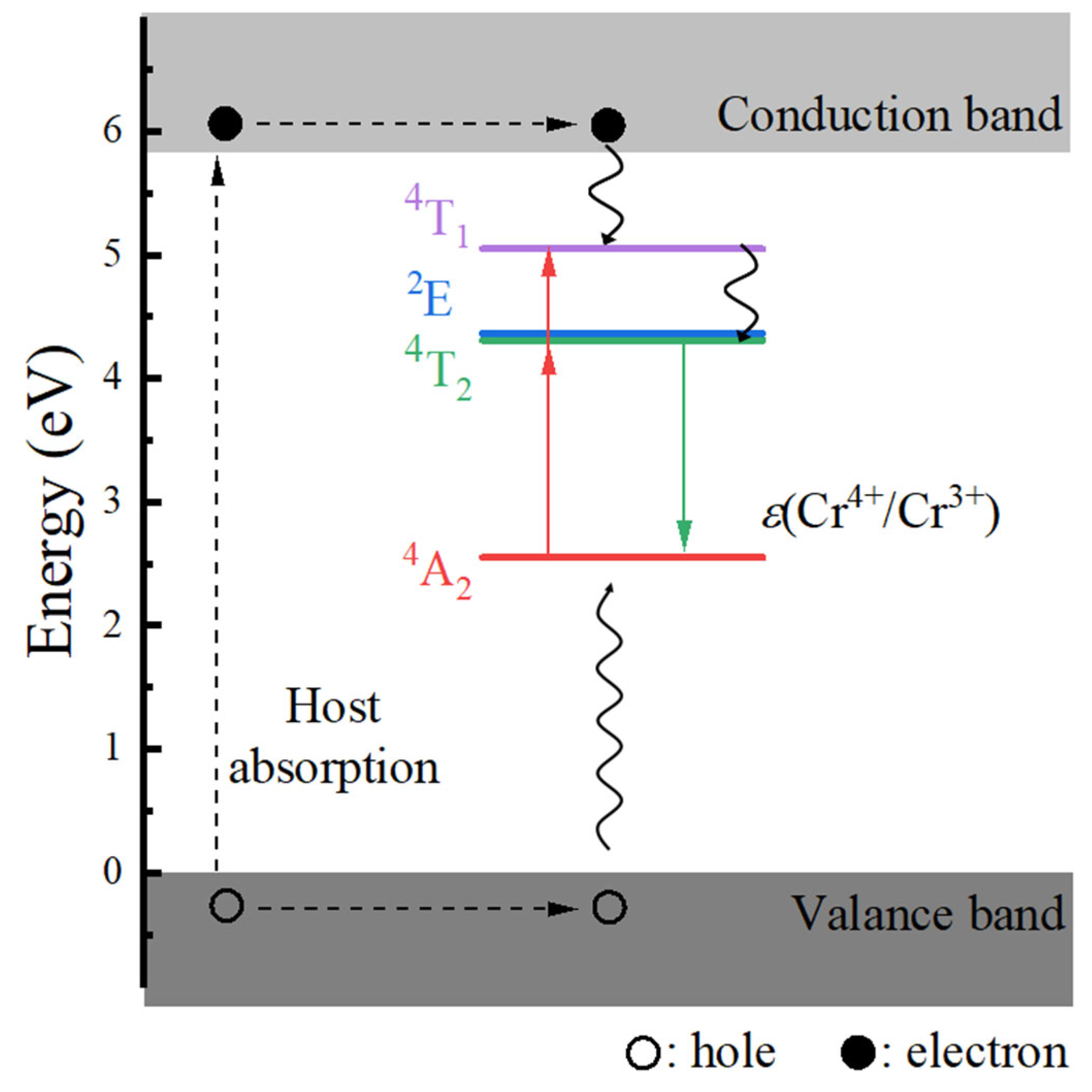
- The host material Ca3Y2Ge3O12 features a direct band gap of 5.82 eV, allowing sufficient space to accommodate the multiple energy levels of Cr3+ dopants. The calculated positions of the ground 4A2 and excited 4T2 energy levels within the band gap underscore the isolated nature of Cr3+ optical centers from the host’s electronic structure. This discovery further supports the observed higher quantum efficiency.
- The calculated energies for the excitation, emission, and Stokes shift associated with the optical transitions 4A2-2E and 4A2-4T2 show a much better agreement with the experimental values. The energy comparison of the optical transitions 4A2-2E and 4A2-4T2 indicates that Cr3+ ions are located in a weak CF. The identification of three distinct excitation pathways that induce the 4T2→4A2 luminescence suggests that the excitations of Cr3+ ions to the 4T1 and 4T2 states are more efficient.
- Our accurate prediction of thermal excitation energy has paved a direct path to providing fundamental analysis of the thermal quenching process in phosphors doped with 3d3 ions, using the configuration coordinate diagram.
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, L.L.; Wang, D.D.; Hao, Z.D.; Zhang, X.; Pan, G.H.; Wu, H.J.; Zhang, J.H. Cr3+-doped broadband NIR garnet phosphor with enhanced luminescence and its application in NIR spectroscopy. Adv. Opt. Mater. 2019, 7, 1900185. [Google Scholar] [CrossRef]
- He, S.; Zhang, L.L.; Wu, H.; Wu, H.J.; Pan, G.H.; Hao, Z.D.; Zhang, X.; Zhang, L.G.; Zhang, H.; Zhang, J.H. Efficient super broadband NIR Ca2LuZr2Al3O12: Cr3+, Yb3+ garnet phosphor for pc-LED light source toward NIR spectroscopy applications. Adv. Opt. Mater. 2020, 8, 1901684. [Google Scholar] [CrossRef]
- Basore, E.T.; Xiao, W.G.; Liu, X.F.; Wu, J.H.; Qiu, J.R. Broadband near-infrared garnet phosphors with near-unity internal quantum efficiency. Adv. Opt. Mater. 2020, 8, 2000296. [Google Scholar] [CrossRef]
- Mao, M.Q.; Zhao, T.L.; Zeng, H.T.; Wang, L.; Huang, F.; Tang, X.Y.; Xie, R.J. Broadband near-infrared (NIR) emission realized by the crystal-field engineering of Y3-xCaxAl5-xSixO12: Cr3+ (x = 0–2.0) garnet phosphors. J. Mater. Chem. C 2020, 8, 1981–1988. [Google Scholar] [CrossRef]
- Zou, X.K.; Wang, X.J.; Zhang, H.R.; Kang, Y.Y.; Yang, X.; Zhang, X.J.; Molokeev, M.S.; Lei, B.F. A highly efficient and suitable spectral profile Cr3+-doped garnet near-infrared emitting phosphor for regulating photomorphogenesis of plants. Chem. Eng. J. 2021, 428, 132003. [Google Scholar] [CrossRef]
- Zhang, Q.Q.; Li, G.G.; Dang, P.P.; Liu, D.J.; Huang, D.Y.; Lian, H.Z.; Lin, J. Enhancing and tuning broadband near-infrared (NIR) photoluminescence properties in Cr3+-doped Ca2YHf2Al3O12 garnet phosphors via Ce3+/Yb3+-codoping for LED applications. J. Mater. Chem. C 2021, 9, 4815–4824. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, J.N.; Zhang, L.L.; Wu, H.; Wu, H.J.; Pan, G.H.; Liu, F.; Zhang, J.H. Cr3+ activated garnet phosphor with efficient blue to far-red conversion for pc-LED. Adv. Opt. Mater. 2021, 9, 2101134. [Google Scholar] [CrossRef]
- Mao, N.; Liu, S.Q.; Song, Z.; Liu, Q.L. A broadband near-infrared phosphor Ca3Y2Ge3O12: Cr3+ with garnet structure. J. Alloys Compd. 2021, 863, 158699. [Google Scholar] [CrossRef]
- Cui, J.; Li, P.L.; Cao, L.W.; Wang, X.J.; Yao, Y.; Zhang, M.Y.; Zheng, M.J.; Yang, Z.B.; Suo, H.; Wang, Z.J. Achievement of broadband near-infrared phosphor Ca3Y2Ge3O12: Cr3+, Ce3+ via energy transfer for food analysis. J. Lumin. 2021, 237, 118170. [Google Scholar] [CrossRef]
- Adachi, S. Photoluminescence spectroscopy and crystal-field parameters of Cr3+ ion in red and deep red-emitting phosphors. ECS J. Solid State Sci. Technol. 2019, 8, R164–R168. [Google Scholar] [CrossRef]
- Adachi, S. Review-Photoluminescence properties of Cr3+-activated oxide phosphors. ECS J. Solid State Sci. Technol. 2021, 10, 026001. [Google Scholar] [CrossRef]
- Brik, M.G.; Srivastava, A.M. Review-A Review of the electronic structure and optical properties of ions with d3 electron configuration (V2+, Cr3+, Mn4+, Fe5+) and main related misconceptions. ECS J. Solid State Sci. Technol. 2018, 7, R3079–R3085. [Google Scholar] [CrossRef]
- Xiang, J.M.; Zheng, J.M.; Zhao, X.Q.; Zhou, X.; Chen, C.H.; Jin, M.K.; Guo, C.F. Synthesis of broadband NIR garnet phosphor Ca4ZrGe3O12: Cr3+, Yb3+ for NIR pc-LED applications. Mater. Chem. Front. 2022, 6, 440–449. [Google Scholar] [CrossRef]
- Shang, L.B.; Chen, Q.L.; Jing, W.G.; Ma, C.-G.; Duan, C.K.; Du, J.F. First-principles study of transition metal dopants as spin qubits. Phys. Rev. Mater. 2022, 6, 086201. [Google Scholar] [CrossRef]
- Shang, L.B.; Liu, M.Z.; Duan, C.K. Does Cr3+ occupy tetrahedral sites and luminesce in oxides? A first-principles exploration. J. Phys. Chem. Lett. 2022, 13, 10635–10641. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.B.; Quan, Q.S.; Wei, X.T.; Duan, C.K. Understanding near-infrared luminescence in Cr-doped La-gallogermanates through first-principles calculations and ligand-field theory. Phys. Rev. B 2023, 106, 155136. [Google Scholar] [CrossRef]
- Kurboniyon, M.S.; Lou, B.B.; Zafari, U.; Rahimi, F.; Srivastava, A.M.; Yamamoto, T.; Brik, M.G.; Ma, C.G. First-principles study of geometric and electronic structures, and optical transition energies of Mn4+ impurity ions: K2SiF6 as a prototype. J. Lumin. 2023, 263, 120103. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Janotti, A.; Van de Walle, C.G. First-principles calculations for point defects in solids. Rev. Mod. Phys. 2014, 86, 253–305. [Google Scholar] [CrossRef]
- Durrant, T.; Murphy, S.; Watkins, M.; Shluger, A. Relation between image charge and potential alignment corrections for charged defects in periodic boundary conditions. J. Chem. Phys. 2018, 149, 024103. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Razaznejad, B.; Lundqvist, B.I. Potential-energy surfaces for excited states in extended systems. J. Chem. Phys. 2004, 120, 4593–4602. [Google Scholar] [CrossRef] [PubMed]
- Canning, A.; Chaudhry, A.; Boutchko, R.; Gronbech-Jensen, N. First-principles study of luminescence in Ce-doped inorganic scintillators. Phys. Rev. B 2011, 83, 125115. [Google Scholar] [CrossRef]
- Adachi, S. Luminescence spectroscopy of Cr3+ in an oxide: A strong or weak crystal-field phosphor? J. Lumin. 2021, 234, 117965. [Google Scholar] [CrossRef]
- Liberman, D.A. Slater transition-state band-structure calculations. Phys. Rev. B 2000, 62, 6851–6853. [Google Scholar] [CrossRef]
- Chen, Q.L.; Shang, L.B.; Ma, C.-G.; Duan, C.K. Angular Jahn-Teller effect and photoluminescence of the tetrahedral coordinated Mn2+ activators in solids—A first-principles study. Inorg. Chem. 2022, 61, 13471–13480. [Google Scholar] [CrossRef]
- Levy, D.; Barbier, J. Normal and inverse garnets: Ca3Fe2Ge3O12, Ca3Y2Ge3O12 and Mg3Y2Ge3O12. Acta Crystallogr. C 1999, 55, 1611–1614. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Van de Walle, C.G.; Neugebauer, J. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95, 3851–3879. [Google Scholar] [CrossRef]
- Xu, Y.N.; Ching, W.Y.; Brickeen, B.K. Electronic structure and bonding in garnet crystals Gd3Sc2Ga3O12, Gd3Sc2Al3O12, and Gd3Ga5O12 compared to Y3Al5O12. Phys. Rev. B 2000, 61, 1817–1824. [Google Scholar] [CrossRef]
- Butler, P.H. Point Group Symmetry Applications, Method and Tables; Plenum Press: New York, NY, USA, 1981. [Google Scholar]
- Brik, M.G.; Avram, N.M. (Eds.) Optical Properties of 3D-Ions in Crystals; Tsinghua University Press: Beijing, China; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Dorenbos, P. Charge transfer bands in optical materials and related defect level. Opt. Mater. 2017, 69, 8–22. [Google Scholar] [CrossRef]
- Senden, T.; van Dijk-Moes, R.J.A.; Meijerink, A. Quenching of the red Mn4+ luminescence in Mn4+-doped fluoride LED phosphors. Light-Sci. Appl. 2018, 7, 8. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Parameter | Calc. | Expt. a |
|---|---|---|---|
| Ca3Y2Ge3O12 | a = b = c | 12.9381 | 12.8059 |
| O (x, y, z) | 0.9644, 0.0557, 0.1604 | 0.9637, 0.0567, 0.1609 | |
| V(host) | 2165.7508 | 2100.0533 | |
| Y3+-6O2− | 2.245 | 2.234 | |
| Ge4+-4O2− | 1.789 | 1.766 | |
| Ca2+-4O(1)2− | 2.486 | 2.469 | |
| Ca2+-4O(2)2− | 2.596 | 2.560 | |
| Ca3Y2Ge3O12:Cr3+ | V(Cr3+/Y3+) | 2155.4599 | - |
| Cr3+/Y3+-6O2− | 2.062 | - | |
| V(Cr3+/Ge4+) | 2187.9207 | - | |
| Cr3+/Ge4+-4O2− | 1.932 | - | |
| V(Cr3+/Ca2+) | 2148.4651 | - | |
| Cr3+/Ca2+-4O(1)2− | 2.142 | - | |
| Cr3+/Ca2+-4O(2)2− | 2.576 | - |
| Excitation | Emission | ZPL | Stokes Shift | |
|---|---|---|---|---|
| 2E | 1.80 | 1.78 | 1.79 | 0.02 |
| 4T2 | ||||
| Scheme 1 | 1.82 | 1.55 | 1.76 | 0.27 |
| Scheme 2 | 1.82 | 1.39 | 1.71 | 0.43 |
| Expt. a | 1.83 | 1.55 | - | 0.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, W.; Lou, B.; Kurboniyon, M.S.; Buryi, M.; Rahimi, F.; Srivastava, A.M.; Brik, M.G.; Wang, J.; Ma, C. Unraveling Broadband Near-Infrared Luminescence in Cr3+-Doped Ca3Y2Ge3O12 Garnets: Insights from First-Principles Analysis. Materials 2024, 17, 1709. https://doi.org/10.3390/ma17071709
Zou W, Lou B, Kurboniyon MS, Buryi M, Rahimi F, Srivastava AM, Brik MG, Wang J, Ma C. Unraveling Broadband Near-Infrared Luminescence in Cr3+-Doped Ca3Y2Ge3O12 Garnets: Insights from First-Principles Analysis. Materials. 2024; 17(7):1709. https://doi.org/10.3390/ma17071709
Chicago/Turabian StyleZou, Wei, Bibo Lou, Mekhrdod S. Kurboniyon, Maksym Buryi, Farhod Rahimi, Alok M. Srivastava, Mikhail G. Brik, Jing Wang, and Chonggeng Ma. 2024. "Unraveling Broadband Near-Infrared Luminescence in Cr3+-Doped Ca3Y2Ge3O12 Garnets: Insights from First-Principles Analysis" Materials 17, no. 7: 1709. https://doi.org/10.3390/ma17071709
APA StyleZou, W., Lou, B., Kurboniyon, M. S., Buryi, M., Rahimi, F., Srivastava, A. M., Brik, M. G., Wang, J., & Ma, C. (2024). Unraveling Broadband Near-Infrared Luminescence in Cr3+-Doped Ca3Y2Ge3O12 Garnets: Insights from First-Principles Analysis. Materials, 17(7), 1709. https://doi.org/10.3390/ma17071709