Different applications of carbon-based HMs as adsorbents and support for catalysts to remove pollutants from gas streams were reported. Thus, they were used to remove VOCs from air, CO2 capture and NO and SO2 removal from flue gas, separation of CO2 and CH4 and for selective catalytic reduction (SCR) of NO with ammonia. In the next subsections, the currently reported applications of carbon-based HMs will be presented.
3.1. Carbon-based HMs as adsorbents
The dynamic adsorption of butane, toluene, formaldehyde, acetaldehyde and isopropanol using carbon-based HMs was studied [
18]. These structures were prepared by dip-coating ceramic HMs with a phenolic resole of low viscosity (100 cP) that were carbonized at 900 ºC in N
2. High adsorption efficiencies were obtained even with high flow rate and low concentration streams. Adsorption efficiencies and capacities could be controlled by adjusting wall thickness. In addition, it was shown that space velocity should be used instead of surface velocity to characterize adsorption performance.
A comparative study on the adsorption of n-butane from air on an activated carbon monolith and in granular form was carried out by Crittenden
et al. [
34]. The carbon square channel monolith from MAST Carbon had a cell density of around 89 cells/cm
2, with a cell size of 0.63 mm and a wall thickness of 0.43 mm. Granules with sizes between 600 and 1400 μm were obtained by crushing samples of the monolith. Results were obtained using a dynamic flow apparatus and showed that it was possible to manufacture an activated carbon monolith that had a capacity and dynamic mass transfer performance equal to that of the equivalent mass of granules. The pressure drop, however, was less than 6% of that of the granule beds.
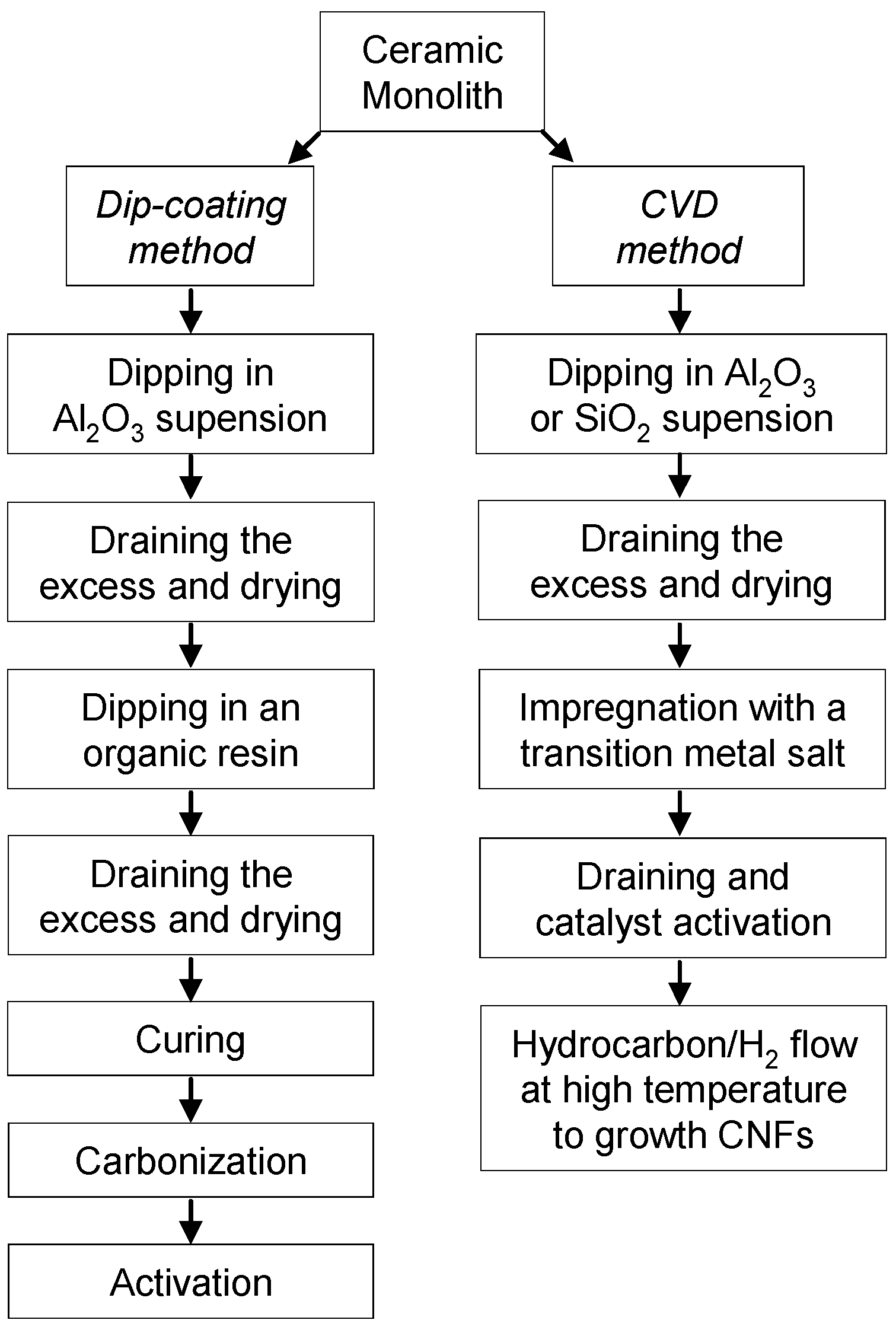
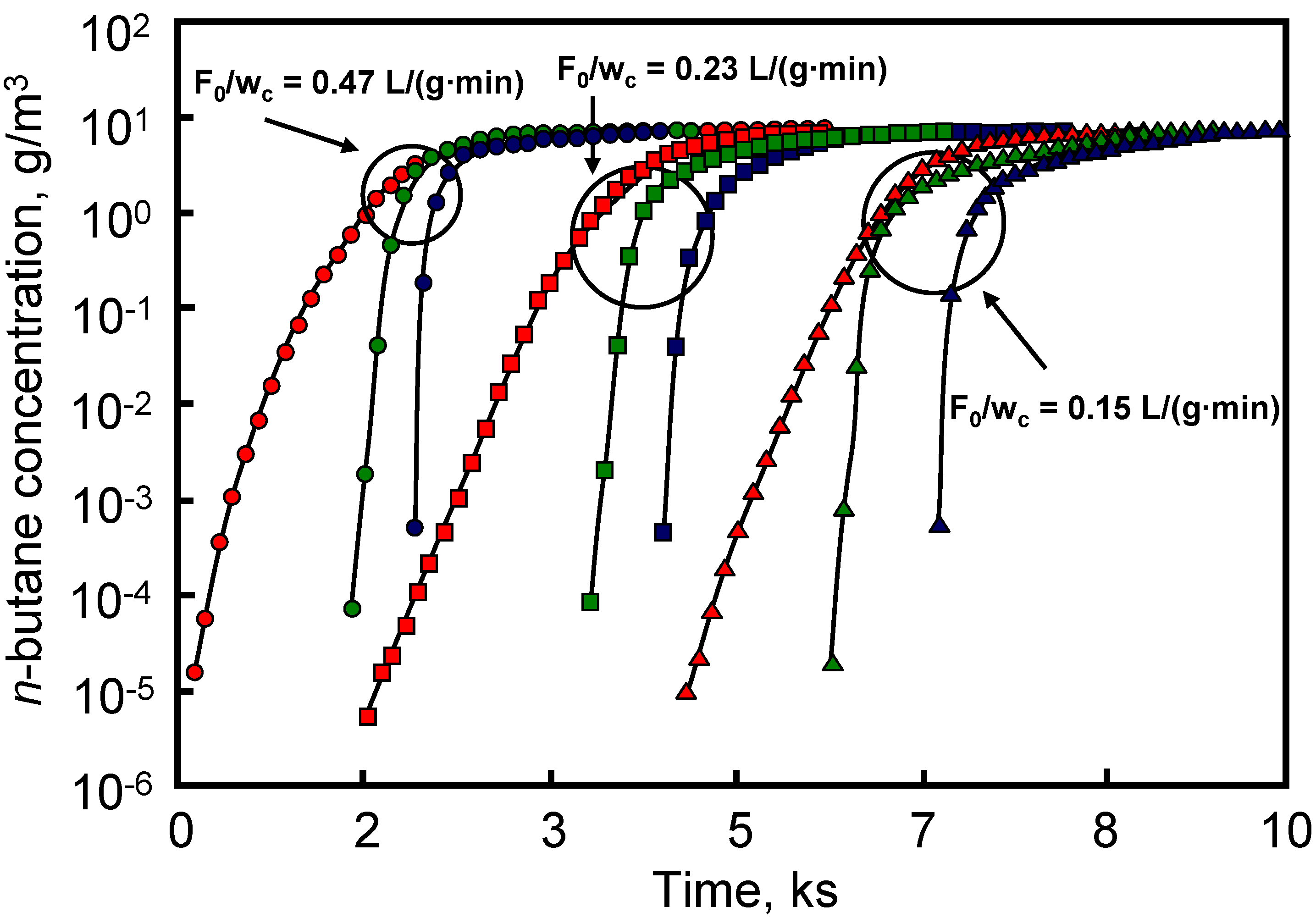
The effect of cell density, stacking and total length of carbon-coated HMs on the breakthrough curves of n-butane diluted (7 g/m
3) in an air flow of 1500 cm
3/min was reported [
40]. The carbon-coated HM was prepared by dip-coating the ceramic monolith in a phenol novolac resin. Carbonization was carried out at 700 ºC and the resulting material was steam activated at the same temperature.
Table 4 shows the width values of the breakthrough profile (time difference for outlet concentrations of C/C
0 = 0.95 and C/C
0 = 0.05). For each cell density value, the increase in total length produced an increase in the breakthrough profile width. In addition, for each total length the increase in cell density produced a decrease in the breakthrough profile width.
Table 4.
Breakthrough profile width t
0.95-t
0.05 (min). From reference [
40], with permission from Elsevier.
Table 4.
Breakthrough profile width t0.95-t0.05 (min). From reference [40], with permission from Elsevier.
| Cell density – individual piece length | Total length (cm) |
|---|
| | 5 | 10 | 15 |
|---|
| 31 cells/cm2 – 5 cm | 31.7 | 41.7 | 46.7 |
| 62 cells/cm2 – 5 cm | 24.3 | 36.2 | 45.0 |
| 93 cells/cm2 – 2.5 cm | 15.3 | 22.4 | 31.4 |
| 140 cells/cm2 – 1 cm | 16.5 | 20.2 | 31.9 |
The performance of activated carbon-packed beds was compared with that of the carbon-coated HMs and results are depicted in
Figure 8. This Figure shows that carbon-coated HMs had a sharper breakthrough profile, with the breakthrough point occurring later than on the activated carbon-packed beds. This behavior was associated with the short diffusion length in the monoliths. This is interesting with respect to the application of the monoliths in gas mask canisters, because breakthrough time is delayed with regard to that found for activated carbon-packed beds.
Figure 8.
Breakthrough profiles for carbon-packed beds and monoliths (31 and 62 cells/cm
2) (
![Materials 03 01203 i005]()
, Norit R1 4.3 g;
![Materials 03 01203 i006]()
, Norit R1 8.6 g;
![Materials 03 01203 i007]()
, Norit R1 13 g;
![Materials 03 01203 i008]()
, (31 cells/cm
2)-5 cm-5 cm;
![Materials 03 01203 i009]()
, (31 cells/cm
2)-5 cm-10 cm;
![Materials 03 01203 i010]()
, (31 cells/cm
2)-5 cm-15 cm;
![Materials 03 01203 i011]()
, (62 cells/cm
2)-5 cm-5 cm;
![Materials 03 01203 i012]()
, (62 cells/cm
2)-5 cm-10 cm;
![Materials 03 01203 i013]()
, (62 cells/cm
2)-5 cm-15 cm. From reference [
40], with permission from Elsevier.
Figure 8.
Breakthrough profiles for carbon-packed beds and monoliths (31 and 62 cells/cm
2) (
![Materials 03 01203 i005]()
, Norit R1 4.3 g;
![Materials 03 01203 i006]()
, Norit R1 8.6 g;
![Materials 03 01203 i007]()
, Norit R1 13 g;
![Materials 03 01203 i008]()
, (31 cells/cm
2)-5 cm-5 cm;
![Materials 03 01203 i009]()
, (31 cells/cm
2)-5 cm-10 cm;
![Materials 03 01203 i010]()
, (31 cells/cm
2)-5 cm-15 cm;
![Materials 03 01203 i011]()
, (62 cells/cm
2)-5 cm-5 cm;
![Materials 03 01203 i012]()
, (62 cells/cm
2)-5 cm-10 cm;
![Materials 03 01203 i013]()
, (62 cells/cm
2)-5 cm-15 cm. From reference [
40], with permission from Elsevier.
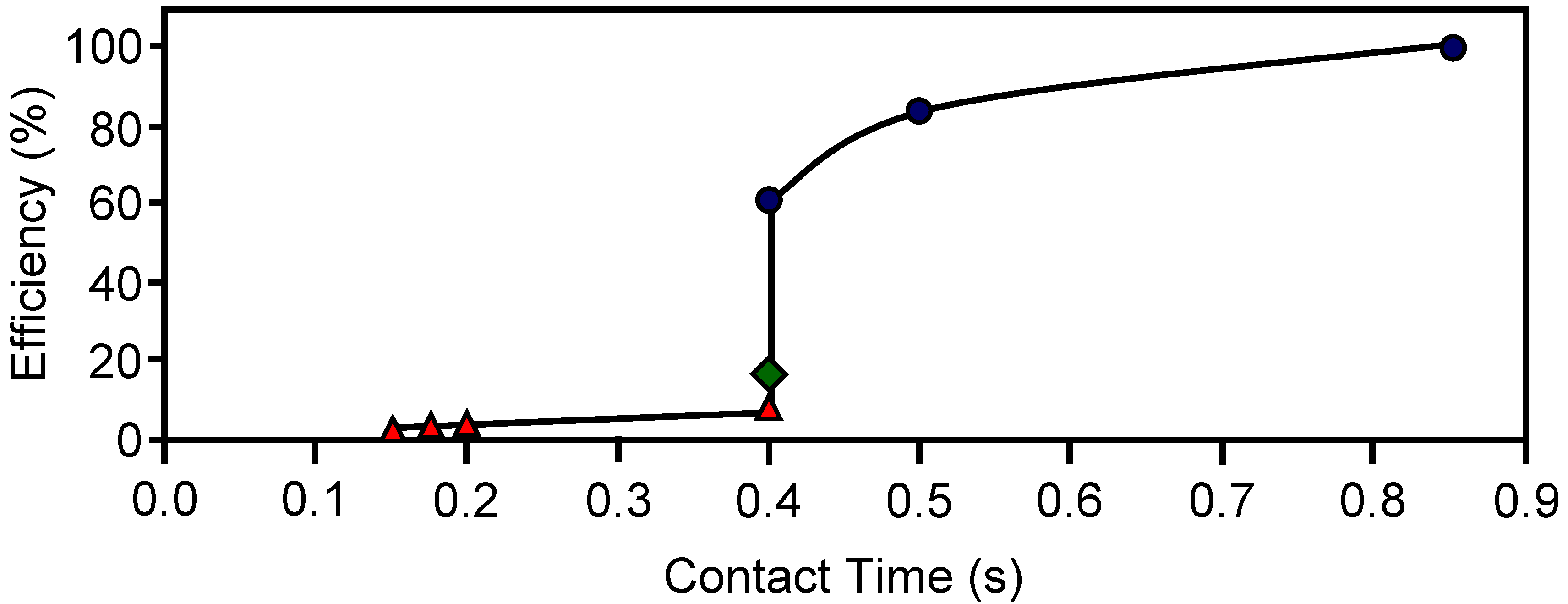
Carbon-based HMs prepared from different activated carbons mixed with a silicate (binder) were used to remove o-dichlorobenzene from air streams at different temperatures and linear velocities. The adsorption capacity of these HMs was also studied under static conditions [
41,
42,
43]. The carbon-based HMs had square section channels with cell density of 8 cells/cm
2 and a wall thickness of 0.9 mm. Adsorption capacity at 30 ºC could be related with the micropore volume of the carbon-based HMs. The dynamic adsorption efficiency is depicted in
Figure 9. When adsorption was carried out at 30 ºC the efficiency decreased from 100 to 60% while contact time diminished from 0.9 to 0.4 s. Therefore, the efficiency of these HMs could be improved by reducing the open channel width.
Figure 9 also shows a severe decrease in efficiency when adsorption temperature increased from 30 to 150 ºC.
The static adsorption of different organic compounds and water vapor on various HMs prepared from commercially available activated carbons, alumina and titania using a silicate clay as binder was also reported [
44]. When the pretreatment temperature of the carbon-based HM increased, the organic vapor adsorption rate also increased, whereas the water adsorption rate decreased. However, for the alumina and titania HMs, the increase in pretreatment temperature did not affect the organic vapor adsorption rate, but decreased the water adsorption rate. These results were explained as to the result of changes in surface hydrophobicity with pretreatment temperature.
Figure 9.
The dynamic adsorption efficiency towards o-DCB for different contact times and temperatures: 30 ºC (
![Materials 03 01203 i014]()
), 100 ºC (
![Materials 03 01203 i015]()
) and 150 ºC (
![Materials 03 01203 i016]()
). From reference [
43], with permission from Elsevier.
Figure 9.
The dynamic adsorption efficiency towards o-DCB for different contact times and temperatures: 30 ºC (
![Materials 03 01203 i014]()
), 100 ºC (
![Materials 03 01203 i015]()
) and 150 ºC (
![Materials 03 01203 i016]()
). From reference [
43], with permission from Elsevier.
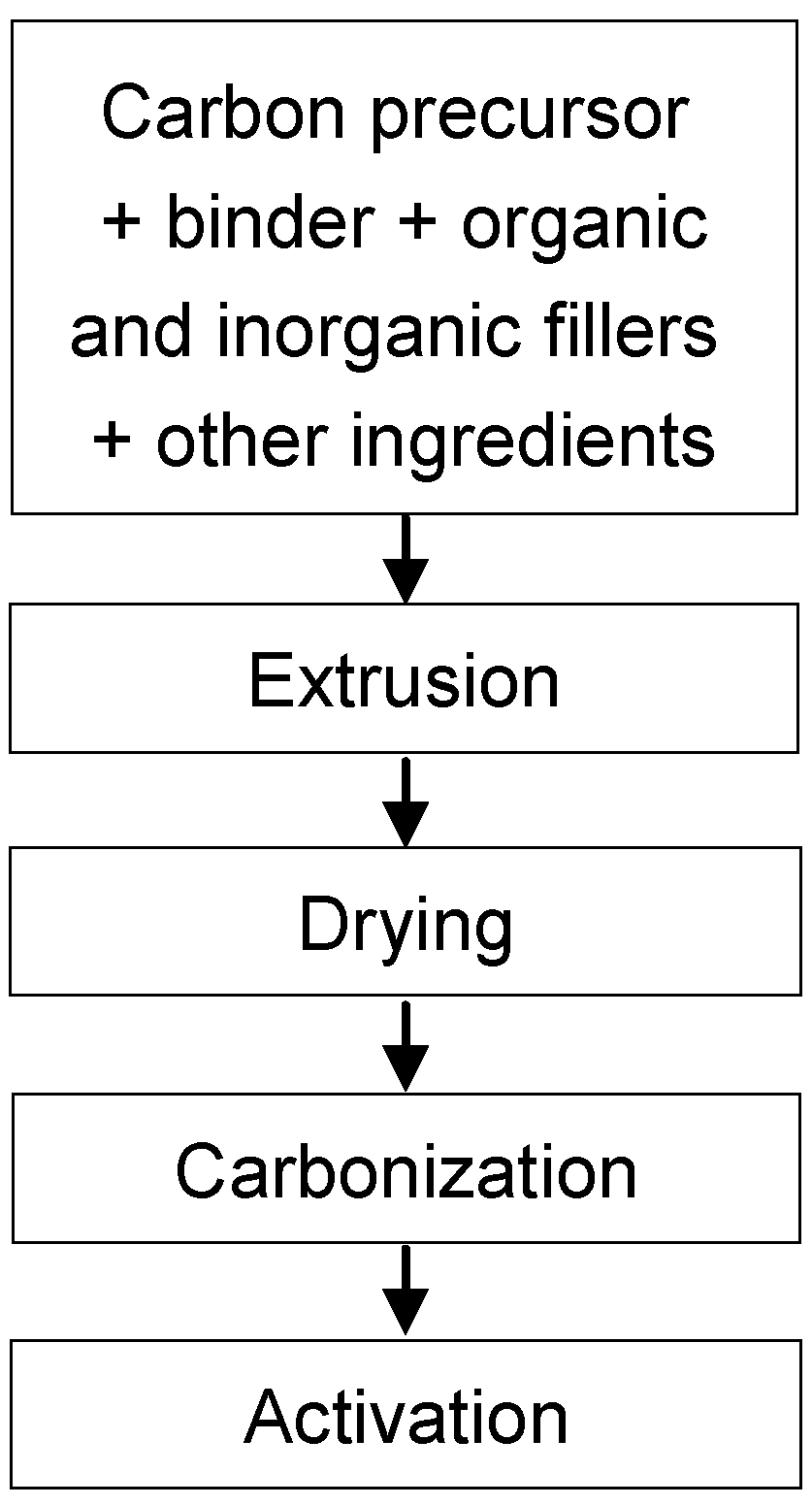
The dynamic adsorption/desorption of o-xylene on an integral carbon honeycomb monolith, which was prepared from a bituminous coal and using some additives to obtain the adequate rheological properties for extrusion, was studied [
45]. The o-xylene adsorption was carried out at 27 ºC from a N
2 flow (100 cm
3/min) containing 0.36% of the pollutant. Total adsorption capacity was around 550 μmol/g, similar to that reported for powder-activated carbons. The adsorption capacity remained constant after several adsorption/desorption cycles.
An integral carbon honeycomb monolith from MAST Carbon was used to adsorb CO
2, CH
4 and N
2 [
46]. Adsorption equilibrium was measured over a pressure range between 0 and 700 kPa and at temperatures between 26 and 149 ºC. Results obtained were well fitted by the multisite Langmuir model. The selectivity toward carbon dioxide decreased when the pressure increased. The diffusion of simple gases in the porous structure of the activated carbon honeycomb monolith was studied by diluted breakthrough experiments. The diffusivity coefficients obtained resulted from a combination of macro and micropore diffusivities and they had and exponential dependence with temperature.
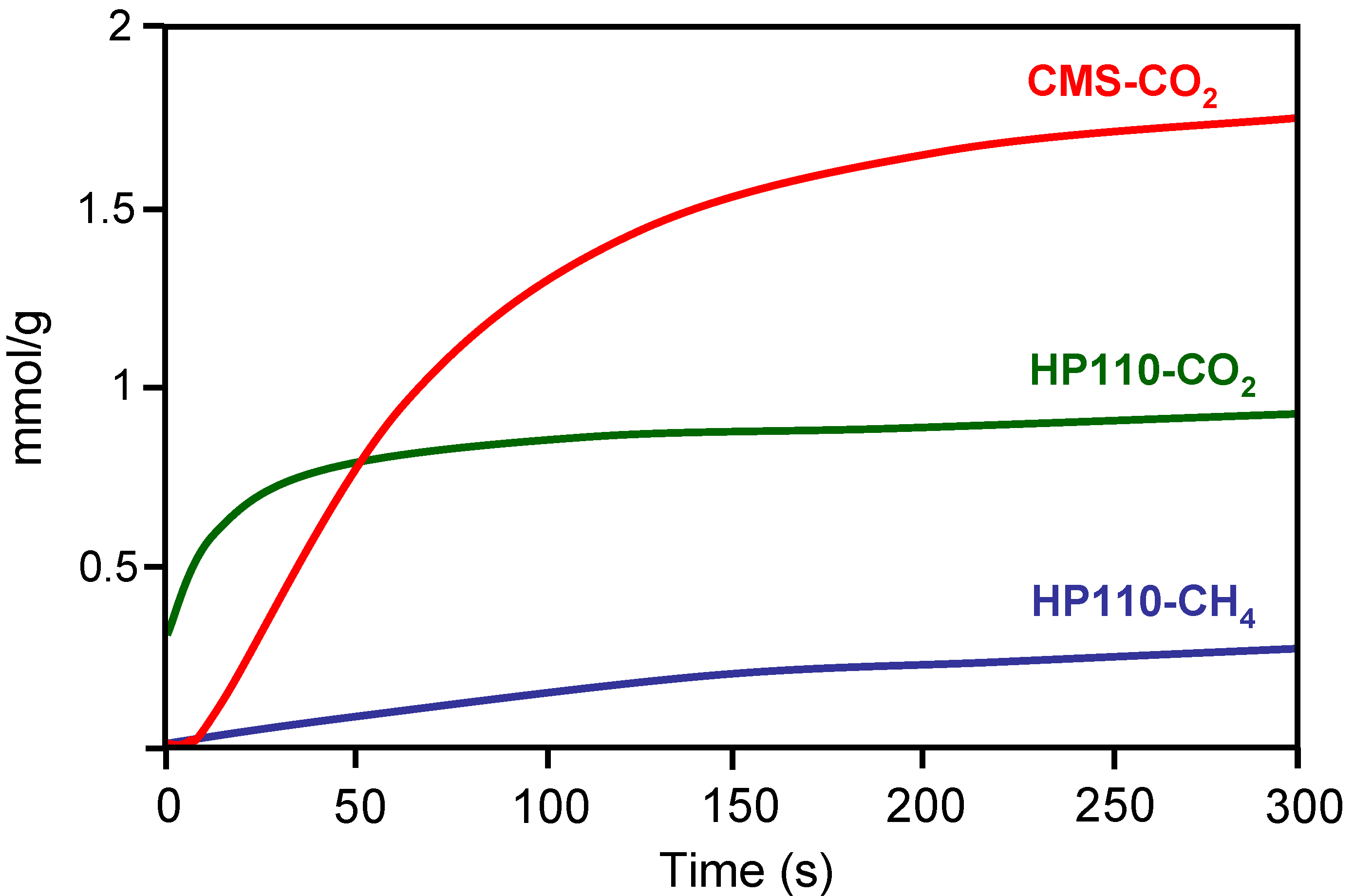
The molecular sieve properties of a carbon-based HM was compared with that of a commercial carbon molecular sieve (CMS) [
24]. For this purpose, the CH
4 and CO
2 adsorption kinetics were studied. The carbon-based HM was prepared by impregnation with a petroleum pitch of a cellulose-based corrugated paper. After stabilization in air (300 ºC), the resulting material was carbonized at 1000 ºC in N
2 flow and CO
2 activated at 870 ºC to different burn-off degrees. CH
4 and CO
2 adsorption kinetics was carried out at constant volume in a high-pressure thermobalance at 0.1 MPa and 25 ºC. Results obtained with one of the carbon-based HM and the CMS are depicted in
Figure 10. The CMS showed good molecular sieving properties since they did not adsorb CH
4 but showed a large CO
2 adsorption capacity. However, the carbon-based HM showed faster initial CO
2 adsorption than CMS. According to the authors this is of great significance for the application in pressure swing adsorption (PSA), where it is more appropriate to reach the adsorption stage in a shorter time from an economical point of view.
Figure 10.
Kinetics of CO
2 and CH
4 adsorption on carbon honeycomb HP110 and a commercial carbon molecular sieve CMS. From reference [
24], with permission from Elsevier.
Figure 10.
Kinetics of CO
2 and CH
4 adsorption on carbon honeycomb HP110 and a commercial carbon molecular sieve CMS. From reference [
24], with permission from Elsevier.
An electric swing adsorption (ESA) technology, which used an integral activated carbon HM from MAST Carbon as adsorbent, was employed to remove low molar fractions of CO
2 in flue gas streams [
47]. The HM (3 cm diameter, 30 cells/cm
2 and 0.11 mm wall thickness) had low electrical resistivity that enabled an enhanced CO
2 desorption rate by heating the HM passing an electrical current through it. Authors proposed a mathematical model for the HM used and different ESA cycles for CO
2 captures.
The ESA technology was also applied to remove toluene from a N
2 flow by using an integral activated carbon HM with 16 square cells/cm
2[
48]. The HM electrical resistivity decreased as temperature and uptake increased. The concentration of the desorbed toluene as function of time had the shape of a peak followed by a tail, which was the main drawback of ESA. Authors indicate that this technology is better to use it for treatments at moderate concentrations.
The use of integral carbon fiber HMs for CO
2 capture from flue gas produced by power plants was recently reported [
39]. This work shows the optimal fabrication parameters for the carbon fiber HM based on maximal CO
2 adsorption performance, which reached a value of 103.5 cm
3 STP/g at 0 ºC. Adsorption breakthrough tests showed that the HM could reduce the CO
2 inlet concentration from 9.7% to 0.29% in the outlet.
An integral carbon HM with 4.6 cells/cm
2 and wall thickness of 1.6 mm was prepared from a mixture of a sub-bituminous coal char and carboxymethylcellulose [
49]. This HM was used to study the dynamic adsorption of NO from Ar flow. The NO adsorption capacity of the carbon-based HM was greater than that of powder activated carbons. In addition, the presence of Cu in the HM or O
2 in the gas stream favored the NO adsorption.
3.2. Carbon-based HMs as supports for catalysts
An efficient technology to reduce nitrogen oxide emissions from stationary sources is the SCR with ammonia. The industrial operations are generally carried out using a V
2O
5/TiO
2 catalyst in monolithic form at temperatures between 300 and 500 ºC [
50]. However, in this temperature range SO
2 and particle poisoning are very serious drawbacks. So, there is interest to develop low-temperature catalysts that could be used for simultaneous NO and SO
2 removal from flue gas streams at temperatures below 250 ºC, which would be more economic than individual separation of both contaminants. The use of carbon-based HMs as supports for vanadium catalysts was also reported in the literature [
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62]. These studies were carried out with both dip-coated and integral carbon HMs and they dealt with the effect of the carbon coating pore texture and thickness, oxidation pretreatments, vanadium content, promotion of a second metal, previous sulfation of the vanadium catalyst and presence of SO
2 and H
2O in the gas feed on the catalyst dispersion and activity and selectivity in the SCR of NO.
When vanadium supported on carbon-coated HMs (62 cell/cm
2) were used it was found that a high mesopore volume of the carbon coating increased the surface oxygen complexes fixed after an oxidation treatment. This was essential to obtain a high vanadium dispersion and a high activity in the SCR of NO. Conversely, the micropores did not contribute to enhance the vanadium dispersion [
51].
Then a mesoporous carbon-coated HM was used to deposit different vanadium loadings [
52,
53] that was dispersed as V
2O
5 overlays up to 6 wt % loading. For higher loadings vanadia crystallites appeared. Both activity and selectivity to N
2 in the SCR of NO dropped dramatically for loadings surpassing the monolayer coverage. The turnover frequency of the catalysts was lower than that of TiO
2-supported catalysts. However, the carbon-supported vanadia catalysts were not deactivated by SO
2 at low reaction temperatures, while the corresponding titania-supported catalysts were readily deactivated. Finally, an optimal carbon-coating thickness of around 30–40 μm was found by simulating, with a one-dimensional catalyst model, the effects of thickness on the geometric parameters and conversion.
The effect of oxidation of carbon-coated HMs on vanadium dispersion and catalytic activity in the SCR of NO was also studied [
54,
55,
56]. Preparation of vanadia catalysts was carried out by equilibrium adsorption impregnation with VO
2+ of a dip-coated HM (31 cells/cm
2) that was previously oxidized with HNO
3 and ionic exchanged with a NaOH solution [
54]. This preparation method produced an increase in the catalytic activity due to an increase in vanadium content and dispersion.
In other works [
55,
56] oxidation of the dip-coated HM (62 cell/cm
2) was carried out with different oxidizing agents, and vanadium catalysts were prepared by ion-exchange with VO
2+ ions. Treatments led to high vanadium dispersion except in the case of ozone treatment. For well dispersed catalysts, their activity linearly increased when vanadium loading increased. Selectivity to N
2 was 100% in all catalysts. The acidic character of the surface was of great importance for achieving a high NO reduction efficiency. Thus, a high surface acidity led to a strong NH
3 adsorption on the carbon surface decreasing the NO conversion. Conversely, a low surface acidity did not promote the vanadium fixation and dispersion, resulting in a decrease of NO reduction efficiency.
The addition of a second metal (Fe, Cr, Cu or Mn) as promoter to vanadium catalysts supported on mesoporous carbon-coated HMs resulted in a modest increase in their specific activity [
57]. However the introduction of 400 ppm of SO
2 in the gas feed increased three times the activity of the catalysts, which did not change in 20 hours operation at 150 and 180 ºC (see
Table 5). This was attributed by the authors to the accumulation of NH
4HSO
4 in the micropores but not in the mesopores (predominant in the carbon coating) where most of vanadium was located.
Table 5.
Turnover frequency at steady state in the SCR of NO with NH
3 before and after addition of 400 ppm SO
2. From reference [
57], with permission from Elsevier.
Table 5.
Turnover frequency at steady state in the SCR of NO with NH3 before and after addition of 400 ppm SO2. From reference [57], with permission from Elsevier.
| Reaction temperature (ºC) | TOF (s-1) x 10–4 |
|---|
| With fresh catalyst | 20h Time-on-stream with SO2 |
|---|
| 150 | 1.7 | 4.9 | |
| 180 | 4.9 | 9.6 | |
A vanadia catalyst supported on a carbon-coated HM (31 cells/cm
2) was used to treat the exit gases of a coal-fired power plant [
54]. The catalyst was placed in the outlet pipe where temperature was about 150 ºC. Some values of the operation parameters of the power plant were: gas flow 1.3 × 10
6 m
3/h, 200–700 ppm NO, 100–350 ppm SO
2 and 6–10% O
2. After 24hours, the conversion dropped to 40% of the initial value, and after 200hours, conversion remained constant at 13% of the initial value. Loss of activity was due to poisoning by As and sulfate formation.
Wang
et al. [
58,
59] studied, at laboratory scale, the simultaneous NO and SO
2 removal at low temperatures using a vanadia catalyst supported on an integral activated carbon HM (16 cells/cm
2). Catalysts with 1–2 wt % vanadia and at 200 ºC showed high activity in the simultaneous NO and SO
2 removal. Regeneration of the catalysts produced a high increase in the SCR of NO with NH
3 and a low increase in the SO
2 removal. Some components of the binder used such as silica, alumina, alkaline and alkaline-earth oxides were unfavorable for NO and SO
2 removal.
Sulfation of vanadia supported carbon-coated HM resulted (62 cell/cm
2), after an induction period, in a much higher conversion than the fresh catalyst in the SCR of NO at low temperatures (<200 ºC) [
60]. Sulfates were anchored to carbon in the vicinity of vanadyl sites. This improved the redox properties in the sulfated catalyst, which caused its superior performance compared to the fresh one. When either water or SO
2 was added to the gas feed the sulfated catalyst kept a 100% conversion to N
2 in the low temperature range (200–230 ºC). Conversely, when both water and SO
2 were added simultaneously the conversion and selectivity decreased, because water sped the ammonium sulfate deposition. This sulfated catalyst had higher activity at low temperature than commercial VO
x/TiO
2 catalysts and, in addition, the presence of water in the gas feed did not inhibit its activity [
61].
The influence of the coating (carbon or alumina) of a cordierite HM on the behavior of vanadia catalysts for the SCR of NO with ammonia was recently reported [
62]. Results found showed that both kinds of catalysts had similar activity under steady-state conditions. However, the carbon coating was much thinner, better adhered to the cordierite, provided a higher surface area, and had higher thermal shock and vibration resistance than the alumina coating.
Mn and Cu oxides supported on carbon-based HMs have been also used as catalysts for the SCR of NO with NH
3[
63,
64,
65]. A carbon-coated HM (31 cells/cm
2) was used as support of manganese oxide [
63], which showed good activity, 60–70% NO reduction at 150 °C. Support oxidation enhanced the Mn loading and activity. A high active catalyst (more than 90% conversion at 150–200 ºC) was also obtained with other Mn oxide supported on a dip-coated HM [
64]. The SCR of NO was improved by doping with Ce and Pd. In addition, the tolerance to SO
2 increased after doping with Fe and V.
On the other hand, Mn and Cu were used as catalysts supported on integral carbon HMs (13.7 cells/cm
2 and 0.8 mm wall thickness) [
65]. The most relevant results, according to the authors, were that microwave drying of the impregnated monoliths led to better metal dispersion and distribution and also to differences in the metallic phases formed compared to conventional drying methods. Some of the Cu catalysts were very active at low temperatures and stable upon consecutive cycles and with time.
Catalytic combustion is one of the most important technologies for eliminating VOCs present at low concentration in effluent streams. From an energetic point of view, and to avoid NOx formation, low temperatures (below 200 ºC) are preferred. However, at these conditions, water vapor produced during combustion can be retained on the catalytic support, with negative effects on the activity of the catalyst. Hydrophobic catalyst supports may overcome this effect, so the tunable surface hydrophobicity of carbon materials has been an important consideration for their application as catalyst support in VOCs combustion.
The catalytic combustion of benzene, toluene and xylenes (BTX) using Pd and Pt supported on different carbon-based HMs was reported [
30,
66,
67,
68]. Some supports were prepared from a commercial cordierite HM of 5 cm length, 1 cm diameter, 62 cells/cm
2 and 0.18 mm wall thickness. A carbon layer was deposited on the channel walls, by the dip-coating method, of either the cordierite support or the cordierite support previously modified with alumina to block its macroporosity. Another carbon-based HM was prepared by CNF growing on the above alumina coated cordierite HM. Finally, two commercial integral HMs were also used with the same geometric characteristics than the above cordierite HM. Pd and Pt catalysts were prepared by impregnation with an aqueous solution of the corresponding tetraammine metal (II) nitrates.
Table 6.
Surface characteristics of the carbon-based HMs. Values in parenthesis are given per gram of carbon. From reference [
67], with permission from Elsevier.
Table 6.
Surface characteristics of the carbon-based HMs. Values in parenthesis are given per gram of carbon. From reference [67], with permission from Elsevier.
| Support | SBET (m2/g) | SExternal (m2/g) | VMacro(cm3/g) | VMeso (cm3/g) |
|---|
| WA | 474 (1366) | 4 (12) | 0.325 (0.937) | 0 |
| WB | 460 (1489) | 62 (199) | 0.233 (0.754) | 0.138 (0.447) |
| HPM | (2) | (<1) | non detected | non detected |
The monolithic catalysts were stable during the reaction and no gasification of the carbon-coated monoliths was observed during the catalytic combustion of xylenes in the temperature range studied (120–180 ºC). Pt catalysts were more active than the Pd ones. Pt catalysts with higher metal particle size were more active whereas the opposite was observed with Pd catalysts. Complete xylene combustion was reached in the temperature range studied with total conversion to CO
2 and H
2O [
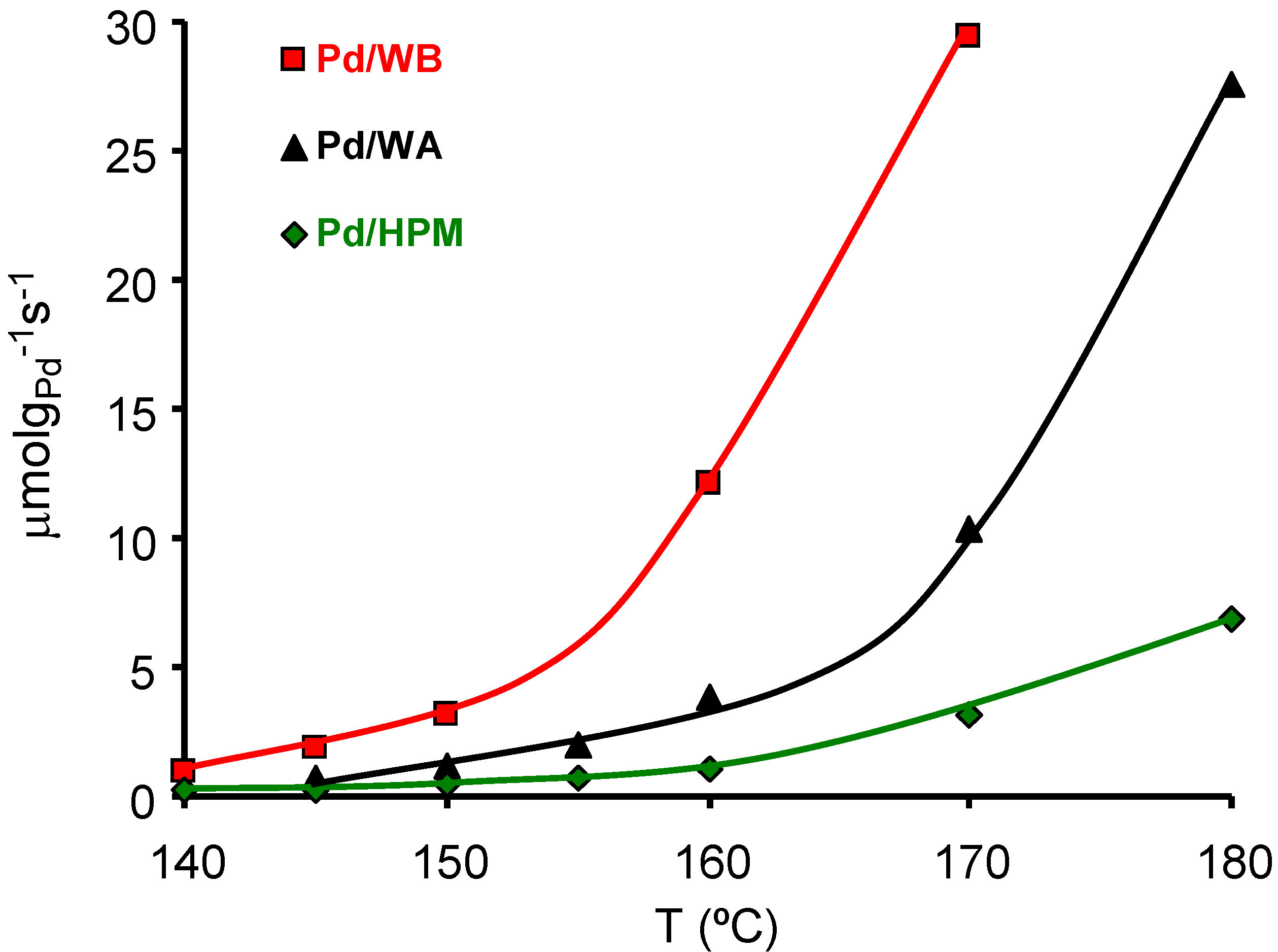
66]. The effect of carbon coating porosity on the Pd catalyzed m-xylene combustion was studied [
67] with different carbon-coated HMs whose surface area and porosity are compiled in
Table 6. Samples WA and WB were two integral carbon HMs with a total carbon content of 34.7 and 30.9 wt %, from MeadWestvaco Corporation. Sample HPM was a carbon-coated HM with a total carbon content of 6.3 wt %. The activity of the Pd catalysts with close Pd content (around 0.4 wt %) supported on the above supports is depicted in
Figure 11. These results show that the carbon external surface area, the macro and mainly mesopores, play an important role in this reaction, improving the contact between the Pd particles and the
m-xylene molecules. Thus, for similar Pd loading and Pd particle size the larger the macro- and mesoporous surface area the higher the activity.
Figure 11.
Activity of the catalysts in the m-xylene combustion (μmol of m-xylene burned per gram of Pd and per second). From reference [
67], with permission of Elsevier.
Figure 11.
Activity of the catalysts in the m-xylene combustion (μmol of m-xylene burned per gram of Pd and per second). From reference [
67], with permission of Elsevier.
The catalytic behavior of Pd and Pt supported on CNF-coated HMs was studied in the low-temperature catalytic combustion of BTX, and compared with the performance of the above metals supported on alumina-coated HMs [
30]. The catalyst supported on the CNF-coated HMs was the most active, independent of the metal catalyst or the tested aromatic compound as indicated by the TOF values obtained at low conversion that are shown in
Table 7. This was due to the fact that the CNF surface was more hydrophobic than that of alumina, and the release of water molecules produced during the combustion was favored. One of Pt catalysts supported showed the best performance, and was already active at 120 ºC in the benzene combustion.
Table 7.
TOF [×10
3] data (s
-1) for the catalysts in the benzene combustion at 150 ºC, and for toluene and m-xylene at 160 ºC. From reference [
30], with permission from Elsevier.
Table 7.
TOF [×103] data (s-1) for the catalysts in the benzene combustion at 150 ºC, and for toluene and m-xylene at 160 ºC. From reference [30], with permission from Elsevier.
| Catalyst | Benzene | Toluene | m-Xylene |
|---|
| Pt/CNF | 5.07 | 2.79 | 1.95 |
| Pd/CNF | 1.38 | 0.62 | 0.56 |
| Pt/Alumina | 0.24 | 0.55 | 0.46 |
| Pd/Alumina | 0.16 | 0.40 | 0.23 |
BTX combustion reactions were catalyzed by Pt and Pd through different kinetic mechanisms, which explained according to the authors why Pt catalysts were always more active than the Pd ones deposited on the same support [
30,
68].










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

 ), nutshell (
), nutshell (  ), wood (
), wood (  ) and peat (
) and peat (  ). From reference [33], with permission from Elsevier.
). From reference [33], with permission from Elsevier.

 , Norit R1 4.3 g;
, Norit R1 4.3 g;  , Norit R1 8.6 g;
, Norit R1 8.6 g;  , Norit R1 13 g;
, Norit R1 13 g;  , (31 cells/cm2)-5 cm-5 cm;
, (31 cells/cm2)-5 cm-5 cm;  , (31 cells/cm2)-5 cm-10 cm;
, (31 cells/cm2)-5 cm-10 cm;  , (31 cells/cm2)-5 cm-15 cm;
, (31 cells/cm2)-5 cm-15 cm;  , (62 cells/cm2)-5 cm-5 cm;
, (62 cells/cm2)-5 cm-5 cm;  , (62 cells/cm2)-5 cm-10 cm;
, (62 cells/cm2)-5 cm-10 cm;  , (62 cells/cm2)-5 cm-15 cm. From reference [40], with permission from Elsevier.
, (62 cells/cm2)-5 cm-15 cm. From reference [40], with permission from Elsevier.

 ), 100 ºC (
), 100 ºC (  ) and 150 ºC (
) and 150 ºC (  ). From reference [43], with permission from Elsevier.
). From reference [43], with permission from Elsevier.


