
Preparation of Phosphate Glass by the Conventional and Microwave Melt-Quenching Methods and Research on Its Performance

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of PG
2.3. Characterization of Materials
2.4. Dissolution Test
2.4.1. Determination of the Slow-Release Dissolution Rate of PG
2.4.2. Effect of Temperature and pH on Dissolution Rate
2.4.3. Measurement of Orthophosphate and Total Phosphorus
2.5. Test of Scale Inhibition
2.5.1. Measurement of the Scale Inhibition Efficiency of PG
2.5.2. Effect of Temperature and pH on Scale Inhibition Efficiency
3. Results and Discussion
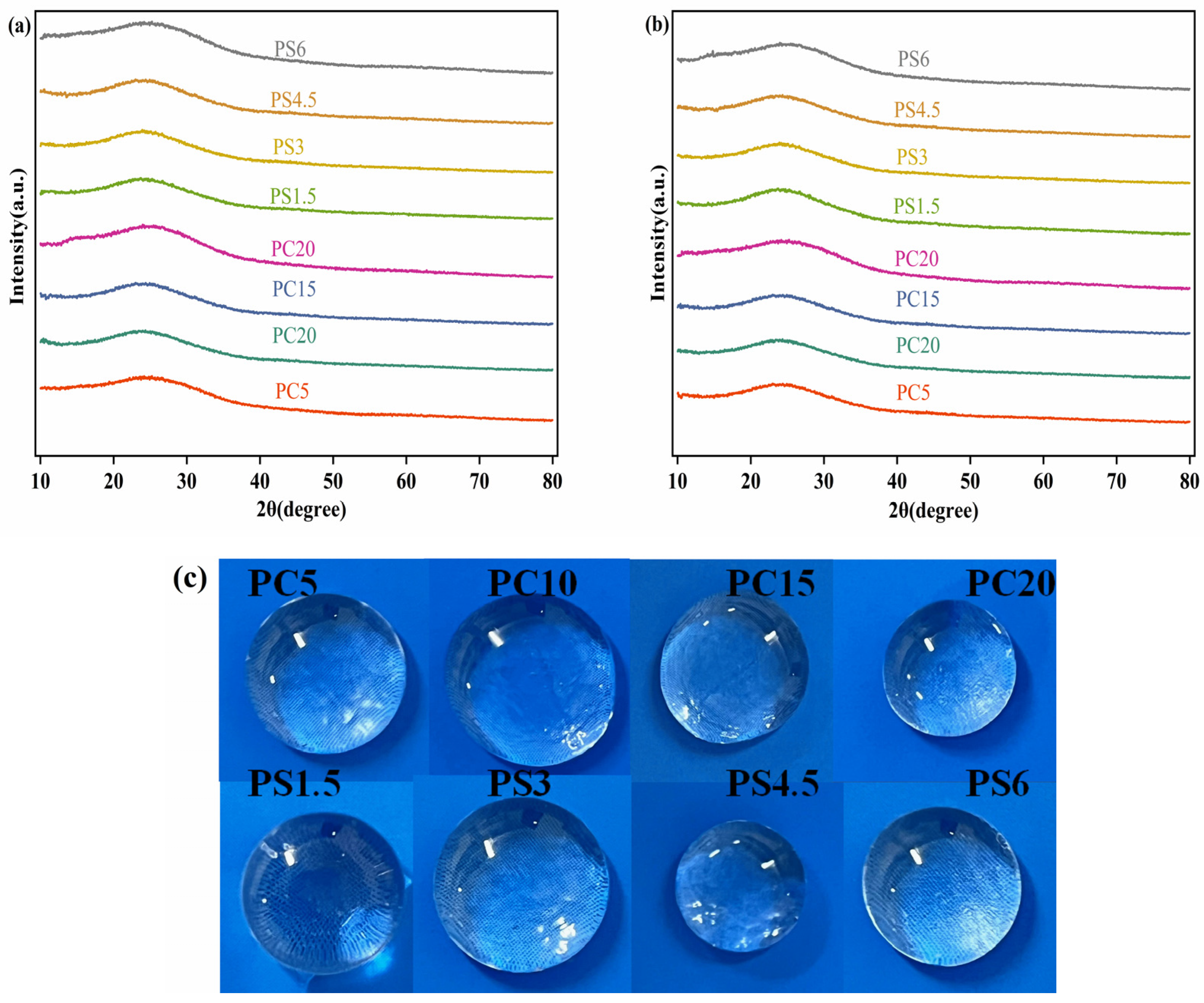
3.1. XRD Characterization
3.2. Structural Characteristics
3.2.1. FTIR Analysis
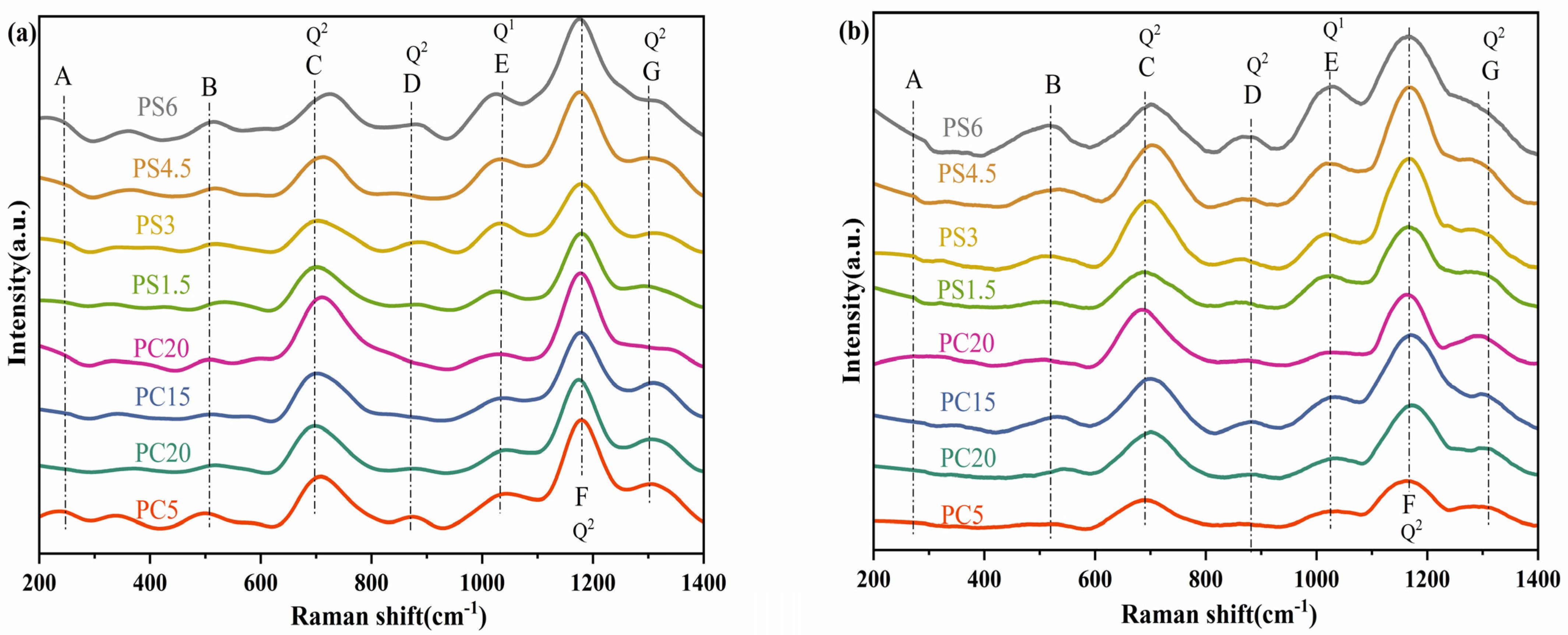
3.2.2. Raman Spectroscopy Analysis
3.3. Physical and Thermal Properties
3.4. Dissolution Behavior
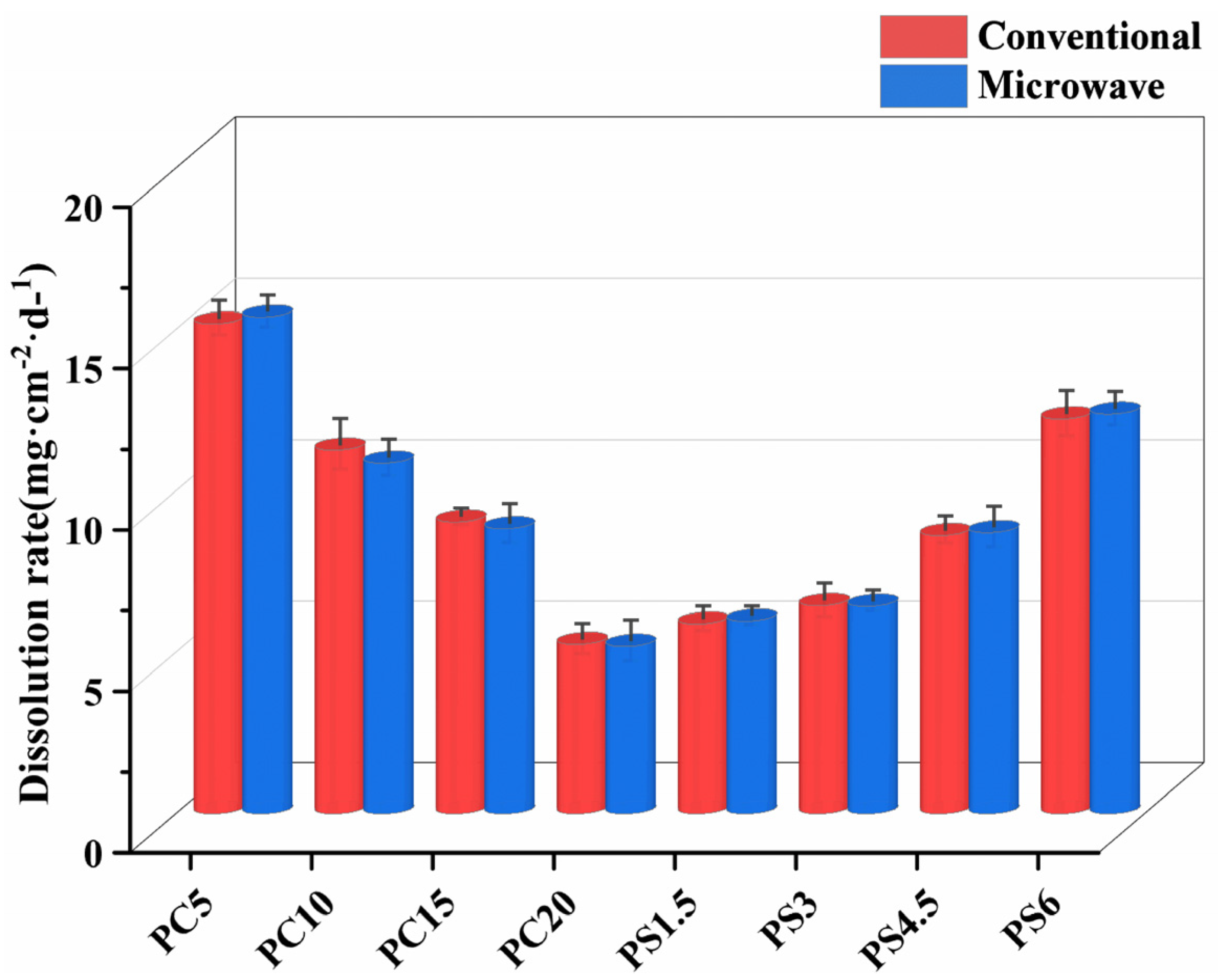
3.4.1. Weight Loss
3.4.2. Effect of pH and Temperature on Dissolution Rate
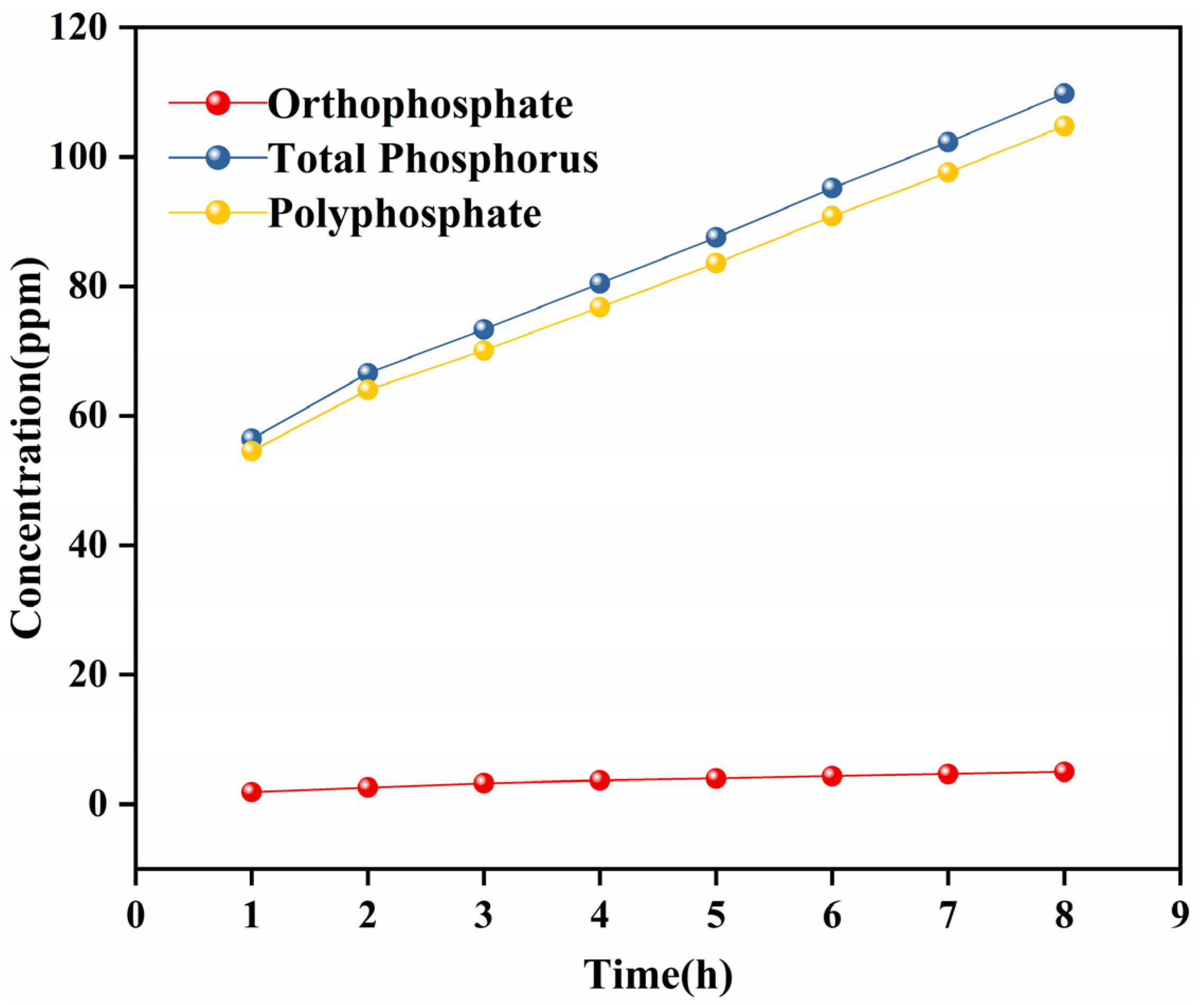
3.4.3. Phosphorus Release
3.5. Evaluation of Scale Inhibition Performance
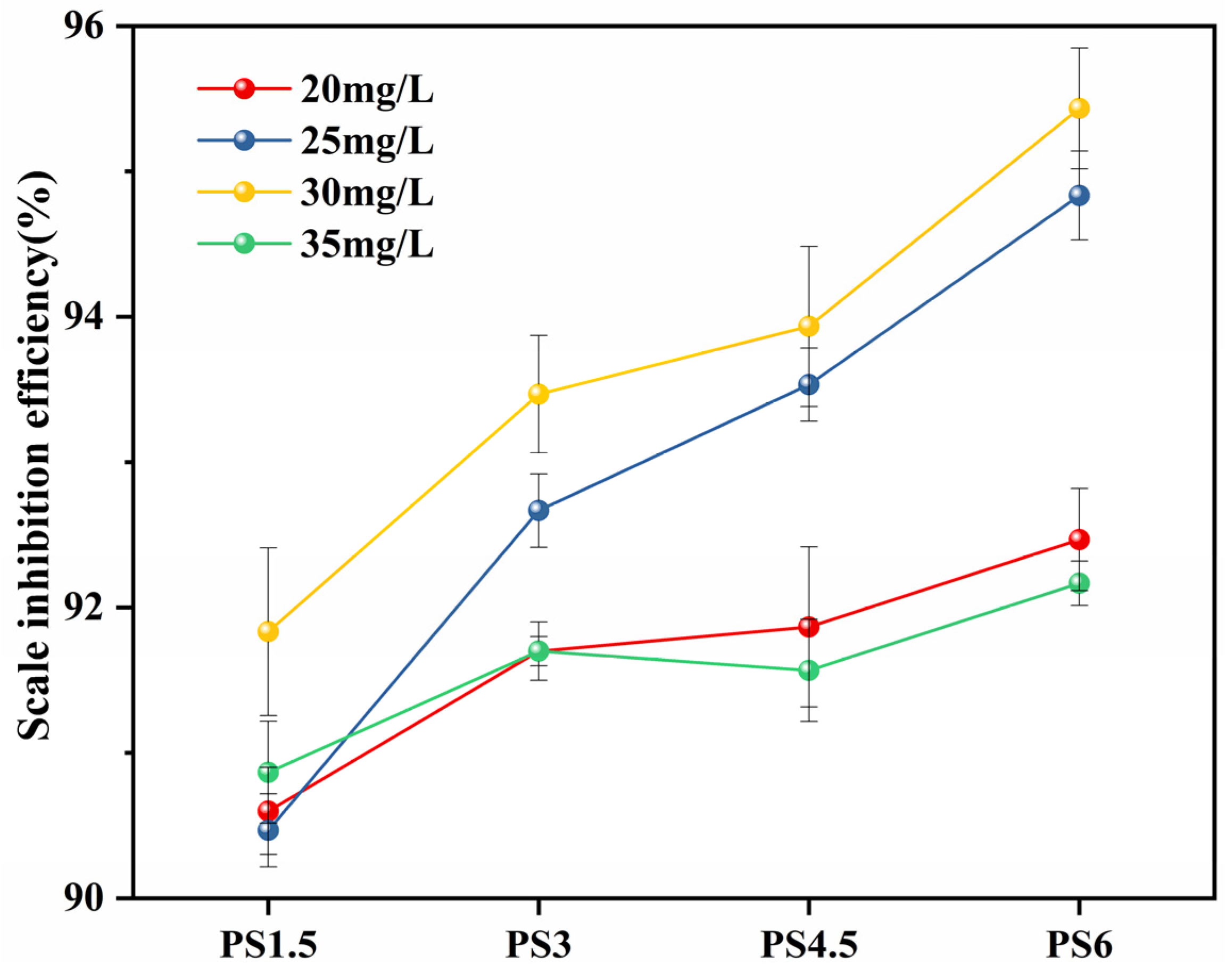
3.5.1. Scale Inhibition Efficiency at Optimal Scale Inhibition Concentration
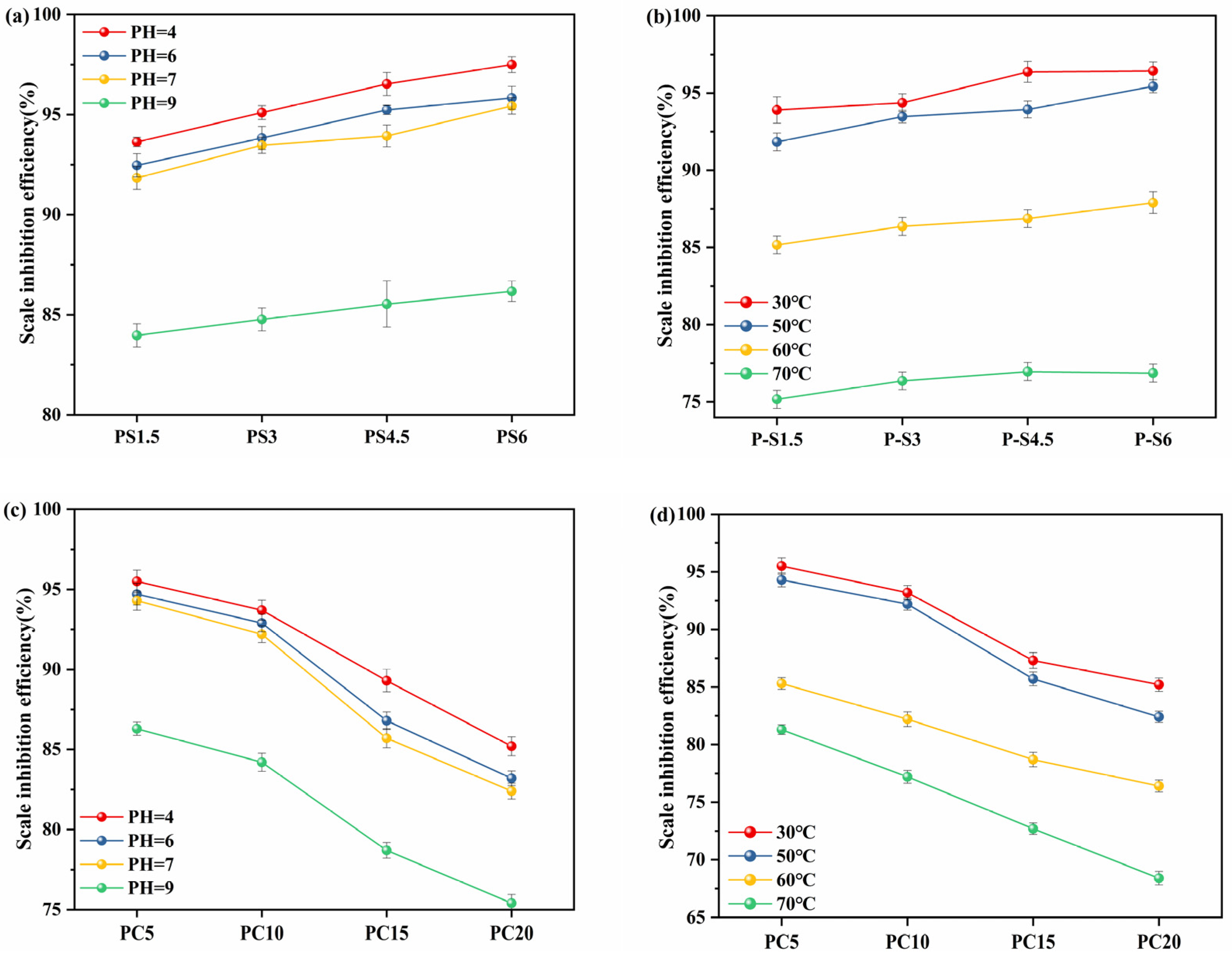
3.5.2. Effect of pH and Temperature on Scale Inhibition
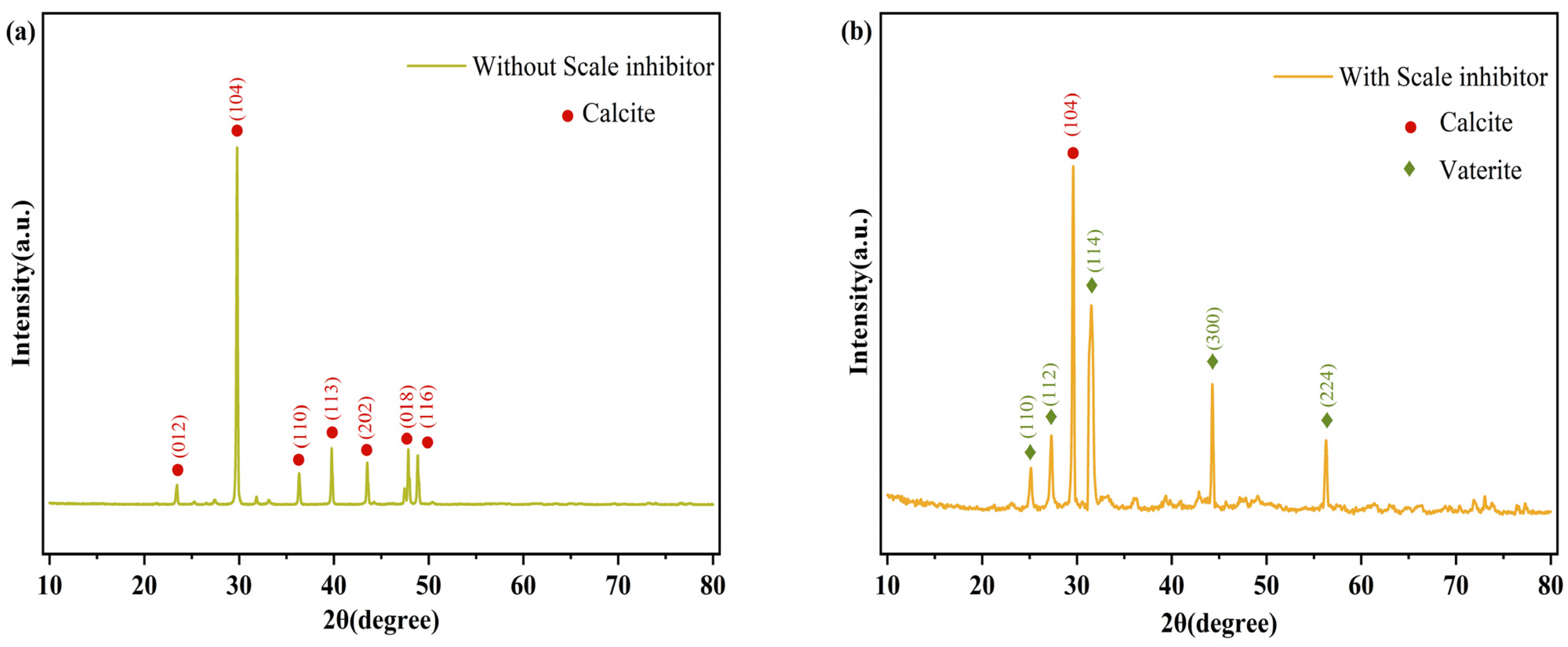
3.5.3. Characterization of CaCO3 Crystals by SEM and XRD
4. Conclusions
- (1)
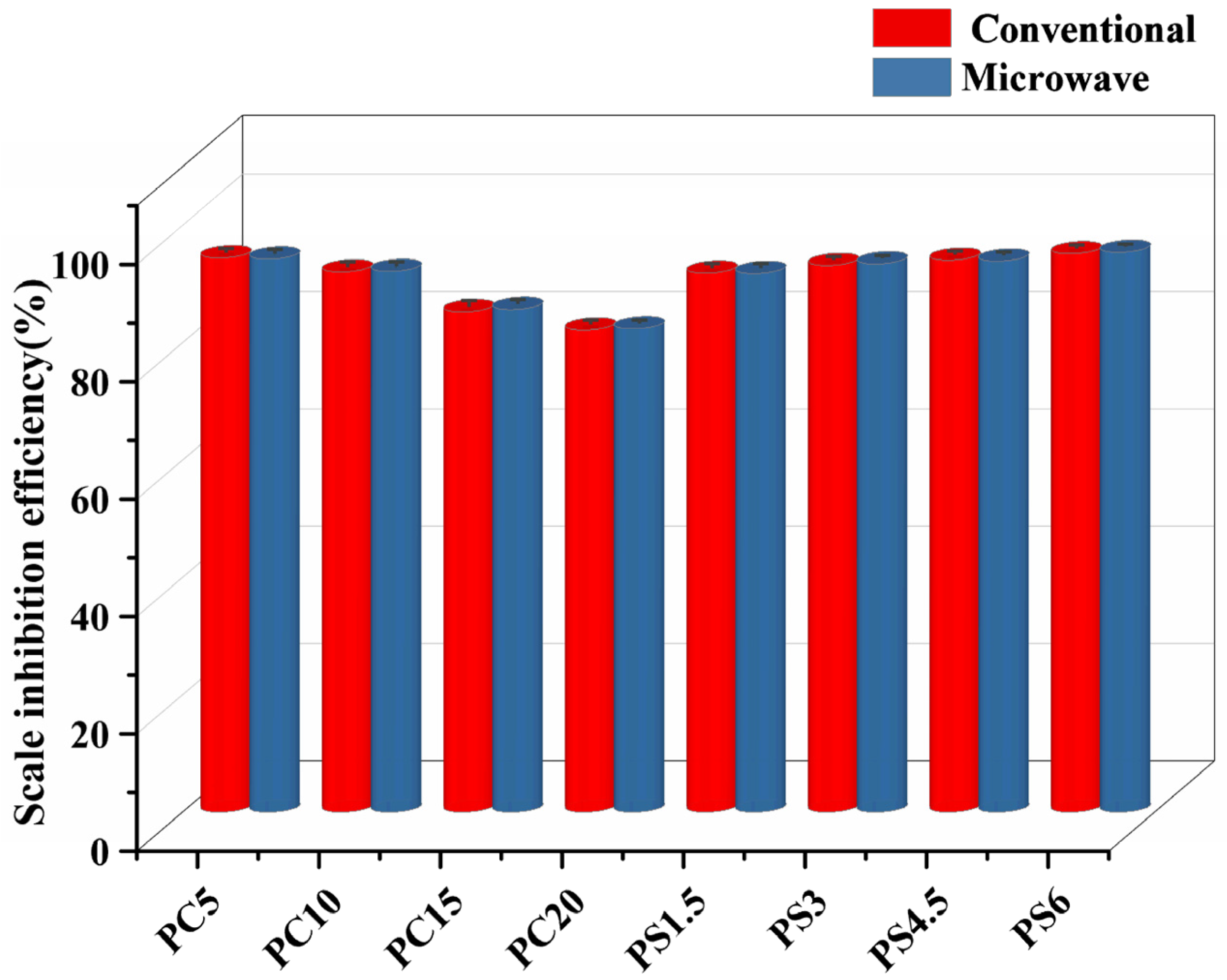
- PG prepared by both methods exhibits similar structural and thermal properties, as well as comparable slow-release dissolution behavior and scale inhibition performance. However, due to microwaves accelerating the melt homogenization process and reducing structural defects, glass synthesized by the microwave-quenching method is slightly denser than that prepared by the conventional melt-quenching method. Under the condition of achieving the same density and performance, when using the microwave melting and quenching method, the melting time is shortened by approximately 78%, and the energy consumption is reduced by about 85%.
- (2)
- Structural and thermal property data: FTIR and Raman spectroscopic analyses confirmed that PG prepared by two methods (traditional melt-quenching and microwave melt-quenching) exhibited essentially identical structural characteristics, primarily consisting of Q2 structural units. Density, glass transition temperature (Tg), and crystallization temperature (Tc) data indicated regular changes in glass density and thermal stability with increasing CaO and SiO2 content. Specifically, the addition of SiO2 enhanced the strength of P-O-P bonds, while the substitution of CaO increased glass cross-linking density and Tg by forming stronger P-O-Ca bonds.
- (3)
- Dissolution behavior data: dissolution experiments conducted at various temperatures (30–70 °C) and pH (4–9) values revealed the slow-release characteristics of PG. Specifically, the decrease in pH accelerated the dissolution rate of PG, while increasing the temperature, although it accelerated the dissolution process and reduced its scale inhibition efficiency. The chemical stability of PG was quantified by measuring weight loss per unit area per unit time. Dissolution in the glass occurs through the breaking of P-O-P chains in the phosphate glass network structure. Initially, dissolution exhibits a high rate of parabolic time dependence, which quickly transitions to a linear dissolution phase.
- (4)
- Scale inhibition performance data: static scale inhibition experiments were conducted to assess the scale inhibition efficiency of PG. The results showed that the PS glass system exhibited a scale inhibition efficiency exceeding 90% at 50 °C, with some samples achieving even higher efficiencies of over 95%. PG inhibits scale formation for two reasons: firstly, the polyphosphate produced by the hydrolysis of PG complexes with Ca2+ in the solution, reduces the concentration of free Ca2+ and prevents the crystallization of CaCO3 into limescale. Secondly, it induces lattice distortion in already-formed CaCO3, altering the crystal form from calcite to vaterite, which results in a looser structure that is more easily removed by flowing water.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Urman, K.; Otaigbe, J.U. New Phosphate Glass/Polymer Hybrids—Current Status and Future Prospects. Prog. Polym. Sci. 2007, 32, 1462–1498. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Zhu, C.; Parsons, A.; Liu, J.; Huang, S.; Ahmed, I.; Rudd, C.; Sharmin, N. Production and Characterisation of Novel Phosphate Glass Fibre Yarns, Textiles, and Textile Composites for Biomedical Applications. J. Mech. Behav. Biomed. 2019, 99, 47–55. [Google Scholar] [CrossRef]
- Li, W.; He, D.; Li, S.; Chen, W.; Hu, L. Investigation on Thermal Properties of a New Nd-Doped Phosphate Glass. Ceram. Int. 2014, 40, 13389–13393. [Google Scholar] [CrossRef]
- Saha, S.; Ntarisa, A.V.; Quang, N.D.; Luan, N.T.; Vuong, P.Q.; Kim, H.J.; Intachai, N.; Kothan, S.; Kaewkhao, J. Scintillation Performance of the Ce3+ -Activated Lithium Phosphate Glass. Radiat. Phys. Chem. 2022, 199, 110285. [Google Scholar] [CrossRef]
- Liu, Q.; Feng, L.; Sun, Y.; Fang, S.; Zhang, Z.; Han, N.; Wang, J.; Zhang, C.; Wang, T. Effects of Phosphate Glass on Cs+ Immobilization in Geopolymer Glass-Ceramics. Ceram. Int. 2022, 49, 6545–6553. [Google Scholar] [CrossRef]
- Gasiorowski, A.; Benavente, J.F. Thermoluminescence Study of Indium Doped Phosphate Glass. Radiat. Phys. Chem. 2024, 217, 111503. [Google Scholar] [CrossRef]
- Huang, Q.; Ding, H.; Zhang, M.; Bai, S.; Dai, S.; Nie, Q.; Wen, L.; Wang, X. High-Q Lasing in Nd3+-Doped Phosphate Glass Microsphere Resonators. Opt. Lett. 2023, 48, 3103–3106. [Google Scholar] [CrossRef]
- Tian, S.; Lun, Y.; Sun, Y.; Chen, D.; Tang, G.; Qian, Q.; Yang, Z. Silicate-Clad Dy3+ Doped Multi-Component Phosphate Glass Core Glass Fiber for Yellow Laser Applications. J. Non-Cryst. Solids 2022, 577, 121313. [Google Scholar] [CrossRef]
- Ahmed, I.; Lewis, M.; Olsen, I.; Knowles, J.C. Phosphate Glasses for Tissue Engineering: Part 1. Processing and Characterisation of a Ternary-Based P2O5–CaO–Na2O Glass System. Biomaterials 2004, 25, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Hossain, K.M.Z.; Hasan, M.S.; Felfel, R.; Ahmed, I. Development of Phosphate-Based Glass Fibers for Biomedical Applications. In Hot Topics in Biomaterials; Future Science Ltd.: London, UK, 2014; pp. 104–115. [Google Scholar]
- Kargozar, S.; Baino, F.; Hamzehlou, S.; Hill, R.G.; Mozafari, M. Bioactive Glasses: Sprouting Angiogenesis in Tissue Engineering. Trends Biotechnol. 2018, 36, 430–444. [Google Scholar] [CrossRef]
- Bitar, M.; Salih, V.; Mudera, V.; Knowles, J.C.; Lewis, M.P. Soluble Phosphate Glasses: In Vitro Studies Using Human Cells of Hard and Soft Tissue Origin. Biomaterials 2004, 25, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Sinanan, A.C.M.; Knowles, J.C.; Hunt, N.P.; Lewis, M.P. Craniofacial Muscle Engineering Using a 3-Dimensional Phosphate Glass Fibre Construct. Biomaterials 2005, 26, 1497–1505. [Google Scholar] [CrossRef]
- Mackevic, I.; Grigorjevaite, J.; Janulevicius, M.; Linkeviciute, A.; Sakirzanovas, S.; Katelnikovas, A. Synthesis and Optical Properties of Highly Efficient Red-Emitting K2LaNb5O15:Eu3+ Phosphors. Opt. Mater. 2019, 89, 25–33. [Google Scholar] [CrossRef]
- Qiao, J.; Ning, L.; Molokeev, M.S.; Chuang, Y.; Zhang, Q.; Poeppelmeier, K.R.; Xia, Z. Site-Selective Occupancy of Eu2+ Toward Blue-Light-Excited Red Emission in a Rb3YSi2O7:Eu Phosphor. Angew. Chem. Int. Ed. 2019, 58, 11521–11526. [Google Scholar] [CrossRef]
- Parsons, A.J.; Burling, L.D.; Scotchford, C.A.; Walker, G.S.; Rudd, C.D. Properties of Sodium-Based Ternary Phosphate Glasses Produced from Readily Available Phosphate Salts. J. Non-Cryst. Solids 2006, 352, 5309–5317. [Google Scholar] [CrossRef]
- Zhao, J.; Nienhuis, E.T.; McCloy, J.S.; Du, J. Structures of Fluoride Containing Aluminosilicate Low Activity Nuclear Waste Glasses: A Molecular Dynamics Simulations Study. J. Non-Cryst. Solids 2020, 550, 120379. [Google Scholar] [CrossRef]
- Knowles, J.C. Phosphate Based Glasses for Biomedical Applications. J. Mater. Chem. 2003, 13, 2395. [Google Scholar] [CrossRef]
- Sharmin, N.; Hasan, M.S.; Parsons, A.J.; Rudd, C.D.; Ahmed, I. Cytocompatibility, Mechanical and Dissolution Properties of High Strength Boron and Iron Oxide Phosphate Glass Fibre Reinforced Bioresorbable Composites. J. Mech. Behav. Biomed. 2016, 59, 41–56. [Google Scholar] [CrossRef]
- Felfel, R.M.; Ahmed, I.; Parsons, A.J.; Palmer, G.; Sottile, V.; Rudd, C.D. Cytocompatibility, Degradation, Mechanical Property Retention and Ion Release Profiles for Phosphate Glass Fibre Reinforced Composite Rods. Mater. Sci. Eng. C 2013, 33, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.S.; Ahmed, I.; Muja, N.; Rudd, C.D.; Bureau, M.N.; Nazhat, S.N. Effect of Phosphate-Based Glass Fibre Surface Properties on Thermally Produced Poly(Lactic Acid) Matrix Composites. J. Mater. Sci-Mater. M. 2011, 22, 2659–2672. [Google Scholar] [CrossRef] [PubMed]
- Demadis, K.D.; Mavredaki, E.; Stathoulopoulou, A.; Neofotistou, E.; Mantzaridis, C. Industrial Water Systems: Problems, Challenges and Solutions for the Process Industries. Desalination 2007, 213, 38–46. [Google Scholar] [CrossRef]
- Suharso, N.; Buhani, N.; Bahri, S.; Endaryanto, T. Gambier Extracts as an Inhibitor of Calcium Carbonate (CaCO3) Scale Formation. Desalination 2011, 265, 102–106. [Google Scholar] [CrossRef]
- Gu, X.; Qiu, F.; Zhou, X.; Qi, J.; Zhou, Y.; Guo, X.; Yang, D. Preparation, Characterization, and Inhibition Efficiency of Quadripolymer for Use as Scale Inhibitor. Int. J. Polym. Anal. Ch. 2012, 17, 321–332. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, Y.; Gu, A.; Ding, J.; Shen, Z. Investigation of Calcium Carbonate Scaling Inhibition and Scale Morphology by AFM. J. Colloid. Interface Sci. 2001, 240, 608–621. [Google Scholar] [PubMed]
- Gao, H.; Tan, T.; Wang, D. Dissolution Mechanism and Release Kinetics of Phosphate Controlled Release Glasses in Aqueous Medium. J. Control. Release 2004, 96, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Neel, E.A.A.; Pickup, D.M.; Valappil, S.P.; Newport, R.J.; Knowles, J.C. Bioactive Functional Materials: A Perspective on Phosphate-Based Glasses. J. Mater. Chem. 2009, 19, 690–701. [Google Scholar] [CrossRef]
- Shi, Q.; Kang, J.; Qu, Y.; Liu, S.; Khater, G.A.; Li, S.; Wang, Y.; Yue, Y. Effect of Rare-Earth Oxides on Structure and Chemical Resistance of Calcium Aluminophosphate Glasses. J. Non-Cryst. Solids 2018, 491, 71–78. [Google Scholar]
- Chatterjee, A.; Ghosh, A. Correlation between Ion Transport and Network Structure of Li2O-P2O5 Glasses. Solid. State Ionics 2018, 314, 1–8. [Google Scholar]
- Al-Zaibani, M.; Shahboub, A.; Ramadan, R.M.; Altawaf, A.; El Agammy, E.F. Structure and physical properties of Li2O–Fe2O3–P2O5 glasses. Phys. Scripta 2021, 96, 125701. [Google Scholar] [CrossRef]
- Ersundu, M.Ç.; Kuzu, B.; Ersundu, A.E. Structural Properties and Dissolution Behavior of New Generation Controlled Release Phosphate Glass Fertilizers. J. Non-Cryst. Solids 2022, 576, 121239. [Google Scholar] [CrossRef]
- Menezes, R.R.; Kiminami, R.H.G.A. Microwave Sintering of Alumina–Zirconia Nanocomposites. J. Mater. Process. Tech. 2008, 203, 513–517. [Google Scholar] [CrossRef]
- Panneerselvam, M.; Rao, K.J. A Microwave Method for the Preparation and Sintering of β′-SiAlON. Mater. Res. Bull. 2003, 38, 663–674. [Google Scholar] [CrossRef]
- Souto, P.M.; Menezes, R.R.; Kiminami, R.H.G.A. Effect of Y2O3 Additive on Conventional and Microwave Sintering of Mullite. Ceram. Int. 2011, 37, 241–248. [Google Scholar] [CrossRef]
- Filho, J.C.; Zilio, S.C.; Messias, D.N.; Pilla, V.; Silva, A.C.A.; Dantas, N.O.; Andrade, A.A. Effects of Aluminum Substitution by Potassium in the P2O5–Al2O3–Na2O–K2O Phosphate Glasses. J. Alloys Compd. 2020, 815, 152359. [Google Scholar] [CrossRef]
- Jha, P.K.; Pandey, O.P.; Singh, K. FTIR Spectral Analysis and Mechanical Properties of Sodium Phosphate Glass–Ceramics. J. Mol. Struct. 2015, 1083, 278–285. [Google Scholar] [CrossRef]
- Baia, L.; Muresan, D.; Baia, M.; Popp, J.; Simon, S. Structural Properties of Silver Nanoclusters–Phosphate Glass Composites. Vib. Spectrosc. 2007, 43, 313–318. [Google Scholar] [CrossRef]
- Glorieux, B.; Salminen, T.; Massera, J.; Lastusaari, M.; Petit, L. Better Understanding of the Role of SiO2, P2O5 and Al2O3 on the Spectroscopic Properties of Yb3+ Doped Silica Sol-Gel Glasses. J. Non-Cryst. Solids 2018, 482, 46–51. [Google Scholar] [CrossRef]
- Logrado, M.; Eckert, H.; Ikeda, H.; Nakane, S.; Yamazaki, H. Silica Incorporation into Sodium Aluminum Phosphate Glasses: Structural Characterization by Raman Spectroscopy and Multinuclear Solid-State NMR. J. Non-Cryst. Solids 2022, 579, 121366. [Google Scholar] [CrossRef]
- Karakassides, M.A.; Saranti, A.; Koutselas, I. Preparation and Structural Study of Binary Phosphate Glasses with High Calcium and/or Magnesium Content. J. Non-Cryst. Solids 2004, 347, 69–79. [Google Scholar] [CrossRef]
- Silva, A.M.B.; Correia, R.N.; Oliveira, J.M.M.; Fernandes, M.H.V. Structural Characterization of TiO2–P2O5–CaO Glasses by Spectroscopy. J. Eur. Ceram. Soc. 2010, 30, 1253–1258. [Google Scholar] [CrossRef]
- Niyogi, S.; Sen Gupta, B. Mechanical properties and pore size distribution in athermal porous glasses. J. Soft Matter. 2021, 17, 9716–9724. [Google Scholar] [CrossRef] [PubMed]
- Menezes, R.R.; Souto, P.M.; Kiminami, R.H.G.A. Microwave Hybrid Fast Sintering of Porcelain Bodies. J. Mater. Process. Tech. 2007, 190, 223–229. [Google Scholar] [CrossRef]
- Menaa, B.; Mizuno, M.; Takahashi, M.; Tokuda, Y.; Yoko, T. Polycarboxylic Acids as Network Modifiers for Water Durability Improvement of Inorganic–Organic Hybrid Tin-Silico-Phosphate Low-Melting Glasses. J. Solid. State Chem. 2006, 179, 492–499. [Google Scholar] [CrossRef]
- Sharmin, N.; Rudd, C.D. Structure, Thermal Properties, Dissolution Behaviour and Biomedical Applications of Phosphate Glasses and Fibres: A Review. J. Mater. Sci. 2017, 52, 8733–8760. [Google Scholar] [CrossRef]
- Oosterbeek, R.N.; Margaronis, K.I.; Zhang, X.C.; Best, S.M.; Cameron, R.E. Non-Linear Dissolution Mechanisms of Sodium Calcium Phosphate Glasses as a Function of pH in Various Aqueous Media. J. Eur. Ceram. Soc. 2021, 41, 901–911. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glass System | Sample Code | Theoretical Composition (mol%) | |||
|---|---|---|---|---|---|
| P2O5 | Na2O | CaO | SiO2 | ||
| PC | PC5 | 50 | 45 | 5 | - |
| PC10 | 50 | 40 | 10 | - | |
| PC15 | 50 | 35 | 15 | - | |
| PC20 | 50 | 30 | 20 | - | |
| PS | PS1.5 | 48.5 | 30 | 20 | 1.5 |
| PS3 | 47 | 30 | 20 | 3 | |
| PS4.5 | 45.5 | 30 | 20 | 4.5 | |
| PS6 | 44 | 30 | 20 | 6 | |
| Peak | Attribution |
|---|---|
| A | Asymmetric stretching vibration of P=O bond [36] |
| B | Symmetric stretching vibration of PO2 in Q2 unit [31] |
| C | Symmetric stretching vibration of Q1 unit [36] |
| D | Asymmetric stretching vibration of P-O-P bond in Q2 unit [31] |
| E | Asymmetric stretching vibration of P-O-P bond in Q2 unit [31] |
| F | Symmetric stretching vibration of P-O-P bond in Q2 unit [31] |
| G | Bending vibration of the PO4 tetrahedron [37] |
| H | Bending vibration of Si-O-Si or Si-O-P bond [35] |
| Peak | Attribution |
|---|---|
| A | Bending vibration of the PO4 tetrahedron [41] |
| B | Bending vibration of P=O bond [31,39] |
| C | Symmetric stretching vibration of P-O-P bond in Q2 unit [39,41] |
| D | Asymmetric stretching vibration of P-O-P bond in Q2 unit [39] |
| E | Symmetric stretching vibration of P-O-P in Q1 unit [39] |
| F | Symmetric stretching vibration of PO2 in Q2 unit [41] |
| G | Asymmetric stretching vibration of PO2 in Q2 unit [39,41] |
| Glass System | Sample Code | ρ(g/cm3) ± 0.001 | Tg (°C) ± 2 | Tc (°C) ± 2 | |||
|---|---|---|---|---|---|---|---|
| Melt-Quenching (m) | Microwave (w) | Melt-Quenching (m) | Microwave (w) | Melt-Quenching (m) | Microwave (w) | ||
| PC | PC5 | 2.477 | 2.476 | 283 | 280 | 383 | 382 |
| PC10 | 2.498 | 2.501 | 332 | 333 | 439 | 439 | |
| PC15 | 2.525 | 2.528 | 388 | 387 | 493 | 492 | |
| PC20 | 2.545 | 2.549 | 425 | 421 | 522 | 520 | |
| PS | PS1.5 | 2.541 | 2.540 | 445 | 446 | 579 | 578 |
| PS3 | 2.543 | 2.545 | 520 | 524 | 651 | 652 | |
| PS4.5 | 2.546 | 2.548 | 536 | 534 | 655 | 654 | |
| PS6 | 2.538 | 2.540 | 564 | 563 | 657 | 655 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Zhou, G. Preparation of Phosphate Glass by the Conventional and Microwave Melt-Quenching Methods and Research on Its Performance. Materials 2025, 18, 1079. https://doi.org/10.3390/ma18051079
Li B, Zhou G. Preparation of Phosphate Glass by the Conventional and Microwave Melt-Quenching Methods and Research on Its Performance. Materials. 2025; 18(5):1079. https://doi.org/10.3390/ma18051079
Chicago/Turabian StyleLi, Baiyi, and Guangdong Zhou. 2025. "Preparation of Phosphate Glass by the Conventional and Microwave Melt-Quenching Methods and Research on Its Performance" Materials 18, no. 5: 1079. https://doi.org/10.3390/ma18051079
APA StyleLi, B., & Zhou, G. (2025). Preparation of Phosphate Glass by the Conventional and Microwave Melt-Quenching Methods and Research on Its Performance. Materials, 18(5), 1079. https://doi.org/10.3390/ma18051079