
Effect of Different Substituents on the Properties of 4-R-1,5-Diaminotetrazolium Pentazolate Salts


Abstract
1. Introduction
2. Calculation Details
3. Results and Discussion
3.1. Interaction Energy and Its Composition Analysis
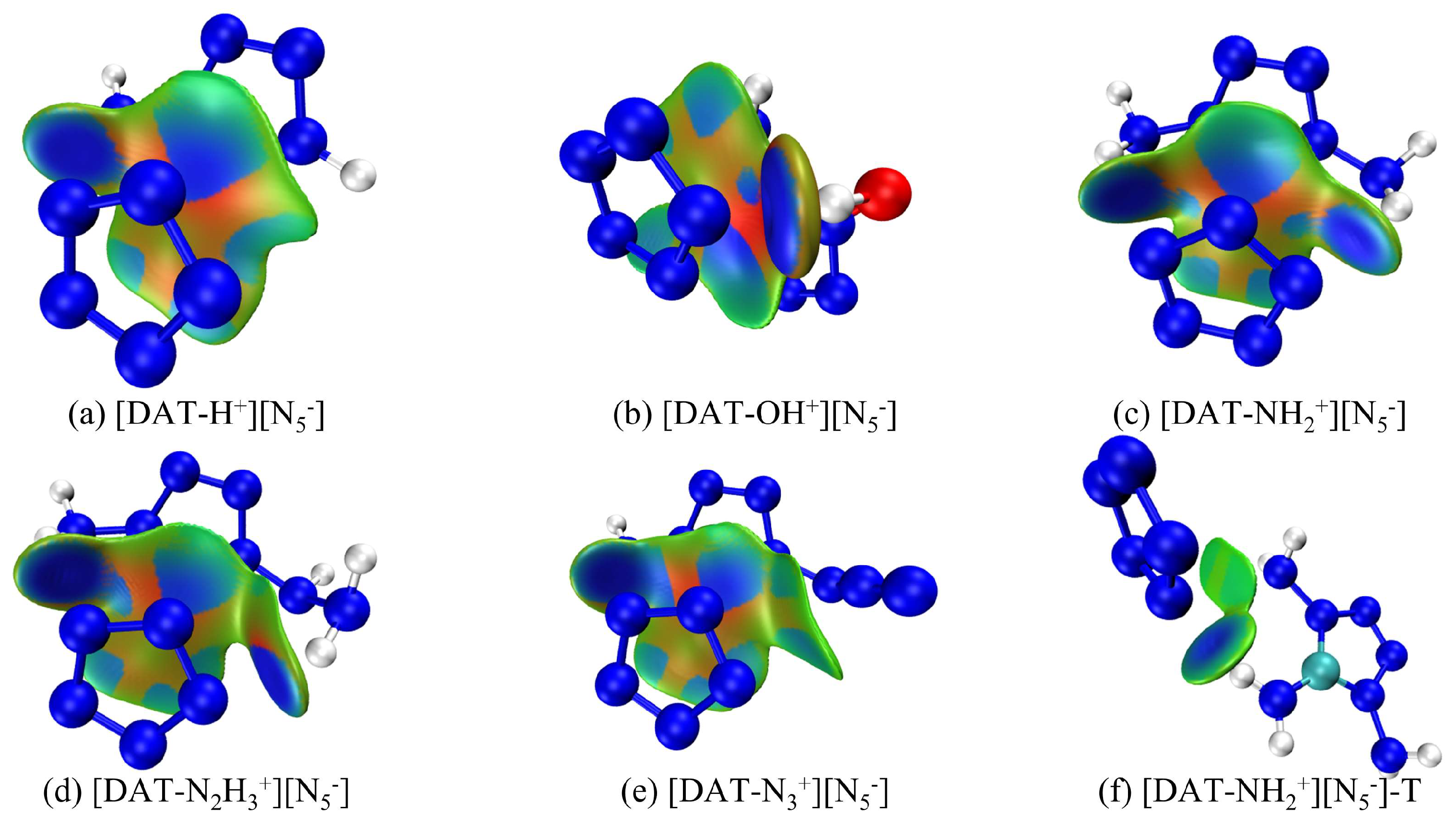
3.2. IGMH Analysis
3.3. AIM Analysis
3.4. Electrostatic Potential Analysis
3.5. Detonation Performance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Christe, K. Polynitrogen chemistry enters the ring. Science 2017, 355, 351. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, Y.; Lin, Q.; Lu, M. Recent advances in the syntheses and properties of polynitrogen pentazolate anion cyclo-N5− and its derivatives. Chem. Soc. Rev. 2018, 47, 7522–7538. [Google Scholar] [CrossRef]
- Lin, Q.; Wang, P.; Xu, Y.; Lu, M. Pentazolate anion cyclo-N5−: Development of a new energetic material. Engineering 2020, 6, 964–966. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, C.; Hu, B.; Yu, C.; Zheng, Z.; Sun, C. A symmetric Co(N5)2(H2O)4·4 H2O high-nitrogen compound formed by cobalt (II) cation trapping of a cyclo-N5− anio. Angew. Chem. Int. Ed. 2017, 56, 4512–4514. [Google Scholar] [CrossRef] [PubMed]
- Laniel, D.; Trybel, F.; Yin, Y.; Fedotenko, T.; Khandarkhaeva, S.; Aslandukov, A.; Aprilis, G.; Abrikosov, A.; Masood, T.B.; Giacobbe, C.; et al. Aromatic hexazine [N6]4− anion featured in the complex structure of the high-pressure potassium nitrogen compound K9N56. Nat. Chem. 2023, 15, 641–646. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Martin, F.A.; Stierstorfer, J. C2N14: An energetic and highly sensitive binary azidotetrazole. Angew. Chem. Int. Ed. 2011, 50, 4227. [Google Scholar] [CrossRef]
- Ugi, I.; Huisgen, R.; Clusius, K.; Vecchi, M. Zur reaktion des benzol-diazonium-ions mit azid nachweis des phenyl-pentazols als zwischenstufe. Angew. Chem. Int. Ed. 1956, 68, 753–754. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W.; Sheehy, J.A.; Boatz, J.A. N5+: A novel homoleptic polynitrogen ion as a high energy density material. Angew. Chem. Int. Ed. 1999, 38, 2004–2009. [Google Scholar] [CrossRef]
- Hirshberg, B.; Gerber, R.B.; Krylov, A.I. Calculations predict a stable molecular crystal of N8. Nat. Chem. 2014, 6, 52–56. [Google Scholar] [CrossRef]
- Peng, F.; Yao, Y.; Liu, H.; Ma, Y.M. Crystalline LiN5 predicted from first-principles as a possible high-energy material. J. Phys. Chem. Lett. 2015, 6, 2363–2366. [Google Scholar] [CrossRef]
- Bazanov, B.; Geiger, U.; Carmieli, R.; Grinstein, D.; Welner, S. Detection of cyclo-N5− in THF solution. Angew. Chem. Int. Ed. 2016, 55, 13233–13235. [Google Scholar] [CrossRef]
- Zhang, C.; Sun, C.; Hu, B.; Yu, C.; Lu, M. Synthesis and characterization of the pentazolate anion cyclo-N5− in (N5)6(H3O)3(NH4)4Cl. Science 2017, 355, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Xu, H. Comment on “Synthesis and characterization of the pentazolate anion cyclo-N5− in (N5)6(H3O)3(NH4)4Cl”. Science 2018, 359, eaao3672. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Zhang, L.; Sun, C.; Zhang, C.; Yang, C.; Chen, J.; Hu, B.C. Response to Comment on “Synthesis and characterization of the pentazolate anion cyclo-N5– in (N5)6(H3O)3(NH4)4Cl”. Science 2018, 359, aas8953. [Google Scholar] [CrossRef]
- Huang, H.; Zhong, J.; Ma, L.; Lv, L.; Francisco, J.S.; Zeng, X. Reconciling the debate on the existence of pentazole HN5 in the pentazolate salt of (N5)6(H3O)3(NH4)4Cl. J. Am. Chem. Soc. 2019, 141, 2984–2989. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Q.; Shen, C.; Lin, Q.; Wang, P.; Lu, M. A series of energetic metal pentazolate hydrates. Nature 2017, 549, 78–81. [Google Scholar] [CrossRef]
- Xu, Y.; Tian, L.; Li, D.; Wang, P.; Lu, M. A series of energetic cyclo-pentazolate salts: Rapid synthesis, characterization, and promising performance. J. Mater. Chem. A 2019, 7, 12468–12479. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, C.; Zheng, Z.; Jiang, C.; Luo, J.; Du, Y.; Hu, B.; Sun, C.; Christe, K.O. Synthesis and characterization of cyclo-pentazolate salts of NH4+, NH3OH+, N2H5+, C (NH2)3+, and N (CH3)4+. J. Am. Chem. Soc. 2018, 140, 16488–16494. [Google Scholar] [CrossRef]
- Xu, Y.; Ding, L.; Yang, F.; Li, D.; Wang, P.; Lin, Q.; Lu, M. LiN5: A novel pentazolate salt with high nitrogen content. Chem. Eng. J. 2022, 429, 132399. [Google Scholar] [CrossRef]
- Hou, T.; Yuan, X.; Jiang, S.; Xu, Z.; Zhang, X.; Lu, M.; Xu, Y. Experimental detection of the diamino-pentazolium cation and theoretical exploration of derived high energy materials. Sci. Rep. 2024, 14, 10120. [Google Scholar] [CrossRef]
- Yu, R.; Liu, Y.; Huang, W.; Tang, Y. A hybrid of tetrazolium and pentazolate: An energetic salt with ultrahigh nitrogen content and energy. Energ. Mater. Front. 2023, 4, 63–67. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Piercey, D.G.; Stierstorfer, J. The 1, 4, 5-triaminotetrazolium cation (CN7H6+): A highly nitrogen-rich moiety. Eur. J. Inorg. Chem. 2012, 2012, 5694–5700. [Google Scholar] [CrossRef]
- Matulis, V.E.; Lyakhov, A.S.; Gaponik, P.N. 1, 5-Diamino-1H-1, 2, 3, 4-tetrazolium picrate: X-ray molecular and crystal structures and ab initio MO calculations. J. Mol. Struct. 2003, 649, 309–314. [Google Scholar] [CrossRef]
- Yocca, S.R.; Zeller, M.; Byrd, E.F.C.; Piercey, D.G. 1, 5-Diaminotetrazole-4 N-oxide (SYX-9): A new high-performing energetic material with a calculated detonation velocity over 10 km s−1. J. Mater. Chem. A 2022, 10, 1876–1884. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T. Molclus Program. Version 1.9.9.7. Available online: http://www.keinsci.com/research/molclus.html (accessed on 23 November 2021).
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on basis sets beautiful: Seasonal plantings of diffuse basis functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef]
- Liakos, D.G.; Neese, F. Improved correlation energy extrapolation schemes based on local pair natural orbital methods. J. Phys. Chem. A 2012, 116, 4801–4816. [Google Scholar] [CrossRef]
- Neese, F.; Valeev, E.F. Revisiting the atomic natural orbital approach for basis sets: Robust systematic basis sets for explicitly correlated and conventional correlated ab initio methods? J. Chem. Theory Comput. 2011, 7, 33–43. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Zheng, J.; Xu, X.; Truhlar, D.G. Minimally augmented Karlsruhe basis sets. Theor. Chem. Acc. 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Weigend, F. A fully direct RI-HF algorithm: Implementation, optimised auxiliary basis sets, demonstration of accuracy and efficiency. Phys. Chem. Chem. Phys. 2002, 4, 4285–4291. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. Phys. Chem. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Szalewicz, K. Symmetry-adapted perturbation theory of intermolecular forces. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 254–272. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1452. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. Intermolecular interaction characteristics of the all-carboatomic ring, cyclo[18]carbon: Focusing on molecular adsorption and stacking. Carbon 2021, 171, 514–523. [Google Scholar] [CrossRef]
- Chan, B.; Deng, J.; Radom, L. G4(MP2)-6X: A cost-effective improvement to G4(MP2). J. Comput. Chem. 2011, 7, 112–120. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Zhu, R.; Zhu, S.; Zhang, S.; Liu, Y.; Liu, G.; Gou, R.; Yang, B. Initial decomposition mechanism of NH3OH+N5– crystal under thermal and shock loading: A first-principles study. J. Phys. Chem. A 2024, 128, 2121–2129. [Google Scholar] [CrossRef]
- Li, T.; Lu, T.; Lei, Q.; Xu, Y.; Lin, Q.; Lu, M.; Lu, Y.; Wang, P. Thermal decomposition kinetics of potential solid propellant combustion catalysts Fe (II), Zn (II), hydroxylammonium, and hydrazinium pentazolates. Propellants Explos. Pyrotech. 2022, 47, e202100140. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Beddall, P.M. Virial field relationship for molecular charge distributions and the spatial partitioning of molecular properties. J. Chem. Phys. 1972, 56, 3320–3329. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of CHO hydrogen bonds on the basis of the charge density. J. Chem. Phys. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Interaction region indicator: A simple real space function clearly revealing both chemical bonds and weak interactions. Chem. Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Politzer, P.; Martinez, J.; Murray, J.S.; Concha, M.C.; Toro-Labb, A. An electrostatic interaction correction for improved crystal density prediction. Mol. Phys. 2009, 107, 2095–2101. [Google Scholar] [CrossRef]
- Rice, B.M.; Hare, J.J.; Byrd, E.F.C. Accurate predictions of crystal densities using quantum mechanical molecular volumes. J. Phys. Chem. A 2007, 111, 10874–10879. [Google Scholar] [CrossRef]
- Politzer, P.; Martinez, J.; Murray, J.S.; Concha, M.C. An electrostatic correction for improved crystal density predictions of energetic ionic compounds. Mol. Phys. 2010, 108, 1391–1396. [Google Scholar] [CrossRef]
- Jenkins, H.D.B.; Roobottom, H.K.; Passmore, J.; Glasser, L. Relationships among ionic lattice energies, molecular (formula unit) volumes, and thermochemical radii. Inorg. Chem. 1999, 38, 3609–3620. [Google Scholar] [CrossRef]
- Jenkins, H.D.B.; Tudela, D.; Glasser, L. Lattice potential energy estimation for complex ionic salts from density measurements. Inorg. Chem. 2002, 41, 2364–2367. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Jacobs, S.J. Chemistry of detonations. I. A simple method for calculating detonation properties of C–H–N–O explosives. J. Chem. Phys. 1968, 48, 23–35. [Google Scholar] [CrossRef]
- Sućeska, M. Evaluation of detonation energy from EXPLO5 computer code results. Propellants Explos. Pyrotech. 1999, 24, 280–285. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | EA,AB | EB,AB | EBSSE | Eint,orca | Eint,SAPT |
|---|---|---|---|---|---|
| [DAT-H+] [N5−] | −718,006.26 | −968,572.03 | 0.19 | −438.76 | −440.03 |
| [DAT-OH+] [N5−] | −718,005.30 | −1,165,736.11 | 0.05 | −500.47 | −502.98 |
| [DAT-NH2+] [N5−] | −718,005.96 | −1,113,698.28 | 0.28 | −445.44 | −447.86 |
| [DAT-N2H3+] [N5−] | −718,005.97 | −1,258,809.31 | 0.01 | −447.63 | −450.16 |
| [DAT-N3+] [N5−] | −718,006.07 | −1,397,493.97 | 0.09 | −451.58 | −454.64 |
| System | Interaction | ρ(r) | ▽2ρ(r) | H(r) | G(r) | V(r) |
|---|---|---|---|---|---|---|
| [DAT-H+] [N5−] | N15-N1 | 0.01341 | 0.05362 | 0.00201 | 0.01140 | −0.00939 |
| C11-N4 | 0.01565 | 0.05842 | 0.00161 | 0.01299 | −0.01138 | |
| N3-H13 | 0.01867 | 0.06528 | 0.00201 | 0.01431 | −0.01229 | |
| [DAT-OH+] [N5−] | H13-N4 | 0.00868 | 0.03168 | 0.00152 | 0.00640 | −0.00488 |
| N2-H7 | 0.05507 | 0.08633 | −0.01455 | 0.03613 | −0.05068 | |
| N15-N1 | 0.01125 | 0.04504 | 0.00182 | 0.00944 | −0.00763 | |
| N3-N8 | 0.01616 | 0.06229 | 0.00162 | 0.01395 | −0.01233 | |
| [DAT-NH2+] [N5−] | H15-N2 | 0.01728 | 0.06135 | 0.00207 | 0.01327 | −0.01120 |
| N5-H17 | 0.01729 | 0.06139 | 0.00207 | 0.01328 | −0.01121 | |
| N3-N10 | 0.01061 | 0.04355 | 0.00177 | 0.00912 | −0.00735 | |
| N10-N4 | 0.01061 | 0.04355 | 0.00177 | 0.00912 | −0.00735 | |
| C13-N1 | 0.01425 | 0.05460 | 0.00161 | 0.01204 | −0.01042 | |
| [DAT-NH2+] [N5−]-T | N6-H9 | 0.01837 | 0.06691 | 0.00260 | 0.01412 | −0.01152 |
| [DAT-N2H3+] [N5−] | H19-N4 | 0.01978 | 0.06938 | 0.00214 | 0.01521 | −0.01307 |
| N1-H15 | 0.02067 | 0.07257 | 0.00195 | 0.01619 | −0.01424 | |
| N3-N10 | 0.01005 | 0.04271 | 0.00179 | 0.00888 | −0.00709 | |
| N10-N2 | 0.01060 | 0.04288 | 0.00188 | 0.00883 | −0.00695 | |
| N5-C13 | 0.01424 | 0.05277 | 0.00154 | 0.01165 | −0.01010 | |
| [DAT-N3+] [N5−] | H15-N1 | 0.01740 | 0.06282 | 0.00208 | 0.01363 | −0.01155 |
| C13-N5 | 0.01556 | 0.05714 | 0.00148 | 0.01280 | −0.01132 | |
| N10-N3 | 0.01106 | 0.04689 | 0.00193 | 0.00980 | −0.00787 | |
| N18-N4 | 0.00954 | 0.04171 | 0.00184 | 0.00859 | −0.00675 |
| System | Nitrogen Content/% | ρ (g·cm−3) a | ∆Hf (kJ·mol−1) b | D (m·s−1) c | P (GPa) d |
|---|---|---|---|---|---|
| [DAT-OH+] [N5−] | 82.35 | 1.638 | 615.56 | 8924.96 | 28.85 |
| [DAT-H+] [N5−] | 90.06 | 1.590 | 669.06 | 8835.06 | 27.39 |
| [DAT-NH2+] [N5−]-T | 90.32 | 1.575 | 763.77 | 8977.92 | 28.16 |
| [DAT-NH2+] [N5−] | 90.32 | 1.583 | 763.15 | 9018.09 | 28.56 |
| [DAT-N2H3+] [N5−] | 90.55 | 1.569 | 888.32 | 9179.52 | 29.70 |
| [DAT-N3+] [N5−] | 92.45 | 1.643 | 1153.71 | 9295.77 | 32.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.; Xu, Z.; Lu, M.; Xu, Y. Effect of Different Substituents on the Properties of 4-R-1,5-Diaminotetrazolium Pentazolate Salts. Materials 2025, 18, 1077. https://doi.org/10.3390/ma18051077
Yuan X, Xu Z, Lu M, Xu Y. Effect of Different Substituents on the Properties of 4-R-1,5-Diaminotetrazolium Pentazolate Salts. Materials. 2025; 18(5):1077. https://doi.org/10.3390/ma18051077
Chicago/Turabian StyleYuan, Xiaofeng, Ze Xu, Ming Lu, and Yuangang Xu. 2025. "Effect of Different Substituents on the Properties of 4-R-1,5-Diaminotetrazolium Pentazolate Salts" Materials 18, no. 5: 1077. https://doi.org/10.3390/ma18051077
APA StyleYuan, X., Xu, Z., Lu, M., & Xu, Y. (2025). Effect of Different Substituents on the Properties of 4-R-1,5-Diaminotetrazolium Pentazolate Salts. Materials, 18(5), 1077. https://doi.org/10.3390/ma18051077