Hydroisomerization of n-Butane over Platinum-Promoted Cesium Hydrogen Salt of 12-Tungstophosphoric Acid


Abstract
:1. Introduction
2. Experimental Section
2.1. Catalyst Syntheses
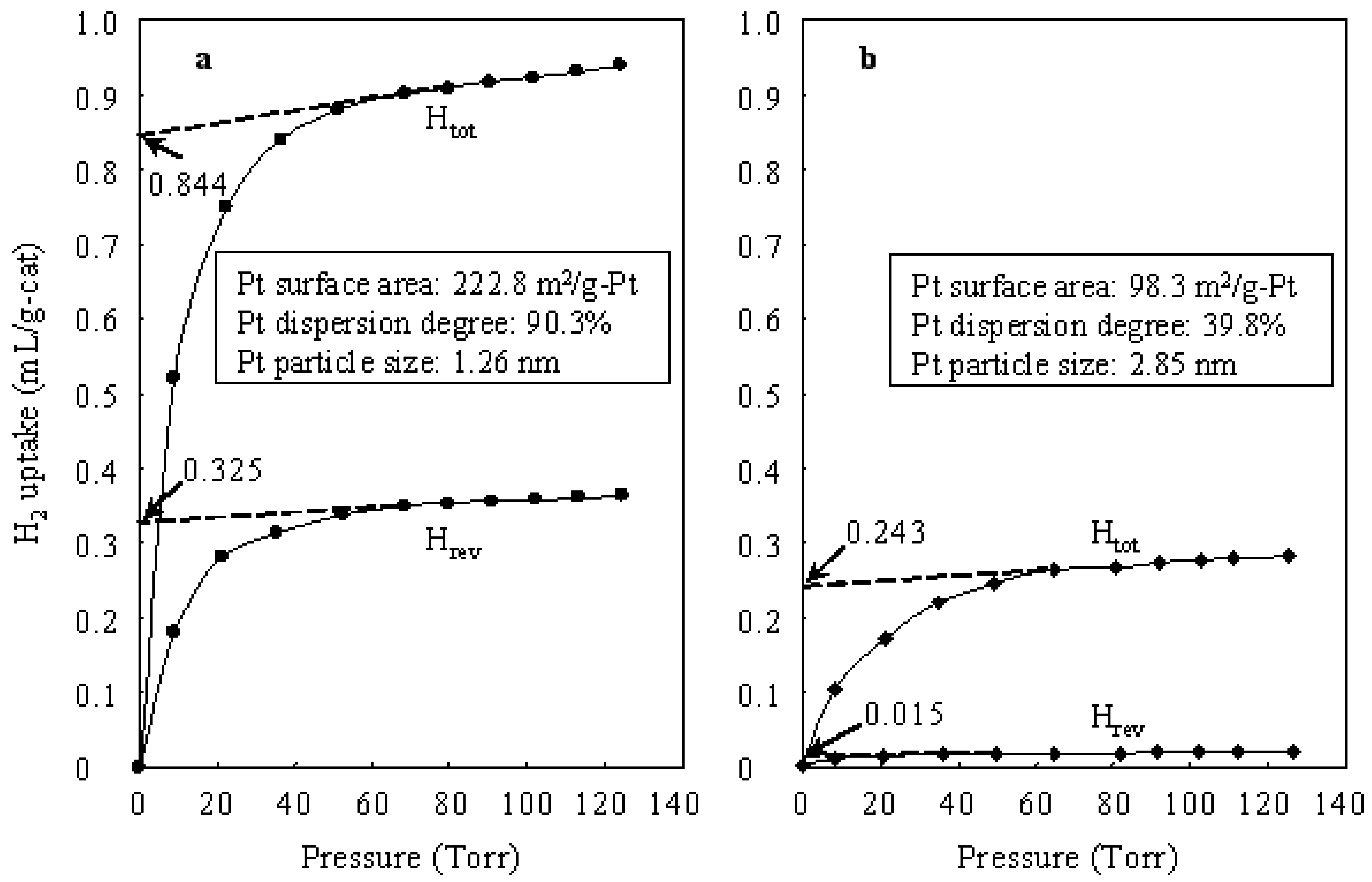
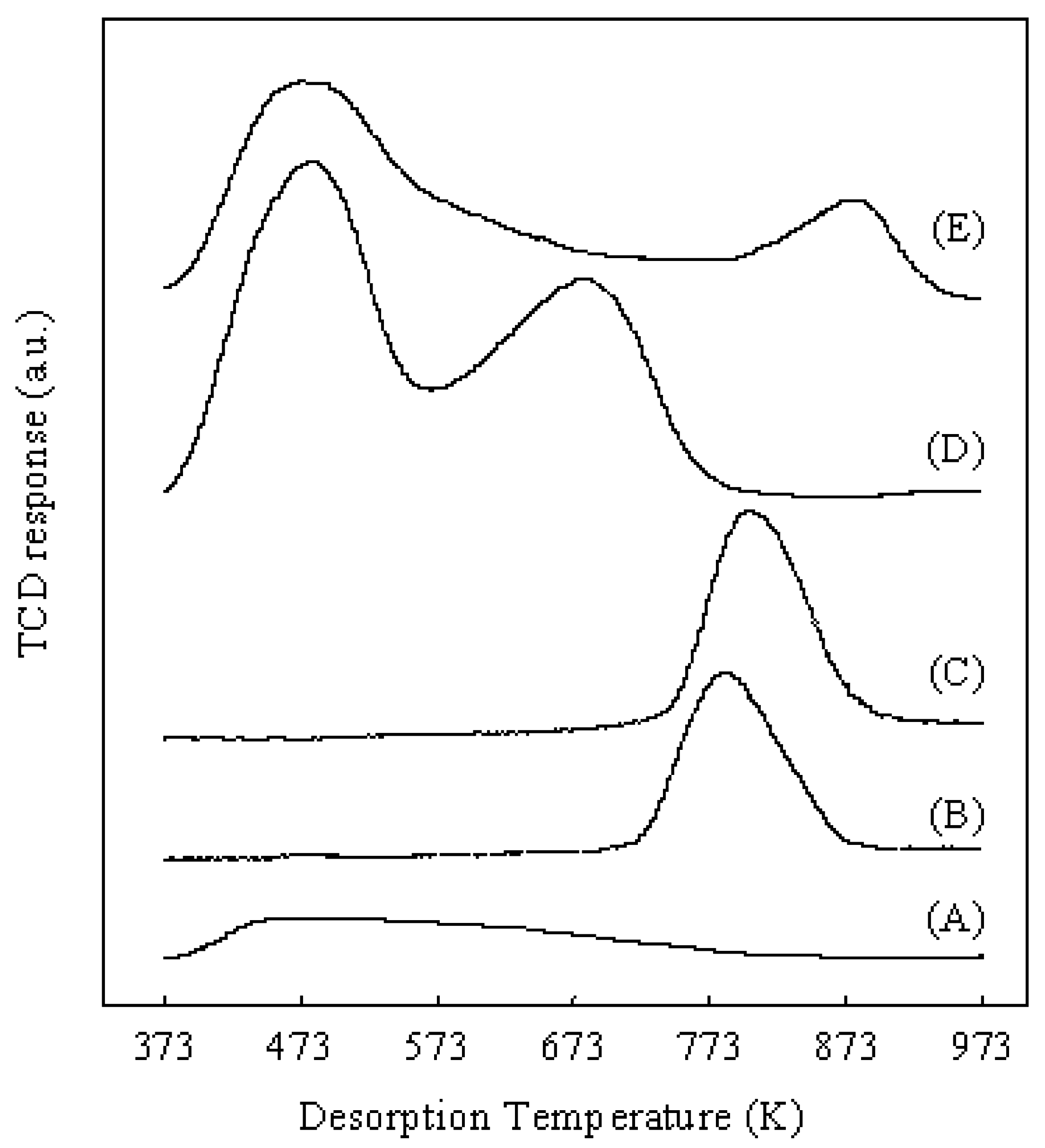
2.2. Characterization
2.3. Catalytic Reaction
3. Results and Discussion
3.1. Hydroisomerization of n-Butane over Various Catalysts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
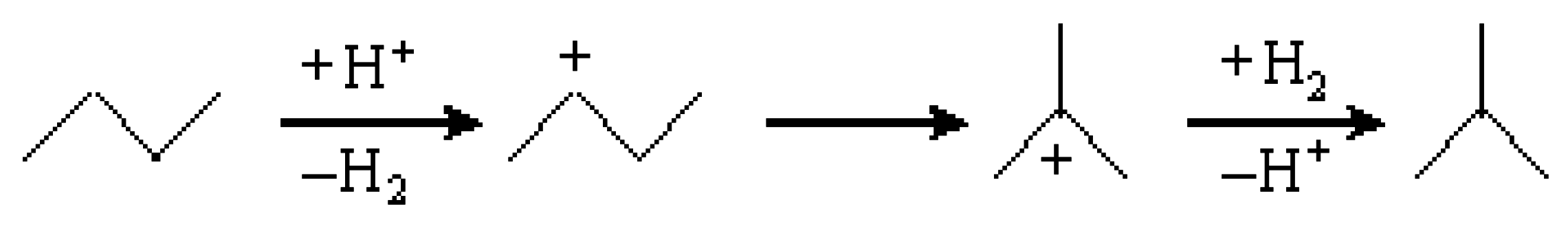{kind=link}
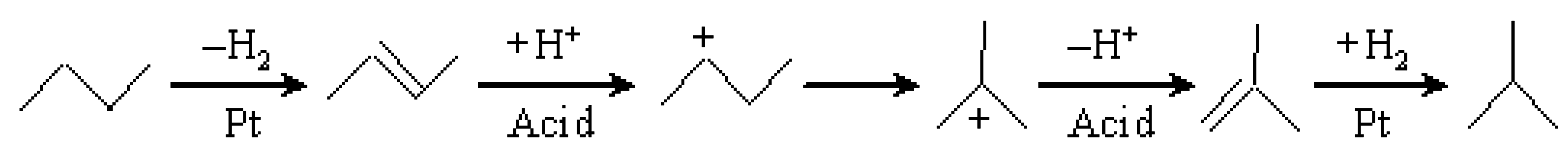{kind=link}
| Catalyst | Time on stream | Conv. (%) | Selectivity (%) | |||||
|---|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | i-C4 | C4= | C5+ | |||
| Pt/Al2O3 | 5 min | 4.2 | 9.7 | 12.6 | 9.3 | 66.2 | 0 | 2.2 |
| 5 h | 2.8 | 8.6 | 9.2 | 8.1 | 71.3 | 0 | 2.8 | |
| Cs2.5 | 5 min | 23.5 | 2.6 | 6.8 | 9.6 | 76.5 | 0.8 | 3.7 |
| 5 h | 9.2 | 1.8 | 5.1 | 7.3 | 82.3 | 0.7 | 2.8 | |
| Pt/Cs2.5 | 5 min | 66.1 | 1.9 | 3.2 | 4.7 | 88.4 | 0.7 | 1.0 |
| 5 h | 42.2 | 1.6 | 2.5 | 4.1 | 90.3 | 0.6 | 0.9 | |
| Pt/Al2O3+Cs2.5 | 5 min | 70.3 | 1.2 | 2.4 | 3.9 | 91.2 | 0.6 | 0.6 |
| 5 h | 64.8 | 0.8 | 2.0 | 3.3 | 92.5 | 0.5 | 0.8 | |



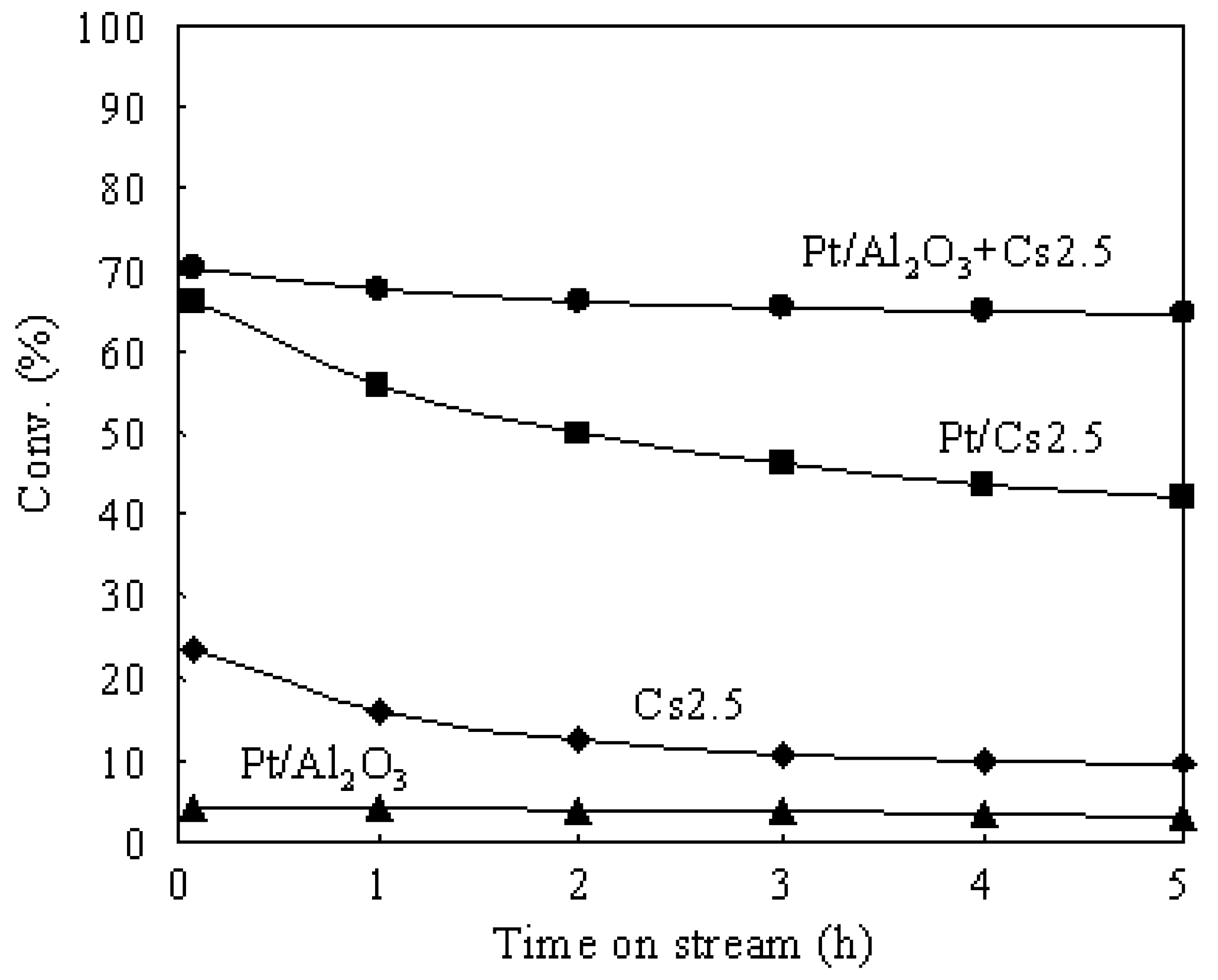
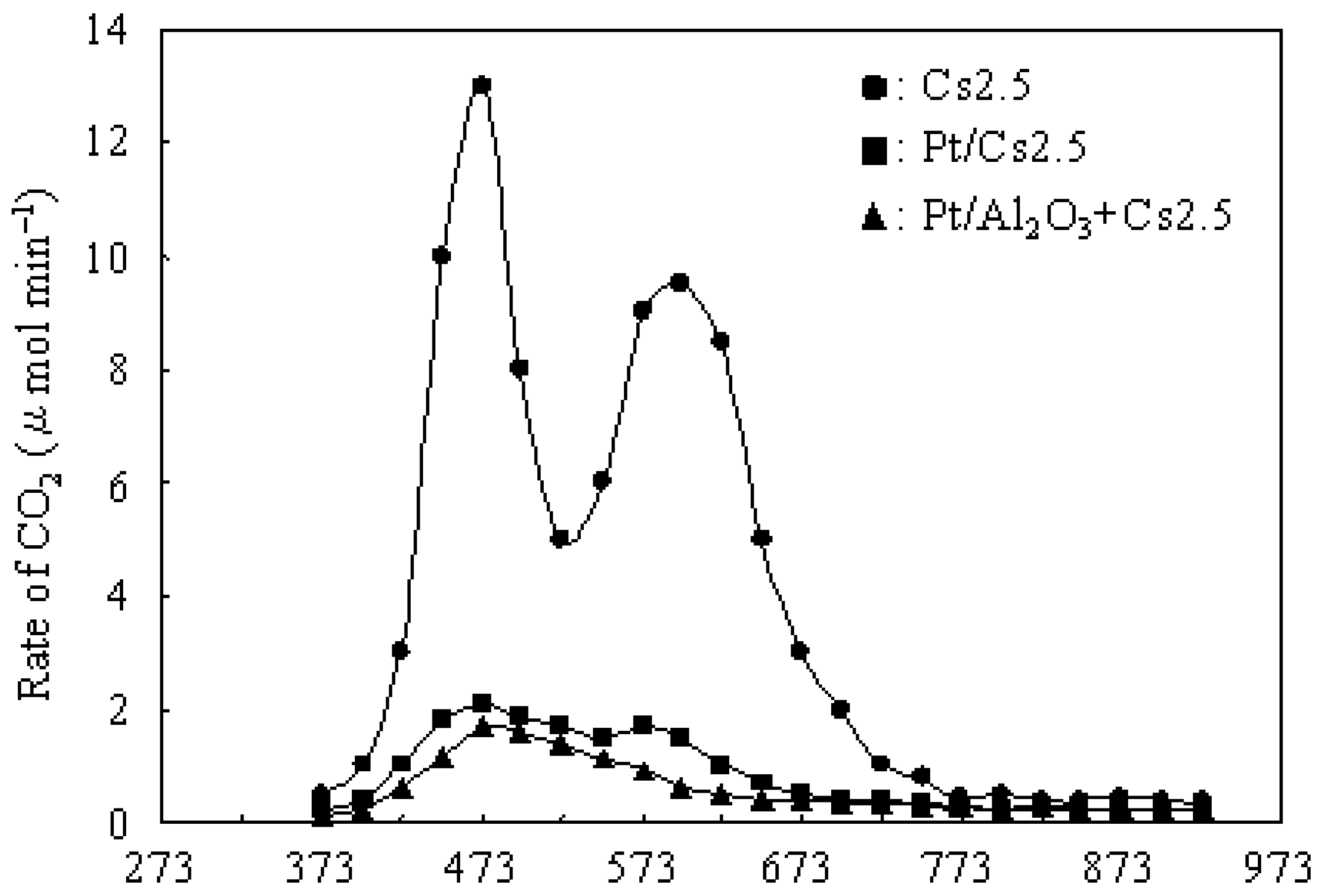
3.2. Deactivation of Various Catalysts in the Hydroisomerization of n-Butane


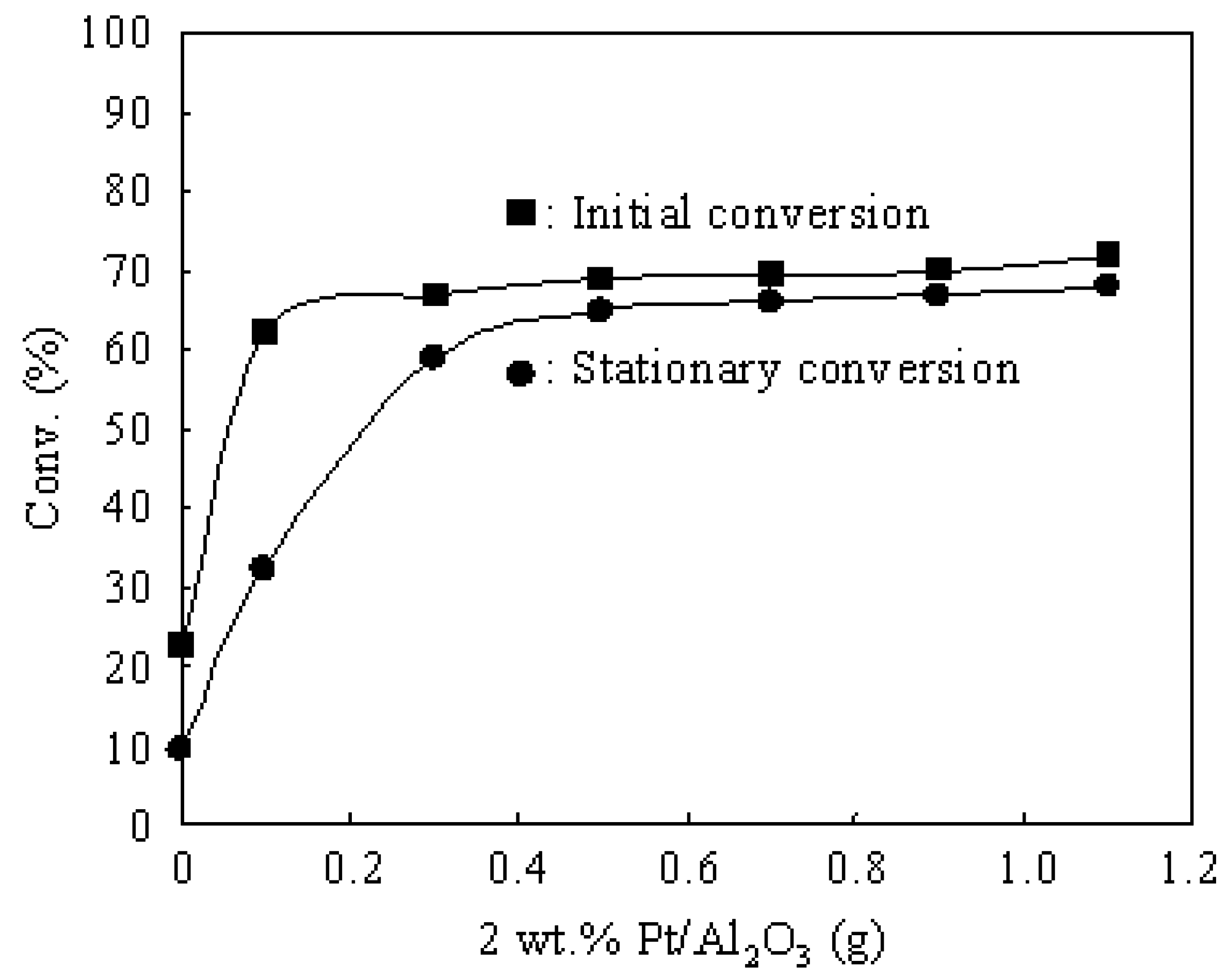
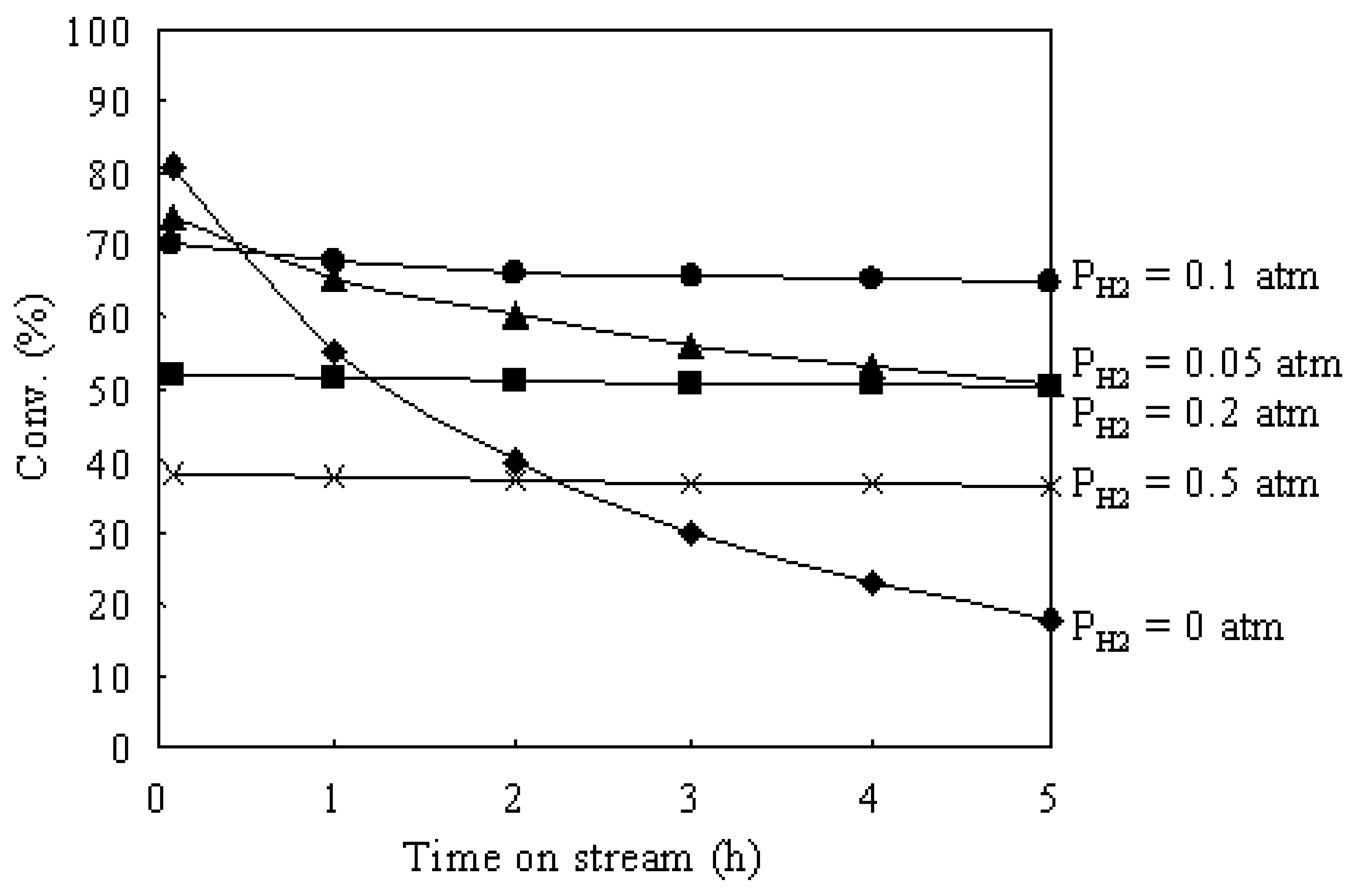
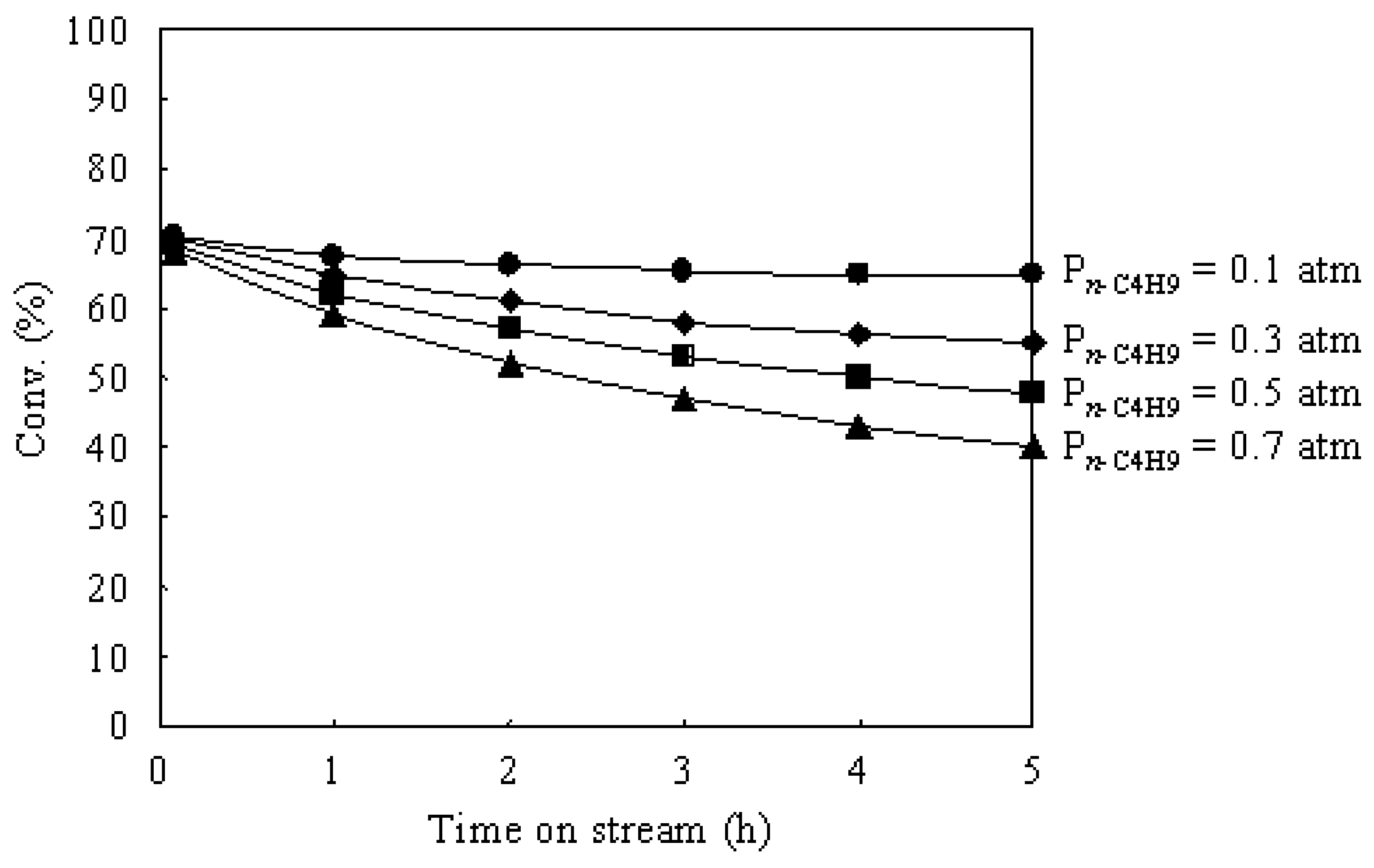
3.3. Hydroisomerization of n-Butane over the Pt/Al2O3+Cs2.5 Catalyst



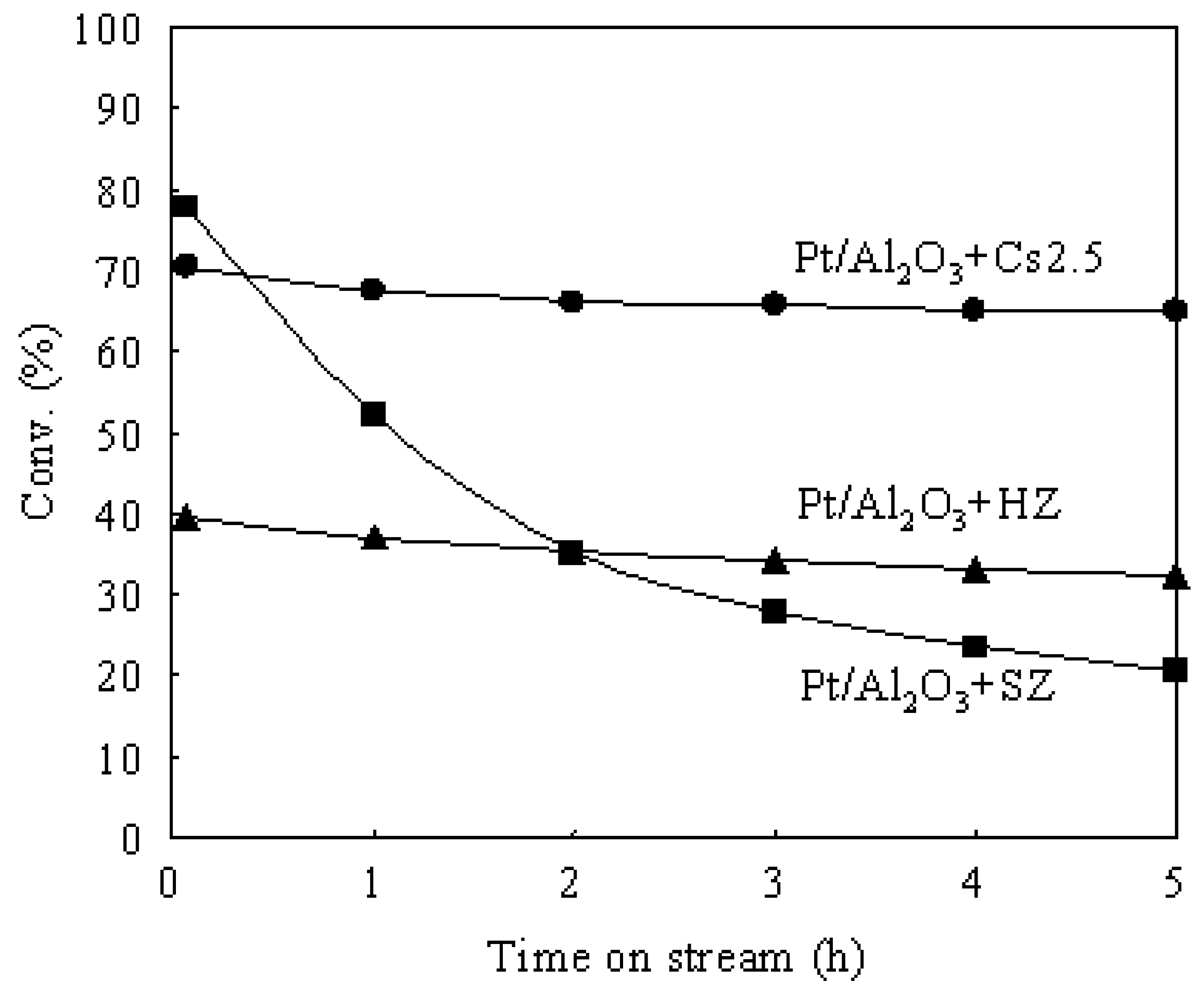
3.4. Comparison of Various Bifunctional Catalysts for the Hydroisomerization of n-Butane
| Catalyst | Time on stream | Conv. (%) | Selectivity (%) | |||||
|---|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | i-C4 | C4= | C5+ | |||
| Pt/Al2O3+Cs2.5 | 5 min | 70.3 | 1.2 | 2.4 | 3.9 | 91.2 | 0.6 | 0.6 |
| 5 h | 64.8 | 0.8 | 2.0 | 3.3 | 92.5 | 0.5 | 0.8 | |
| Pt/Al2O3+SZ | 5 min | 77.6 | 7.7 | 10.6 | 14.3 | 61.2 | 1.3 | 4.8 |
| 5 h | 20.5 | 4.6 | 7.2 | 10.1 | 71.7 | 1.0 | 5.2 | |
| Pt/Al2O3+HZ | 5 min | 39.7 | 2.7 | 3.1 | 5.7 | 87.1 | 0.9 | 0.5 |
| 5 h | 32.2 | 1.8 | 3.3 | 4.6 | 88.7 | 1.1 | 0.4 | |


4. Conclusions
References
- Okuhara, T.; Mizuno, N.; Misono, M. Catalytic chemistry of heteropoly compounds. Adv. Catal. 1996, 41, 113–252. [Google Scholar]
- Okuhara, T.; Mizuno, N.; Misono, M. Catalysis by heteropoly compounds―Recent developments. Appl. Catal. A: Gen. 2001, 222, 63–77. [Google Scholar] [CrossRef]
- Misono, M. Unique acid catalysis of heteropoly compounds (heteropolyoxometalates) in the solid state. Chem. Commun. 2001, 13, 1141–1152. [Google Scholar] [CrossRef]
- Guo, Y.; Hu, C. Heterogeneous photocatalysis by solid polyoxometalates. J. Mol. Catal. A: Chem. 2007, 262, 136–148. [Google Scholar] [CrossRef]
- Liu, Y.; Koyano, G.; Misono, M. Hydroisomerization of n-hexane and n-heptane over platinum-promoted Cs2.5H0.5PW12O40 (Cs2.5) studied in comparison with several other solid acids. Top. Catal. 2000, 11, 239–246. [Google Scholar] [CrossRef]
- Liu, Y.; Na, K.; Misono, M. Skeletal isomerization of n-pentane over Pt-promoted cesium hydrogen salts of 12-tungstophosphoric acid. J. Mol. Catal. A: Chem. 1999, 141, 145–153. [Google Scholar] [CrossRef]
- Liu, Y.; Koyano, G.; Na, K.; Misono, M. Isomerization of n-pentane and n-hexane over cesium hydrogen salt of 12-tungstophosphoric acid promoted by platinum. Appl. Catal. A: Gen. 1998, 166, L263–265. [Google Scholar]
- Liu, Y.; Murata, K.; Hanaoka, T.; Inaba, M.; Sakanishi, K. Syntheses of new peroxo-polyoxometalates intercalated layered double hydroxides for propene epoxidation by molecular oxygen in methanol. J. Catal. 2007, 248, 277–287. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Direct oxidation of benzene to phenol by molecular oxygen over catalytic systems containing Pd(OAc)2 and heteropolyacid immobilized on HMS or PIM. J. Mol. Catal. A: Chem. 2006, 256, 247–255. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Mimura, N. Direct epoxidation of propylene by molecular oxygen over Pd(OAc)2-[(C6H13)4N]3{PO4[W(O)(O2)2]4}-CH3OH catalytic system. Appl. Catal. B: Environ. 2005, 58, 51–59. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Liquid-phase oxidation of benzene to phenol by molecular oxygen over transition metal substituted polyoxometalate compounds. Catal. Commun. 2005, 6, 679–683. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Direct epoxidation of proptlene by molecular oxygen over catalyst system containing palladium and peroxo-Heteropoly compound in methanol. Chem. Commun. 2004, 582–583. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Mimura, N. Selective oxidation of propylene to acetone by molecular oxygen over Mx/2H5-x[PMo10V2O40]/HMS (M = Cu2+, Co2+, Ni2+). Catal. Commun. 2003, 4, 281–285. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Nakajima, H.; Koya, M.; Tomokuni, K. Catalytic oxidation of cyclohexene by molecular oxygen over isopolyoxometalates. Chem. Lett. 2004, 33, 200–201. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M. Epoxidation of propylene with molecular oxygen in methanol over a peroxo-heteropoly compound immobilized on palladium exchanged HMS. Green Chem. 2004, 6, 510–515. [Google Scholar] [CrossRef]
- Adeeva, V.; Sachtler, W.M.H. Mechanism of butane isomerization over industrial isomerization catalysts. Appl. Catal. A: Gen. 1997, 163, 237–243. [Google Scholar] [CrossRef]
- Behaviour, S. Influence of hydrogen chloride addition on the catalytic isomerization activity of chlorinated alumina and chlorinated platinum-alumina solids. J. Chem. Soc. Faraday Trans. 1990, 86, 567–570. [Google Scholar] [CrossRef]
- Na, K.; Okuhara, T.; Misono, M. Catalysis by heteropoly compounds. 34. Skeletal isomerization of n-butane over Pt- or Pd-promoted cesium hydrogen salts of 12-tungstophosphoric acid. J. Catal. 1997, 170, 96–107. [Google Scholar] [CrossRef]
- Na, K.; Iizaki, T.; Okuhara, T.; Misono, M. Molecular design of solid acid catalysts. Isomerization of n-butane catalyzed by acidic cesium salts of 12-tungstophosphoric acid combined with platinum. J. Mol. Catal. A: Chem. 1997, 115, 449–455. [Google Scholar] [CrossRef]
- Ma, Z.; Hua, W.; Ren, Y.; He, H.; Gao, Z. n-Butane isomerization over Cs-salts of H3PW12O40: A mechanistic study by 1C MAS NMR. Appl. Catal. A: Gen. 2003, 256, 243–250. [Google Scholar] [CrossRef]
- Zou, Y.; Yue, B.; Zhang, B.; He, H. Solid synthesis of CsxH3-xPW12O40 salts and their catalytic activity for the isomerization of n-butane. Chem. Lett. 2006, 35, 202–203. [Google Scholar] [CrossRef]
- He, S.; Liu, X.; Xie, S.; Wang, Q.; Xu, Y.; Xu, L. Effect of steam on the acid strength of H3PW12O40/SiO2 · nH2O and its application in skeletal isomerization of n-butane. Catal. Commun. 2003, 4, 585–590. [Google Scholar] [CrossRef]
- Yang, W.; Billy, J.; Taarit, Y.; Vedrine, J.; Essayem, N. H3PW12O40 supported on Cs modified mesoporous silica: catalytic activity in n-butane isomerisation and in situ FTIR study. Comparison with microporous CsxH3-xPW12O40. Catal. Today 2002, 73, 153–165. [Google Scholar] [CrossRef]
- Ebitani, K.; Konishi, J.; Hattori, H. Skeletal isomerization of hydrocarbons over zirconium oxide promoted by platinum and sulfate ion. J. Catal. 1991, 130, 257–267. [Google Scholar] [CrossRef]
- Matsuhashi, H.; Shibata, H.; Nakamura, H.; Arata, K. Skeletal isomerization mechanism of alkanes over solid superacid of sulfated zirconia. Appl. Catal. A: Gen. 1999, 187, 99–106. [Google Scholar] [CrossRef]
- Funamoto, T.; Nakagawa, T.; Segawa, K. Isomerization of n-butane over sulfated zirconia catalyst under supercritical conditions. Appl. Catal. A: Gen. 2005, 286, 79–84. [Google Scholar] [CrossRef]
- Yori, J.C.; D’Amato, M.D.; Costa, G.; Parera, J.M. Isomerization of n-butane on Pt/SO42--ZrO2 and mechanical mixtures of Pt/Al2O3+SO42--ZrO2. J. Catal. 1995, 153, 218–223. [Google Scholar] [CrossRef]
- Tomishige, K.; Okabe, A.; Fujimoto, K. Effect of hydrogen on n-butane isomerization over Pt/SO42--ZrO2 and Pt/SiO2+SO42--ZrO2. Appl. Catal. A: Gen. 2000, 194–195, 383–393. [Google Scholar] [CrossRef]
- Lei, T.; Xu, J.S.; Tang, Y.; Hua, W.M.; Gao, Z. New solid superacid catalysts for n-butane isomerization: γ- Al2O3 or SiO2 supported sulfated zirconia. Appl. Catal. A: Gen. 2000, 192, 181–188. [Google Scholar] [CrossRef]
- Grau, J.A.; Yori, J.C.; Vera, C.R.; Lovey, F.C.; Condo, A.A.; Parera, J.A. Crystal phase dependent metal-support interactions in Pt/SO42--ZrO2 catalysts for hydroconversion of n-alkanes. Appl. Catal. A: Gen. 2004, 265, 141–152. [Google Scholar] [CrossRef]
- Hua, W.M.; Sommer, J. Hydroisomerization of n-butane over sulfated zirconia catalysts promoted by alumina and platinum. Appl. Catal. A: Gen. 2002, 227, 279–286. [Google Scholar] [CrossRef]
- Eibl, S.; Jentoft, R.E.; Gates, B.C.; Knozinger, H. Conversion of n-pentane and of n-butane catalyzed by platinum-containing WOx/TiO2. Phys. Chem. Chem. Phys. 2000, 2, 2565–2573. [Google Scholar] [CrossRef]
- Dorado, F.; Romero, R.; Canizares, P.; Romero, A. Hydroisomerization of n-butane over Pd/HZSM-5 and Pd/H beta with and without binder. Appl. Catal. A: Gen. 2002, 236, 235–243. [Google Scholar] [CrossRef]
- Dorado, F.; Romero, R.; Canizare, P.; Romero, A. Influence of palladium incorporation technique on n-butane hydroisomerization over HZSM-5/bentonite catalysts. Appl. Catal. A: Gen. 2004, 274, 79–85. [Google Scholar] [CrossRef]
- Canizares, P.; Dorado, F.; Sanchez-Herrera. Hydroisomerization of n-butane over hybrid catalysts. Appl. Catal. A: Gen. 2001, 217, 69–78. [Google Scholar] [CrossRef]
- Canizares, P.; Lucas, A.; Dorado, F.; Perez, D. Effect of zeolite pore geometry on isomerization of n-butane. Appl. Catal. A: Gen. 2000, 190, 233–239. [Google Scholar]
- Kumar, N.; Villegas, J.I.; Salmi, T.; Murzin, D.Y.; Heikkila, T. Isomerization of n-butane to isobutane over Pt-SAPO-5, SAPO-5, Pt-H-mordenite and H-mordenite catalysts. Catal. Today 2005, 100, 355–361. [Google Scholar] [CrossRef]
- Baburek, E.; Novakova, J. Effect of platinum in bifunctional isomerization of butane over acid zeolites. Appl. Catal. A: Gen. 2000, 190, 241–251. [Google Scholar] [CrossRef]
- Pieterse, J.A.Z.; Seshan, K.; Lercher, J.A. Structure-activity correlations for TON, FER, and MOR in the hydroisomerization of n-butane. J. Catal. 2000, 195, 326–335. [Google Scholar] [CrossRef]
- Malogolowkin, C.; Poulson, D.F. Stepwise reaction on separate catalytic centers: isomerization of saturated hydrocarbons. Science 1957, 126, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Okuhara, T.; Nishimura, T.; Watanabe, H.; Misono, M. Insoluble heteropoly compounds as highly active catalysts for liquid-phase reactions. J. Mol. Catal. 1992, 74, 247–256. [Google Scholar] [CrossRef]
- Okuhara, T.; Watanabe, H.; Nishimura, T.; Inumaru, K.; Misono, M. Microstructure of cesium hydrogen salts of 12-tungstophosphoric acid relevant to novel acid catalysis. Chem. Mater. 2000, 12, 2230–2238. [Google Scholar] [CrossRef]
- Hino, M.; Arata, K. Synthesis of solid superacid catalyst with acid strength of H0 ≤ −16.04. J. Chem. Soc. Chem. Commun. 1980, 851–852. [Google Scholar] [CrossRef]
- Na, K.; Okuhara, T.; Misono, M. Skeletal isomerization of n-butane over caesium hydrogen salts of 12-tungstophosphoric acid. J. Chem. Soc. Faraday Trans. 1995, 91, 367–373. [Google Scholar] [CrossRef]
- Zhang, A.; Nakamura, I.; Aimoto, K.; Fujimoto, K. Isomerization of n-pentane and other light hydrocarbons on hybrid catalyst. Effect of hydrogen spillover. Ind. Eng. Chem. Res. 1995, 34, 1074–1080. [Google Scholar] [CrossRef]
- Grau, J.M.; Vera, C.R.; Parera, J.M. Preventing self-poisoning in [Pt/Al2O3+SO42--ZrO2] mixed catalysts for isomerization-cracking of heavy alkanes by prereduction of the acid function. Appl. Catal. A: Gen. 2002, 227, 217–230. [Google Scholar]
- Liu, Y.; Hayakawa, T.; Ishii, T.; Kumagai, M.; Yasuda, H.; Suzuki, K.; Hamakawa, S.; Murata, K. Methanol decomposition to synthesis gas at low temperature over palladium support on ceria-zirconia solid solutions. Appl. Catal. A: Gen. 2001, 210, 301–314. [Google Scholar]
- Liu, Y.; Hayakawa, T.; Suzuki, K.; Hamakawa, S.; Tsunoda, T.; Ishii, T.; Kumagai, M. High active copper/ceria catalysts for the steam reforming of methanol. Appl. Catal. A: Gen. 2002, 223, 137–145. [Google Scholar]
- Liu, Y.; Hayakawa, T.; Tsunoda, T.; Suzuki, K.; Hamakawa, S.; Murata, K.; Shiozaki, R.; Ishii, T.; Kumagai, M. Steam reforming of methanol over Cu/CeO2 catalysts studied in comparison with Cu/ZnO and Cu/Zn(Al)O catalysts. Top. Catal. 2003, 22, 205–213. [Google Scholar] [CrossRef]
- Siddiqui, M.R.H.; Holmes, S.; He, H.; Smith, W.; Coker, E.N.; Atkins, M.P.; Kozhevnikov, I.V. Coking and regeneration of palladium-doped H3PW12O40/SiO2 catalysts. Catal. Lett. 2000, 66, 53–57. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V.; Holmes, S.; Siddiqui, M.R.H. Coking and regeneration of H3PW12O40/SiO2 catalysts. Appl. Catal. A: Gen. 2001, 214, 47–58. [Google Scholar]
- Degnan, T.F.; Kennedy, C.R. Impact of catalyst acid/metal balance in hydroisomerization of normal paraffins. AIChE J. 1993, 39, 607–614. [Google Scholar] [CrossRef]
- Essayem, N.; Taarit, Y.B.; Feche, C.; Gayraud, P.Y.; Sapaly, G.; Naccache, C. Comparative study of n-pentane isomerization over solid acid catalysts, heteropolyacid, sulfated zirconia, and mordenite: dependence on hydrogen and platinum addition. J. Catal. 2003, 219, 97–106. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Y.; Misono, M. Hydroisomerization of n-Butane over Platinum-Promoted Cesium Hydrogen Salt of 12-Tungstophosphoric Acid. Materials 2009, 2, 2319-2336. https://doi.org/10.3390/ma2042319
Liu Y, Misono M. Hydroisomerization of n-Butane over Platinum-Promoted Cesium Hydrogen Salt of 12-Tungstophosphoric Acid. Materials. 2009; 2(4):2319-2336. https://doi.org/10.3390/ma2042319
Chicago/Turabian StyleLiu, Yanyong, and Makoto Misono. 2009. "Hydroisomerization of n-Butane over Platinum-Promoted Cesium Hydrogen Salt of 12-Tungstophosphoric Acid" Materials 2, no. 4: 2319-2336. https://doi.org/10.3390/ma2042319
APA StyleLiu, Y., & Misono, M. (2009). Hydroisomerization of n-Butane over Platinum-Promoted Cesium Hydrogen Salt of 12-Tungstophosphoric Acid. Materials, 2(4), 2319-2336. https://doi.org/10.3390/ma2042319