Self-Assembled Hydrogel Nanoparticles for Drug Delivery Applications


Abstract
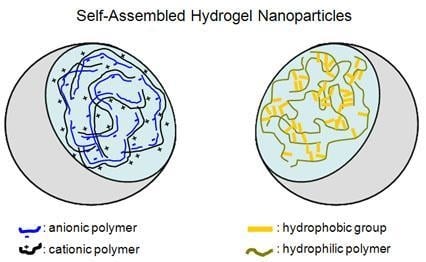
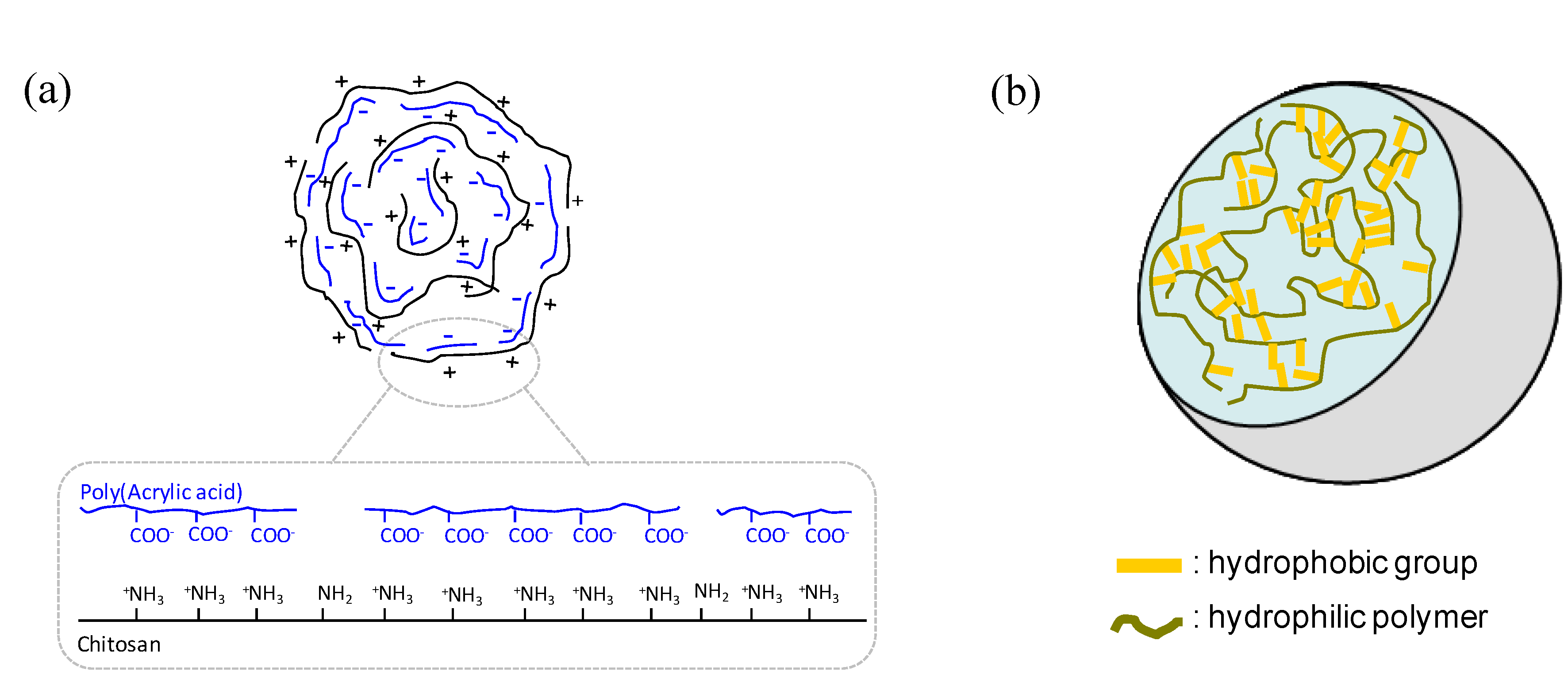
:
1. Introduction
2. Materials, Properties, Methods
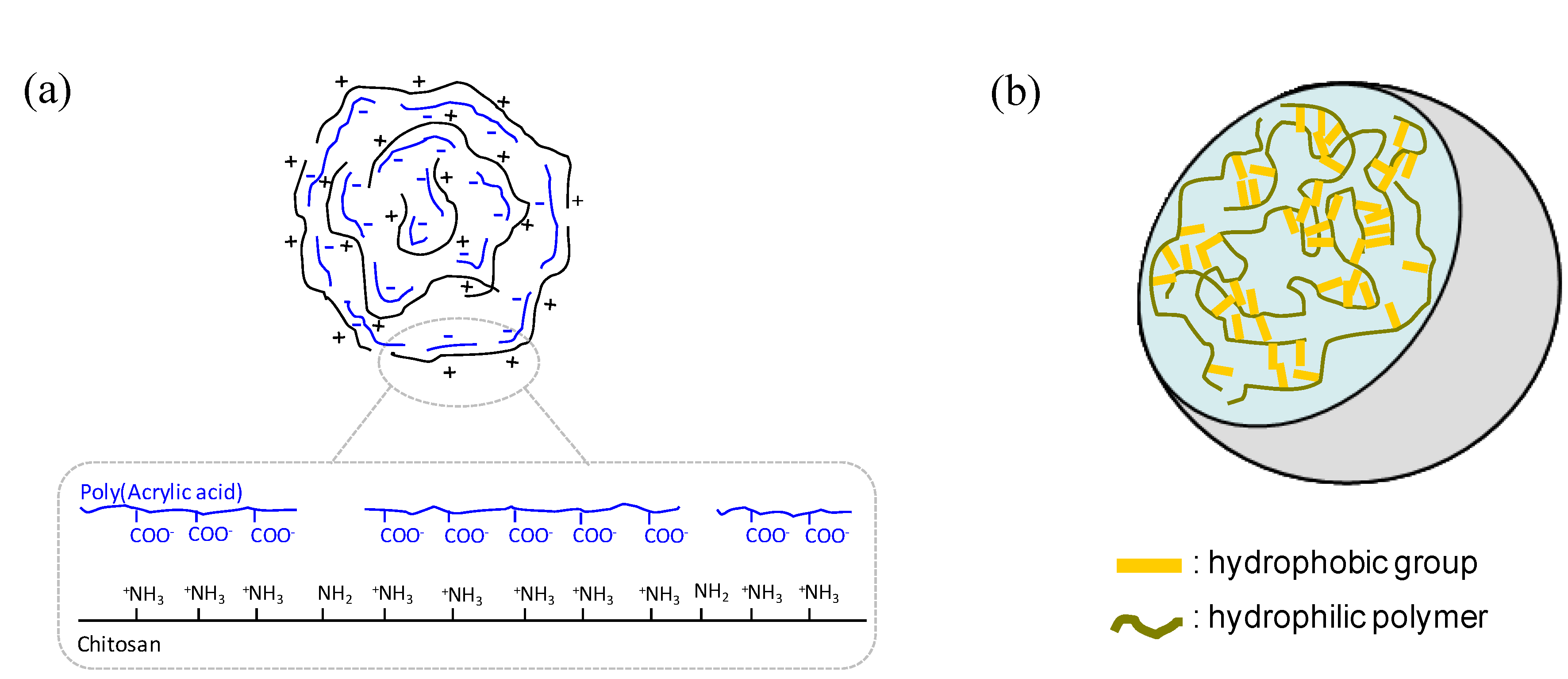
2.1. Materials
2.2. Properties

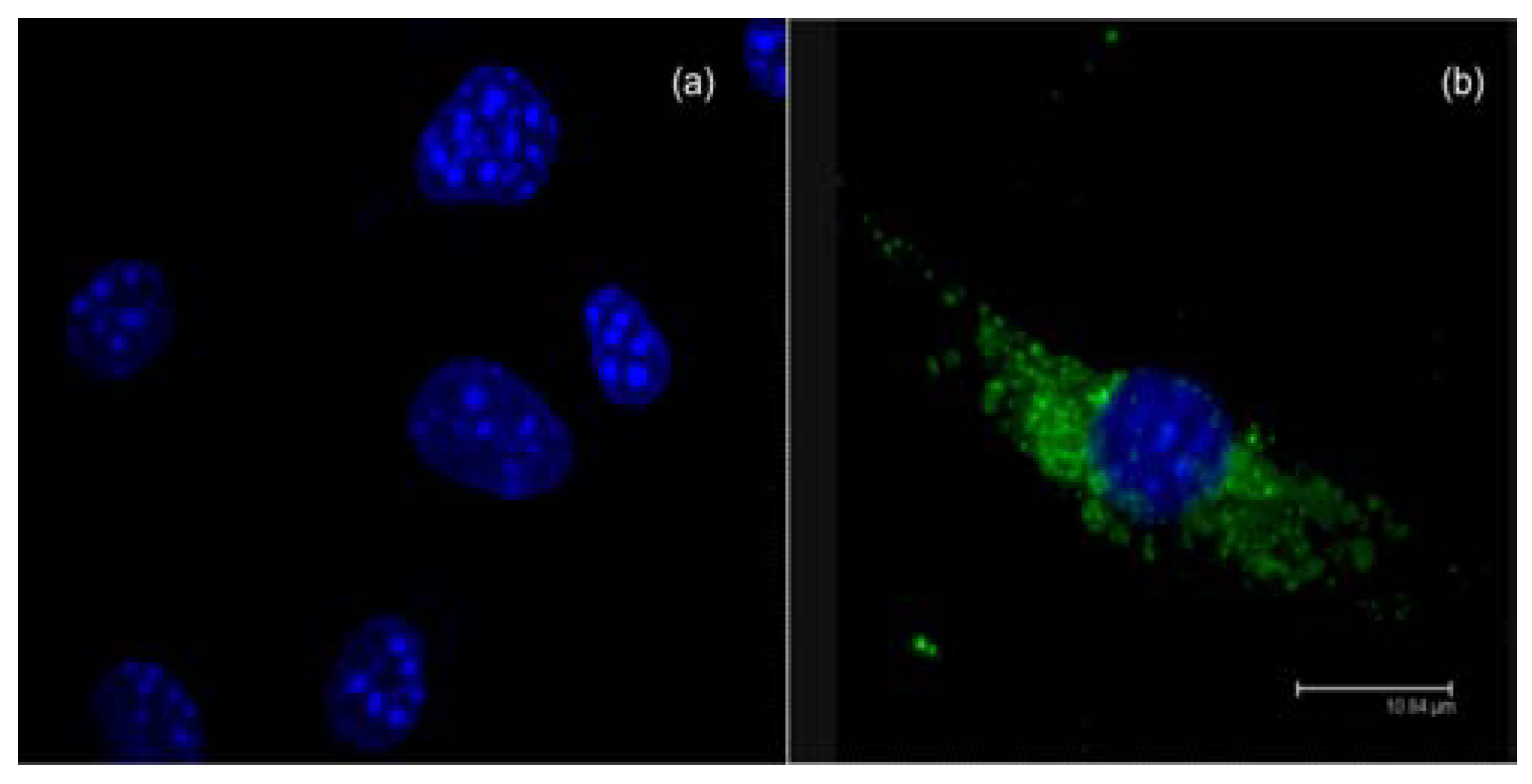
2.3. Methods
3. Drug Loading, Targeting and Release
3.1. Drug loading
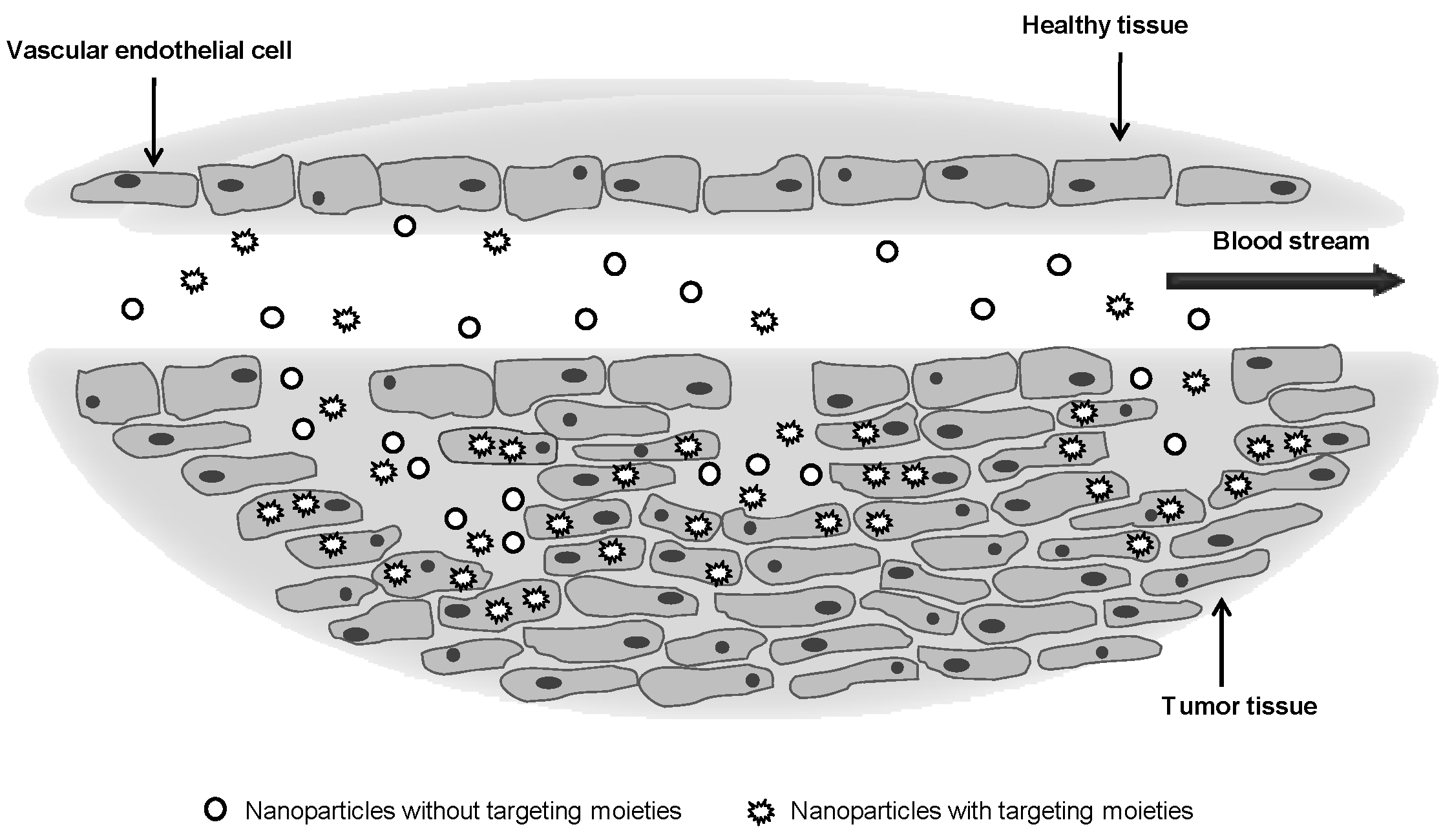
3.2. Targeting
3.3. Drug release
4. Applications
4.1. Small molecular weight drug delivery

{kind=link}
{kind=link}
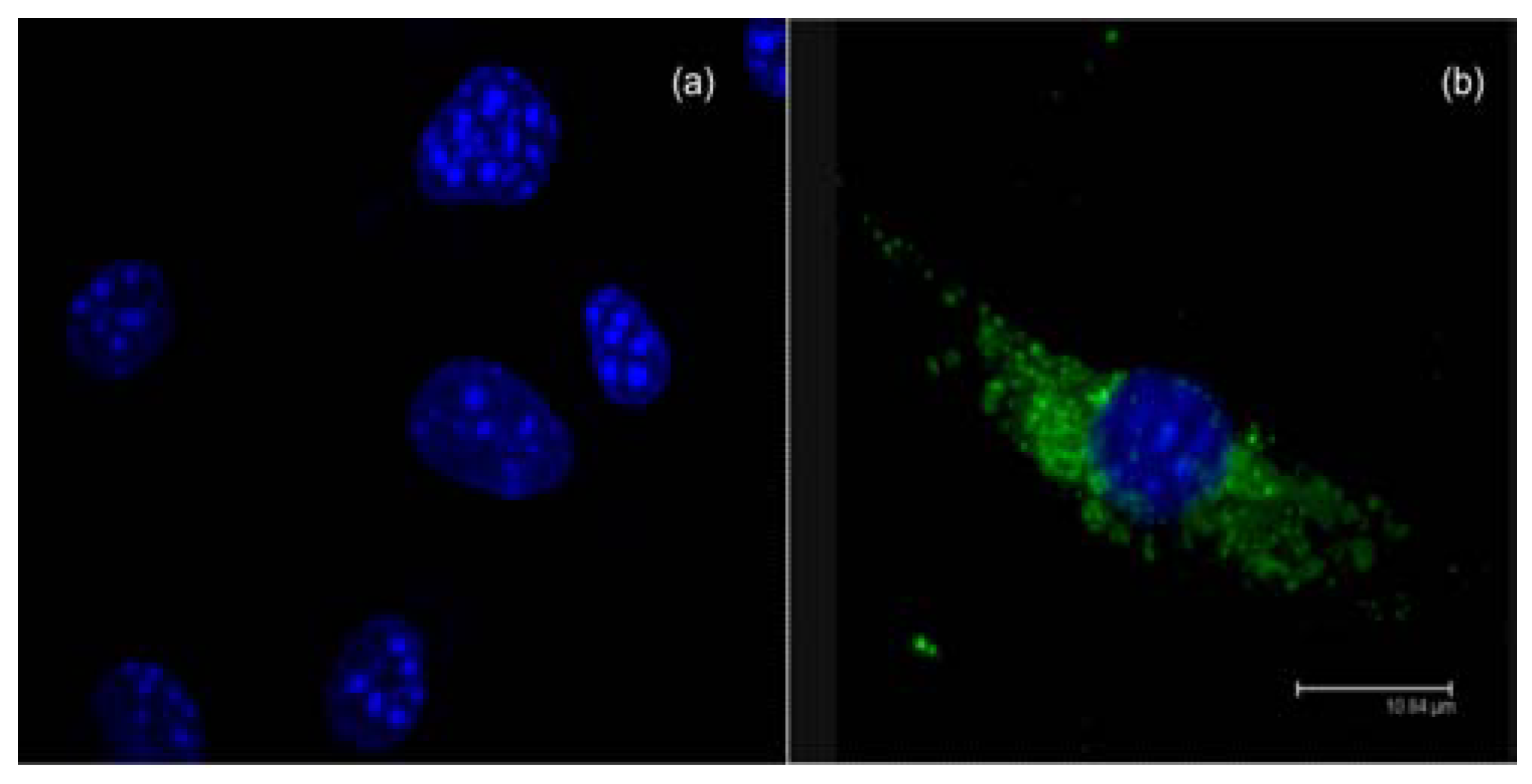{kind=link}
{kind=link}
{kind=link}
| Polymer | Ligand | Target | Cell line | Drug binding | Stimuli | Ref |
|---|---|---|---|---|---|---|
| azo-dextran | aspirin | -- | COS-1 | simple mixing and irradiation with UV light | UV–vis light | [46] |
| carboxymethyl chitosan-linoleic acid | adryamycin (anti-cancer) | -- | HeLa | direct dissolution | -- | [47] |
| pullulan acetate | adriamycin (anti-cancer) | vitamin H (biotin) | HepG2 | dialysis method | -- | [48] |
| pullulan acetate/sulfonamide | adriamycin (anti-cancer) | -- | MCF-7 | dialysis method | pH | [49] |
| poly[(maleilated dextran)-graft-(N-isopropylacrylamide)] | camptothecin (anti-cancer) | -- | L929 | dialysis method | pH, temperature | [50] |
| poly(N-isopropylacrylamide)/chitosan | camptothecin (anti-cancer) | -- | SW480 | direct dissolution | pH | [51] |
| poly[2-(N,N-diethylamino)ethyl methacrylate]-block-PEG | cisplatin (anti-cancer) | -- | SKOV-3 In vivo | solvent displacement method | pH | [52] |
| poly (lactide-co-glycolide)-PEG | curcumin (anti-cancer) | -- | KBM-5, Jurkat, DU145, MDA-MB-231, HCT116,SEG-1 In vivo | nanoprecipitation | -- | [53] |
| polylactide-co-glycolide–PEG–folate | docetaxel (anti-cancer) | folate | SKOV3 | emulsification/solvent diffusion method | -- | [54] |
| poly(D,L-lactic-co-glycolic acid)-block-PEG | docetaxel (anti-cancer) | PSMA aptamer | LNCaP | nanoprecipitation | -- | [55] |
| glycol chitosan-5β-cholanic acid | docetaxel (anti-cancer) | -- | A549 In vivo | dialysis method | -- | [56] |
| poly(l-histidine)-b-PEG-folate (75 wt.%) and poly(L-lactide)-b-PEG-folate (25 wt.%) | doxorubicin (anti-cancer) | folate | MCF-7 | . dialysis method | pH | [57] |
| chitosan-poly(acrylic acid) | doxorubicin (anti-cancer) | -- | HepG2 In vivo | direct dissolution | -- | [58] |
| poly(ε-caprolactone)-PEG-poly(ε-caprolactone) | honokiol (anti-inflammation) | -- | A549 | direct dissolution | -- | [59] |
| ethylcellulose methylcellulose | nimesulide (nonsteroid anti-inflammation) | -- | fresh human blood | desolvation method | -- | [60] |
| poly(ethylene oxide)-modified poly(ε-caprolactone) | tamoxifen | -- | MDA-MB-231 In vivo | solvent displacement | -- | [61] |
| PEG-polycyanoacrylate | paclitaxel (anti-cancer) | transferrin | In vivo | solvent evaporation | -- | [62] |
| poly (lactide-co-glycolide fumarate)/poly(lactide-co-ethylene oxide fumarate) poly (lactide-fumarate)/ poly(lactide-co-ethylene oxide fumarate) | paclitaxel (anti-cancer) | -- | HCT116 In vivo | dialysis method | -- | [63] |
| PEG750-block-poly(ε-caprolactone-co-trimethylenecarbonate) | paclitaxel (anti-cancer) | -- | HeLa In vivo | solvent evaporation | -- | [64] |
| poly (lactide-co-glycolide)/ poly(ε-caprolactone)-PEG | paclitaxel (anti-cancer) | -- | HeLa | simple emulsion or nanoprecipitation method | -- | [65] |
| linoleic acid/poly(β-malic acid) Chitosan | paclitaxel (anti-cancer) | -- | In vivo | sonication and dialysis method | -- | [66] |
| poly(β-amino ester)-graft-PEG | paclitaxel camptothecin (anti-cancer) | -- | SKOV-3 | solvent displacement or dialysis method | pH | [42] |
| glycol chitosan-5β-cholanic acid | protophorphyrin IX (photosensitizer, photodynamic therapy) | -- | SCC7 In vivo Ex vivo | dialysis method | -- | [67] |
| poly(β-benzyl-l-aspartate)-block-poly(vinylpyrrolidone) | prednisone (anti-inflammation) | -- | SW-1990 | dialysis method | pH | [68] |
| poly (10-undecenoic acid-b-N-isopropylacrylamide) | prednisone (anti-inflammation) | -- | ECV304 | dialysis method | pH, temperature | [69] |
| cellulose-g- poly(l-lactide) | prednisone (anti-inflammation) | -- | 3T3 | dialysis method | -- | [70] |
| galactosylated polycaprolactone-g-dextran | prednisone (anti-inflammation) | galactose | HepG2, 3T3 In vivo | dialysis method | -- | [71] |
| poly(ethylene oxide)-modified poly(ε-caprolactone) | saquinavir (HIV-protease inhibitor) | -- | THP-1 | solvent displacement | -- | [72] |
| water soluble chitosan | thymol (anti-microbial) | -- | Staphylococcus aureus Bacillus subtilis Escherichia coli Aspergillus niger | sonication method | -- | [73] |
4.2. Protein, peptide and oligosaccharide delivery
- -
- -
- -
- -
4.3. Vaccine delivery
| Polymer | Antigen | Remarks | route | Ref |
|---|---|---|---|---|
| poly-l-lysine coated polystyrene particles | sOVA-C1 plasmid | Particles of different sizes may target different APCs. | intradermal | [113] |
| poly-(ε-caprolactone) -poly(lactide-co-glycolide) | diphtheria toxoid | Correlations between polymer characteristic (e.g., hydrophobicity ) and route of administration, indicate that such characteristics can have interesting implications in immune responses. | intranasal intramuscular | [114] |
| methoxyPEG–poly(lactide-co-glycolide) | recombinant hepatitis B surface antigen (HBsAg) | Delivery of HBsAg encapsulated within a nanoparticle is a superior way for generating faster immune responses, as compared to the non-encapsulated counterpart. | intraperitoneal | [115] |
| poly lactic acid-PEG | recombinant hepatitis B surface antigen (HBsAg) | Different compositions of PLA and PEG polymers were synthesized to stabilize the antigen. A comparison of their efficacy in the generation of an effective immune responses is shown. | nasal | [5] |
| poly(γ-glutamic acid)-graft-L-phenylalanine | japanese encephalitis (JE) vaccine | A single dose of JE vaccine with nanoparticles enhanced the neutralizing antibody titer. | intraperitoneal | [116] |
| poly(γ-glutamic acid)-graft-l-phenylalanine | influenza hemagglutinin (HÁ) vaccine | Subcutaneous immunization with a mixture of HA vaccine and nanoparticles induced higher mononuclear cell proliferation and production of IFN-γ, IL-4, and IL-6 upon HA restimulation. | subcutaneous | [117] |
| poly (d,l-lactide-co-glycolide)–polyethyleneimine | DNA encoding Mycobacterium tuberculosis latency antigen Rv1733c | The polyplexes were able to mature human dendritic cells and stimulated the secretion of cytokines, comparable to levels observed after lipopolysaccharide stimulation. | intramuscularly endotracheal | [118] |
| hydrophobically modified poly(γ-glutamic acid) | gp120 (human immunodeficiency virus -1) | The protein-encapsulated nanoparticles induced cytotoxic T lymphocyte. Efficient uptake by immature dendritic cells (DC) and induction of DC maturation was observed. | intranasal | [25] |
| chitosan | DNA vaccine encoding mite dust allergen Der p 2 (Der p 2-pDNA) | Chitosan-DNA nanoparticles can generate a higher level expression of gene in vivo, therefore can preferentially activate specific Th1 immune responses thus preventing subsequent sensitization of Th2 cell-regulated specific IgE responses. | oral | [119] |
| chitosan | plasmid DNA encoding surface protein of Hepatitis B virus (pRc/CMV-HBs(S)) | Administration of nanoparticles resulted in serum anti-HBsAg titer and induced sIgA titre in mucosal secretions. Chitosan nanoparticles were able to induce humoral mucosal and cellular immune responses. | nasal | [120] |
| chitosan | DNA plasmids expressing different M. tuberculosis epitopes | Chitosan nanoparticles protect DNA from degradation by nucleases, induce dendritic cells maturation and increased IFN-γ secretion from T cells. | pulmonary | [121] |
| chitosan | pcDNA3-VP1, encoding VP1, major structural protein of coxsackievirus (CVB3) | Nasal administrated chitosan–DNA produced higher levels of serum IgG and mucosal secretory IgA. Strong cytotoxic T lymphocyte activities helped to effectively eliminate CVB3 viruses. | intranasal | [122] |
| low molecular weight chitosan (LMWC) | plasmid DNA encoding human cholesteryl ester transfer protein C-terminal fragment (CEPT) | LMWC had lower binding affinity to DNA, but mediated higher transfection efficiency. Polyplexes could elicit significant systemic immune responses, modulate the plasma lipoprotein profile and attenuate the progression of atherosclerosis. | intranasal | [123] |
| mono-N-carboxymethyl chitosan (MCC) N-trimethyl chitosan (TMC) | tetanus toxoid | Surface charge and particle size exert an important influence in the production of an enhanced immune response. | intranasal | [112] |
4.4. Gene delivery
| Polymer | Therapeutic agent | Ligand | Remarks | Ref |
|---|---|---|---|---|
| chitosan | sense or antisense oligodeoxynucleotides (ODNs) against malarial topoisomerase II gene | -- | Antisense-nanoparticles demonstrate a significant higher inhibition of human malaria parasite, as comparison with sense-nanoparticles and free ODNs. More easily dissociated complexes mediate a faster onset of action. | [146] |
| folate-N-trimethyl chitosan | pDNA | folate | Folate conjugation increased intracellular uptake , transfection efficiency and induce endosomal escape. | [147] |
| folic acid-chitosan | pDNA (pVR1412) | folate | Nanoparticle with positive zeta potentials interact with the cell membrane allowing their endocytosis. | [148] |
| galactosylated 6-amino-6-deoxychitosan | pDNA (pCMV-Luc) | galactose | The increase of transfection efficiency of Gal-6ACT was therefore likely due to improvements in intracellular trafficking and not due to the increase of cellular uptake., | [149] |
| chitosan/hyaluronic acid | pDNA(pEGFP-C1, pβ-gal) | hyaluronan | Polyplexes were able to provide high transfection without affecting cell viability, entering the corneal epithelial cells by CD44 receptor–mediated endocytic uptake. | [150] |
| mannosylated chitosan | pDNA (pGL3-Luc) | mannose | Cellular uptake mediated by mannose recognition. Reduced toxicity and high transfection efficiency. | [151] |
| chitosan –IL-1Ra folate- IL-1Ra- Chitosan | IL-1Ra plasmid DNA | folate | Folate-chitosan-DNA nanoparticles containing the IL-1 Ra gene prevent bone damage and inflammation in rat adjuvant-induced arthritis model that overexpress folate receptors. | [152] |
| PEG-Chitosan | pDNA (pRE-luciferase; pREP4;pCMV-luciferase) | transferrin KNOB protein | The transfection efficiency was much impressive with KNOB (130-fold improvement), in HeLa cells. Chitosan exhibited limited buffering capacity. The clearance of the PEGylated nanoparticles was slightly slower than that of the unmodified nanoparticles. | [153] |
| chitosan | plasmid pGL3-Luc | -- | Polyplexes are endocytosed and possibly released from endosomes due to swelling of both lysosomes and polyplexes, causing the endosome rupture. | [154] |
| chitosan | pDNA (pAAV-tetO-CMV-mEpo and pCMVβ) | -- | Oral gene therapy was efficient in delivering genes to enterocytes. | [155] |
| thiolated chitosan | pDNA (pEGFP-N2) | -- | Improved gene delivery in vitro as well as in vivo. The extended pDNA release and subsequent gene expression were achieved by oxidation of introduced thiol groups to crosslink the thiolated chitosan. | [156] |
| quaternized (trimethylated) chitosan oligomer | pDNA (pEGFcp1-GFP) | -- | Transfection efficiency decreases increasing the degree of quaternization. The polymer effectively transfers the GFP gene into cells both in vitro and in vivo. | [157] |
| 6-N,N,N-trimethyltriazole chitosan | pDNA (EGFP-N1 | -- | The presence of the trimethyltriazole group led to significantly increased cellular uptake, which resulted in higher transfection efficiency in HEK 293 and MDA-MB-468 cells. | [158] |
| methoxy PEG–PEI–chitosan | pDNA (pVRfat-1) | -- | The mPEG increased the slow-releasing ability and water solubility, while PEI improved the transfection efficiency. | [159] |
| chitosan/ poly(γ-glutamic acid) | pDNAs (pEGFP-N2, pGL4.13 and pEGFP-N2) | -- | The incorporation of γ-PGA in the chitosan nanoparticles facilitates the dissociation of chitosan and DNA, increasing transfection efficiency. Trypsin-cleavable proteins in cellular membrane affect internalization of polyplexes. | [160] |
| methylated collagen | pDNA (pRELuc) | -- | Methylated collagen improved DNA binding ability and the stability of the complexes at physiological conditions, as compared with unmodified native collagen. | [161] |
| cationized gelatin | plasmid DNA of transforming growth factor-βR (TGF-βR) siRNA expression vector | -- | The injection of polyplexes significantly decreased the level of TGF-βR and α-smooth muscle actin over-expression, the collagen content of mice kidney, and the fibrotic area of renal cortex, in contrast to free plasmid DNA injection. | [162] |
| PEG–modified thiolated gelatin | pDNA (EGFP-N1) | -- | Nanoparticles released encapsulated plasmid DNA in response to varying concentrations of glutathione. | [163] |
| Polymer | Therapeutic agent | Ligand | Remarks | Ref |
|---|---|---|---|---|
| PEG-PEI (NanoGelTM) | antisense oligonucleotide (ODN) targeting the mdr1 gene | transferrin insulin | Transport efficacy across the blood-brain barrier is increased by modification with transferrin or insulin. Improvement of ODN accumulation in the brain (15 fold). | [9] |
| lactoferrin-PEI | pDNA | lactoferrin | Selectivity for bronchial epithelial cells. Lower cellular toxicity of polyplexes and higher transfection efficiency (5-fold higher), as compared with PEI/pDNA complexes. | [170] |
| RGD-PEG-PEI | siRNA inhibiting vascular endothelial growth factor receptor-2 | RGD | Selective tumor uptake, siRNA sequence-specific inhibition of protein expression within the tumor and inhibition of both tumor angiogenesis and growth rate. | [44] |
| PEI-g-PEG-RGD | pDNA (pCMV-sFlt-1) | RGD | Efficient inhibition on proliferation of endothelial cells that expressed sFlt-1 predominantly bound to exogenous VEGF and blocked the binding of VEGF to the full-length Flt-1 receptor. | [171] |
| siRNA-PEG-LHRH/PEI | siRNA (VEGF-vascular endothelial growth factor) | luteinizing hormone- releasing hormone (LHRH) | Enhancement of cellular uptake, as compared to those without LHRH, resulting in increased VEGF gene silencing efficiency via receptor-mediated endocytosis. | [172] |
| EGF-PEG-PEI | pDNA (pCMVLuc) | epidermal growth factor (EGF) peptides | EGF-containing polyplexes were 10- to 100-fold more efficient than polyplexes without EGF. | [173] |
| PEI | pDNA | Peptide (NL4-10K) | Polyplexes displayed no toxicity in neuronal cells. Enhancement of gene expression (up to 1000-fold) and transfection efficiency (59-fold higher), in dorsal root ganglia, compared to nontargeting polyplexes. | [174] |
| PEI-g-Clenbuterol | pDNA (pCMVLuc) | β2-adrenoceptor (clenbuterol) | Specific cellular uptake into alveolar (transfection efficiency 14-fold higher than for unmodified PEI) but not bronchial epithelial cells. | [175] |
| folate–PEG–PEI | pDNA (pCMV-Luc or pcDNA/rev-caspase-3) | folate | Higher transfection efficiency than other commercially available transfection agents. | [176] |
| PEI-PEG-Fab’ | pDNA (pCMVLuc) | anti- glutamic acid decarboxylase (GAD) | Selectivity toward the islet cells. High transfection efficiency in GAD-expressing mouse insulinoma cells. | [177] |
| HerPEI | pDNA(pcDNA3-CMV-Luc) | anti-HER2 | The HerPEI polyplexes showed significantly greater transfection activity (up to 20-folds) than nonderivatized PEI-based polyplexes in the HER2 overexpressing breast cancer cells. | [178] |
| mannose-PEI | pDNA | mannose | Dendritic cells transfected with polyplexes containing adenovirus particles are effective in activating T cells of T cell receptor transgenic mice in an antigen-specific fashion. | [179] |
| methoxypolyethyleneglycol-PEI-cholesterol | pDNA (pmIL-12) | -- | Inhibition of tumor growth enhanced when combined with specific chemotherapeutic agents. | [180] |
| dextran-PEI | pDNA | -- | Stability of the complex in the presence of BSA. The transfection efficiency depended on the molecular weight of dextran and the grafting degree. | [181] |
| acid-labile PEI | pDNA (pCMV-Luc) | -- | The acid-labile PEI was much less toxic and showed comparable transfection efficiency to that of PEI25K. Polyplexes may be rapidly degraded in acidic endosome. | [182] |
| disulfide-crosslinked low molecular weight linear PEI- sodium hyaluronate | pDNA (pBR322, pEGFP-C1) | -- | Polyplexes achieved significantly higher transfection efficiency than other polymer systems, especially in the presence of serum. | [183] |
| galactosylated PLL | pDNA (pCAT) | galactose | Hepatoma cell line revealed high gene expression. After intravenous injection, polyplexes were rapidly eliminated from the circulation and preferentially taken up by the liver’s parenchymal cells. | [184] |
| Lactosylated PEG-siRNA/PLL | RNAi | lactose | pH-responsive and targetable polyplexes exhibited significant gene silencing human hepatoma cells. | [185] |
| AWBP-PEG-PLL | pDNA (pMNK) | Artery wall binding peptide (AWBP) | High transfection efficiency in bovine aorta endothelial cells and smooth muscle cells. | [186] |
| Antibody-PLL | pDNA(pSV-b-galactosidase) | Anti JL1 | Polyplexes internalization into Molt 4 cells and human leukemia T cells. Higher in vitro transfection efficiency than polyplexes without targeting ligand. | [187] |
| RGD-PEG-block-PLL | pDNA | RGD | Synergistic effect of cyclic RGD peptide and disulfide cross-links to exert the smooth release of pDNA in the intracellular environment via reductive cleavage. Enhanced transfection efficiency against HeLa cells, due to a change in their intracellular trafficking route. | [188] |
| Polymer | Therapeutic agent | Remarks | Ref |
|---|---|---|---|
| poly(imidazole/ 2-dimethylaminoethylamino)phosphazene | pDNA | Imidazole effect on cytotoxicity and transfection efficiency. Evaluation of half-lives under neutral and acidic conditions. | [144] |
| poly[α-(4-aminobutyl)-L-glycolic acid] (PAGA) | pDNA( pCAGGS-Il10, pCAGGS-Il4) | Combined administration of mouse Il4 and Il10 plasmids prevents the development of autoimmune diabetes in non-obese diabetic mice. | [191] |
| poly(4-hydroxy-L-proline ester) | pDNA(CMV-βGal) | The minimum viability of cells incubated with poly(4-hydroxy-L-proline ester) was 85%, which is excellent when compared to the cases of polylysine (20%) and polyethylenimine (2%). | [192] |
| poly(amido amine)s containing multiple disulfide linkages | pDNA | Buffer capacity of poly(amido amine)s in the pH range 7.4-5.1. High transfection efficiency and gene expression, in the presence of serum. | [193] |
| cationic amphoteric polyamidoamine | pDNA (pEGFP) | Evaluation of toxicity and hemolytic activity in the pH range 4.0-7.4. Circulation time and organ accumulation assessment. Study of complex stability and transfection efficiency | [194] |
| three blocks of amino acids Ac-(AF)6-H5-K15-NH2 (FA32) | Doxorubicin, pCMV-luciferase, pCMV-p53 | Co-delivery of drug and gene using nanoparticles was demonstrated via confocal imaging, luciferase expression in the presence of doxorubicin, and synergy in cytotoxic effect towards HepG2 cells. | [166] |
| N,N-diethylethylenediamine-polyurethane | pDNA (pCMV-βgal) | Cytotoxicity was substantially lower and transfection efficiency comparable to the well-known gene carrier poly(2-dimethylaminoethyl methacrylate | [195] |
5. Conclusion
References
- Dubernet, C.; Couvreur, P. Nanoparticles in cancer therapy and diagnosis. Adv. Drug Delivery Rev. 2002, 54, 631–651. [Google Scholar] [CrossRef]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Delivery Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar] [PubMed]
- Sun, T.-M.; Du, J.-Z.; Yan, L.-F.; Mao, H.-Q.; Wang, J. Self-assembled biodegradable micellar nanoparticles of amphiphilic and cationic block copolymer for siRNA delivery. Biomaterials 2008, 29, 4348–4355. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Goyal, A.K.; Gupta, P.N.; Khatri, K.; Mishra, N.; Mehta, A.; Mangal, S.; Vyas, S.P. Synthesis, characterization and evaluation of novel triblock copolymer based nanoparticles for vaccine delivery against hepatitis B. J. Control. Release 2009, 136, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Nanjwade, B.K.; Bechra, H.M.; Derkar, G.K.; Manvi, F.V.; Nanjwade, V.K. Dendrimers: Emerging polymers for drug-delivery systems. Eur. J. Pharm. Sci. 2009, 38, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L.; Gallardo, V.; Ruiz, M.A.; Delgado, A.V. Ftorafur loading and controlled release from poly(ethyl-2-cyanoacrylate) and poly(butylcyanoacrylate) nanospheres. Int. J. Pharm. 2007, 337, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, H.; Maruyama, K.; Okinaga, K.; Sekine, T.; Ishida, O.; Ogiwara, N.; Johkura, K.; Yonemura, Y. Intracellular targeting therapy of cisplatin-encapsulated transferrin-polyethylene glycol liposome on peritoneal dissemination of gastric cancer. Int. J. Cancer 2002, 99, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.V.; Batrakova, E.V.; Kabanov, A.V. Nanogels for oligonucleotide delivery to the brain. Bioconjug. Chem. 2004, 15, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Plard, J.P.; Bazile, D. Comparison of the safety profiles of PLA(50) and Me.PEG-PLA(50) nanoparticles after single dose intravenous administration to rat. Colloids Surf. B 1999, 16, 173–183. [Google Scholar] [CrossRef]
- Peracchia, M.T.; Fattal, E.; Desmaele, D.; Besnard, M.; Noel, J.P.; Gomis, J.M.; Appel, M.; d'Angelo, J.; Couvreur, P. Stealth PEGylated polycyanoacrylate nanoparticles for intravenous administration and splenic targeting. J. Control. Release 1999, 60, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Hans, M.L.; Lowman, A.M. Biodegradable nanoparticles for drug delivery and targeting. Curr. Opin. Solid State Mater. Sci. 2002, 6, 319–327. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs1. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Brannon-Peppas, L.; Blanchette, J.O. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Delivery Rev. 2004, 56, 1649–1659. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Nanomedicine: Current status and future prospects. FASEB J. 2005, 19, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.H.; Jacobs, C.; Kayser, O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv. Drug Delivery Rev. 2001, 47, 3–19. [Google Scholar] [CrossRef]
- Hornig, S.; Bunjes, H.; Heinze, T. Preparation and characterization of nanoparticles based on dextran-drug conjugates. J. Colloid Interface Sci. 2009, 338, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Vila, A.; Sanchez, A.; Tobio, M.; Calvo, P.; Alonso, M.J. Design of biodegradable particles for protein delivery. J. Control. Release 2002, 78, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, C.; Torrado, E.; Martins, T.; Pereira, P.; Pedrosa, J.; Gama, M. Dextrin nanoparticles: Studies on the interaction with murine macrophages and blood clearance. Colloids Surf. B 2010, 75, 483–489. [Google Scholar] [CrossRef]
- Storm, G.; Belliot, S.; Daemen, T.; Lasic, D.D. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Adv. Drug Delivery Rev. 1995, 17, 31–48. [Google Scholar] [CrossRef]
- Luttmann, W.; Bratke, K.; Kupper, M.; Myrtek, D. Immunology; Academic Press (Elsevier): London, UK, 2006. [Google Scholar]
- Roser, M.; Fischer, D.; Kissel, T. Surface-modified biodegradable albumin nano- and microspheres. II: Effect of surface charges on in vitro phagocytosis and biodistribution in rats. Eur. J. Pharm. Biopharm. 1998, 46, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.E., III; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Panyam, J.; Sahoo, S.K.; Prabha, S.; Bargar, T.; Labhasetwar, V. Fluorescence and electron microscopy probes for cellular and tissue uptake of poly(d,l-lactide-co-glycolide) nanoparticles. Int. J. Pharm. 2003, 262, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Akagia, T.; Wang, X.; Utob, T.; Baba, M.; Akashi, M. Protein direct delivery to dendritic cells using nanoparticles based on amphiphilic poly(amino acid) derivatives. Biomaterials 2007, 28, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Emerging biological materials through molecular self-assembly. Biotechnol. Adv. 2002, 20, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Non-covalent interactions in polysaccharide systems. Macromol. Biosci. 2006, 6, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, C.; Martins, J.A.; Gama, F.M. Self-assembled nanoparticles of dextrin substituted with hexadecanethiol. Biomacromolecules 2007, 8, 392–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehvar, R. Dextrans for targeted and sustained delivery of therapeutic and imaging agents. J. Control. Release 2000, 69, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.K.; Jain, D.; Shrivastava, P.K.; Piyush, T. Flurbiprofen- and suprofen-dextran conjugates: Synthesis, characterization and biological evaluation. Tropical J. Pharm. Res. 2009, 8, 221–229. [Google Scholar] [CrossRef]
- Mu, L.; Feng, S.S. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol): PLGA nanoparticles containing vitamin E TPGS. J. Control. Release 2003, 86, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, Y.M. Taxol-loaded block copolymer nanospheres composed of methoxy poly(ethylene glycol) and poly(ε-caprolactone) as novel anticancer drug carriers. Biomaterials 2001, 22, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Govender, T.; Riley, T.; Ehtezazi, T.; Garnett, M.C.; Stolnik, S.; Illum, L.; Davis, S.S. Defining the drug incorporation properties of PLA-PEG nanoparticles. Int. J. Pharm. 2000, 199, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Govender, T.; Stolnik, S.; Garnett, M.C.; Illum, L.; Davis, S.S. PLGA nanoparticles prepared by nanoprecipitation: Drug loading and release studies of a water soluble drug. J. Control. Release 1999, 57, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Panyam, J.; Williams, D.; Dash, A.; Leslie-pelecky, D.; Labhasetwar, V. Solid-state solubility influences encapsulation and release of hydrophobic drugs from PLGA/PLA nanoparticles. J. Pharm. Sci. 2004, 93, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Zambaux, M.F.; Bonneaux, F.; Gref, R.; Dellacherie, E.; Vigneron, C. Preparation and characterization of protein C-loaded PLA nanoparticles. J. Control. Release 1999, 60, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Ishida, E.; Hashimoto, K.; Kobayashi, H.; Aoki, T.; Yasuda, J.; Obata, K.; Kikuchi, H.; Ishida, T.; Kiwada, H.; Harashima, H. Tumor targeting of doxorubicin by anti-MT1-MMP antibody-modified PEG liposomes. Int. J. Pharm. 2007, 342, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Kirpotin, D.B.; Drummond, D.C.; Shao, Y.; Shalaby, M.R.; Hong, K.; Nielsen, U.B.; Marks, J.D.; Benz, C.C.; Park, J.W. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006, 66, 6732–6740. [Google Scholar] [CrossRef] [PubMed]
- Tarragó-Trani, M.T.; Storrie, B. Alternative routes for drug delivery to the cell interior: Pathways to the golgi apparatus and endoplasmic reticulum. Adv. Drug Delivery Rev. 2007, 59, 782–797. [Google Scholar] [CrossRef]
- Watson, P.; Jones, A.T.; Stephens, D.J. Intracellular trafficking pathways and drug delivery: Fluorescence imaging of living and fixed cells. Adv. Drug Delivery Rev. 2005, 57, 43–61. [Google Scholar] [CrossRef]
- Shen, Y.; Tang, H.; Zhan, Y.; Kirk, E.A.V.; Murdoch, W.J. Degradable poly(β-amino ester) nanoparticles for cancer cytoplasmic drug delivery. Nanomed. Nanotechno. Biol. Med. 2009, 5, 192–201. [Google Scholar] [CrossRef]
- Chapman, A.P. PEGylated antibodies and antibody fragments for improved therapy: A review. Adv. Drug Delivery Rev. 2002, 54, 531–545. [Google Scholar] [CrossRef]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mittal, G.; Sahana, D.K.; Bhardwaj, V.; Kumar, M.N.V.R. Estradiol loaded PLGA nanoparticles for oral administration: Effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J. Control. Release 2007, 119, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, S.; Sharma, A.K.; Garg, B.S.; Gandhi, R.P.; Gupta, K.C. Photoregulation of drug release in azo-dextran nanogels. Int. J. Pharm. 2007, 342, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-L.; Liu, C.-G. Self-aggregated nanoparticles from linoleic acid modified carboxymethyl chitosan: Synthesis, characterization and application in vitro. Colloids Surf. B 2009, 69, 178–182. [Google Scholar] [CrossRef]
- Na, K.; Bum Lee, T.; Park, K.H.; Shin, E.K.; Lee, Y.B.; Choi, H.K. Self-assembled nanoparticles of hydrophobically-modified polysaccharide bearing vitamin H as a targeted anti-cancer drug delivery system. Eur. J. Pharm. Sci. 2003, 18, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Na, K.; Lee, E.S.; Bae, Y.H. Adriamycin loaded pullulan acetate/sulfonamide conjugate nanoparticles responding to tumor pH: pH-Dependent cell interaction, internalization and cytotoxicity in vitro. J. Control. Release 2003, 87, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wu, H.; Fan, L.; Zhang, H.; Zhang, H.; Chunhu, G. Study of dual responsive poly[(maleilated dextran)-graft-(N-isopropylacrylamide)] hydrogel nanoparticles: Preparation, characterization and biological evaluation. Polym. Int. 2009, 58, 1023–1033. [Google Scholar] [CrossRef]
- Li Fan, H.W.; Zhang, H.; Li, F.; Yang, T.-H.; Gu, C.-H.; Yang, Q. Novel super pH-sensitive nanoparticles responsive to tumor extracellular pH. Carbohydr. Polym. 2008, 73, 390–400. [Google Scholar] [CrossRef]
- Xu, P.; Kirk, E.A.V.; Murdoch, W.J.; Zhan, Y.; Isaak, D.D.; Radosz, M.; Shen, Y. Anticancer efficacies of cisplatin-releasing pH-responsive nanoparticles. Biomacromolecules 2006, 7, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Nair, H.B.; Sung, B.; Kunnumakkara, A.B.; Yadav, V.R.; Tekmal, R.R.; Aggarwal, B.B. Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem. Pharm. 2010, 79, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, F.; Ghahremani, M.H.; Ostad, S.N.; Atyabi, F.; Seyedabadi, M.; Malekshahi, M.R.; Amini, M.; Dinarvand, R. Folate-receptor-targeted delivery of docetaxel nanoparticles prepared by PLGA-PEG-folate conjugate. J. Drug Targeting 2008, 16, 415–423. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sheri, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.Y.; Kim, I.S.; Kwon, I.C.; Kim, Y.H. Tumor targetability and antitumor effect of docetaxel-loaded hydrophobically modified glycol chitosan nanoparticles. J. Control. Release 2008, 128, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Na, K.; Bae, Y.H. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J. Control. Release 2005, 103, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ding, Y.; Ding, D.; Sun, M.; Zhang, L.; Jiang, X.; Yang, C. Hollow chitosan/poly(acrylic acid) nanospheres as drug carriers. Biomacromolecules 2007, 8, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Gong, C.; Shi, S.; Fu, S.; Men, K.; Zeng, S.; Zheng, X.; Gou, M.; Chen, L.; Qiu, L.; Qian, Z. Self-assembled honokiol-loaded micelles based on poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone) copolymer. Int. J. Pharm. 2009, 369, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Ravikumara, N.R.; Madhusudhan, B.; Nagaraj, T.S.; Hiremat, S.R.; Gargi, R. Preparation and evaluation of nimesulide-loaded ethylcellulose and methylcellulose nanoparticles and microparticles for oral delivery. J. Biomat. Appl. 2009, 24, 47–64. [Google Scholar] [CrossRef]
- Shenoy, D.B.; Amiji, M.M. Poly(ethylene oxide)-modified poly(ε-caprolactone) nanoparticles for targeted delivery of tamoxifen in breast cancer. Int. J. Pharm. 2005, 293, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gu, W.; Huang, J.; Sui, H.; Zhou, Z.; Yang, Y.; Yan, Z.; Li, Y. In vitro and in vivo evaluation of actively targetable nanoparticles for paclitaxel delivery. Int. J. Pharm. 2005, 288, 361–368. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Mercado, A.E.; Xu, W.; Jabbari, E. Cytotoxicity of paclitaxel in biodegradable self-assembled core-shell poly (lactide-co-glycolide ethylene oxide fumarate) nanoparticles. Pharm. Res. 2008, 25, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Magotteaux, N.; Ucakar, B.; Lecouturier, N.; Brewster, M. Novel self-assembling PEG-p-(CL-co-TMC) polymeric micelles as safe and effective delivery system for paclitaxel. Eur. J. Pharm. Biopharm. 2009, 73, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Lecouturier, N.; Marchand-brynaert, J.; Feron, O. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: In vitro and in vivo evaluation. J. Control. Release 2009, 133, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; He, M.; Yin, L.; Bao, J.; Shi, L.; Wang, B.; Tang, C.; Yin, C. Biodegradable nanoparticles based on linoleic acid and poly(β-malic acid) double grafted chitosan derivatives as carriers of anticancer drugs. Biomacromolecules 2009, 10, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, K.; Oh, Y.-K.; Kwon, S.-H.; Her, S.; Kim, I.-S.; Choi, K.; Lee, S.J.; Kim, H.; Lee, S.G.; Kim, K.; Kwon, I.C. Tumor specificity and therapeutic efficacy of photosensitizer-encapsulated glycol chitosan-based nanoparticles in tumor-bearing mice. Biomaterials 2009, 30, 2929–2939. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zeng, R.; Li, C.; Qiao, R. Self-assembled polypeptide-block-poly(vinylpyrrolidone) as prospective drug-delivery systems. Colloids Surf. B 2009, 74, 284–292. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, X.Z.; Cheng, H.; Chen, W.Q.; Cheng, S.X.; Zhuo, R.X. Self-assembled thermo- and pH responsive micelles of poly(10-undecenoic acid-b-N-isopropylacrylamide) for drug delivery. J. Control. Release 2006, 116, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Xu, Q.; Li, Y.; Mo, S.; Cai, S.; Liu, L. The synthesis of biodegradable graft copolymer cellulose-graft-poly(l-lactide) and the study of its controlled drug release. Colloids Surf. B 2008, 66, 26–33. [Google Scholar] [CrossRef]
- Wu, D.Q.; Lu, B.; Chang, C.; Chen, C.S.; Wang, T.; Zhang, Y.Y.; Cheng, S.X.; Jiang, X.J.; Zhang, X.Z.; Zhuo, R.X. Galactosylated fluorescent labeled micelles as a liver targeting drug carrier. Biomaterials 2009, 30, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Shah, L.K.; Amiji, M.M. Intracellular delivery of saquinavir in biodegradable polymeric nanoparticles for HIV/AIDS. Pharm. Res. 2006, 23, 2638–2645. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Du, Y.; Wang, X.; Feng, T.; Science, E.; Key, H.P. Self-aggregation of water-soluble chitosan and solubilization of thymol as an antimicrobial agent. J. Biomed. Mater. Res. A 2009, 90, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Budhian, A.; Siegel, S.J.; Winey, K.I. Controlling the in vitro release profiles for a system of haloperidol-loaded PLGA nanoparticles. Int. J. Pharm. 2008, 346, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Kubota, T.; Choi, T.; Takahashi, M.; Ayano, E.; Kanazawa, H.; Higaki, M. Polymeric nanoparticles encapsulating betamethasone phosphate with different release profiles and stealthiness. Int. J. Pharm. 2009, 375, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Avgoustakis, K.; Beletsi, A.; Panagi, Z.; Klepetsanis, P.; Karydas, A.G.; Ithakissios, D.S. PLGA-mPEG nanoparticles of cisplatin: In vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J. Control. Release 2002, 79, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Sun, C.; Zhang, X.; Wu, Z.; Wang, Z.; Li, C. Hollow and degradable polyelectrolyte nanocapsules for protein drug delivery. Acta Biomater. 2010, 6, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Rieux, A.; Fievez, V.; Garinot, M.; Schneider, Y.J.; Preat, V. Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J. Control. Release 2006, 116, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Cui, F.; Ding, J.; Tang, C.; Yin, C. Chitosan graft copolymer nanoparticles for oral protein drug delivery: Preparation and characterization. Biomacromolecules 2006, 7, 2722–2727. [Google Scholar] [CrossRef] [PubMed]
- Mi, F.-L.; Wu, Y.-Y.; Lin, Y.-H.; Sonaje, K.; Ho, Y.-C.; Chen, C.-T.; Juang, J.-H.; Sung, H.-W. Oral delivery of peptide drugs using nanoparticles self-assembled by poly(γ-glutamic acid) and a chitosan derivative functionalized by trimethylation. Bioconjugate Chem. 2008, 19, 1248–1255. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Chen, C.-T.; Liang, H.-F.; Kulkarni, A.R.; Lee, P.-W.; Chen, C.-H.; Sung, H.-W. Novel nanoparticles for oral insulin delivery via the paracellular pathway. Nanotechnology 2007, 18, 1–11. [Google Scholar]
- Sonaje, K.; Lin, Y.H.; Juang, J.H.; Wey, S.P.; Chen, C.T.; Sung, H.W. In vivo evaluation of safety and efficacy of self-assembled nanoparticles for oral insulin delivery. Biomaterials 2009, 30, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Damge, C.; Maincent, P.; Ubrich, N. Oral delivery of insulin associated to polymeric nanoparticles in diabetic rats. J. Control. Release 2007, 117, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kishida, T.; Hasegawa, U.; Ueda, Y.; Imanishi, J.; Yamagishi, H.; Akiyoshi, K.; Otsuji, E.; Mazda, O. Nanogel DDS enables sustained release of IL-12 for tumor immunotherapy. Biochem. Biophys. Res. Commun. 2008, 367, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, U.; Sawada, S.-I.; Shimizu, T.; Kishida, T.; Otsuji, E.; Mazda, O.; Akiyoshi, K. Raspberry-like assembly of cross-linked nanogels for protein delivery. J. Control. Release 2009, 140, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Bessa, P.C.; Machado, R.; Dopler, D.; Banerjee, A.; Redl, H.; Griensven, M.V.; Reis, R.L.; Casal, M. Thermoresponsive self-assembled elastin-based nanoparticles for delivery of BMPs. J. Control. Release 2009, in press. [Google Scholar]
- Park, J.H.; Kwon, S.; Nam, J.O.; Park, R.W.; Chung, H.; Seo, S.B.; Kim, I.S.; Kwon, I.C.; Jeong, S.Y. Self-assembled nanoparticles based on glycol chitosan bearing β-cholanic acid for RGD peptide delivery. J. Control. Release 2004, 95, 579–588. [Google Scholar] [CrossRef]
- Campos, A.M.D.; Sa, A. Chitosan nanoparticles: A new vehicle for the improvement of the delivery of drugs to the ocular surface. Application to cyclosporin A. Int. J. Pharm. 2001, 224, 159–168. [Google Scholar] [PubMed]
- El-Shabouri, M.H. Positively charged nanoparticles for improving the oral bioavailability of cyclosporin-A. Int. J. Pharm. 2002, 249, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kuno, Y.; Sugimoto, S.; Takeuchi, H.; Kawashima, Y. Surface-modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctions. J. Control. Release 2005, 102, 373–381. [Google Scholar] [PubMed]
- Prego, C.; Torres, D.; Fernandez-Megia, E.; Novoa-Carballal, R.; Quinoa, E.; Alonso, M.J. Chitosan-PEG nanocapsules as new carriers for oral peptide delivery. Effect of chitosan pegylation degree. J. Control. Release 2006, 111, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wu, B.; Zhang, Q.; Chen, J.; Zhu, J.; Zhang, W.; Rong, Z.; Chen, H.; Jiang, X. Brain delivery of vasoactive intestinal peptide enhanced with the nanoparticles conjugated with wheat germ agglutinin following intranasal administration. J. Control. Release 2007, 121, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.L.Z.; Wang, Y.; Ye, W.-H.; Yoon, H.S.; Chan, S.Y.; Yang, Y.-Y. Efficient intracellular delivery of functional proteins using cationic polymer core/shell nanoparticles. Biomaterials 2008, 29, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Aktas, Y.; Yemisci, M.; Andrieux, K.; Gursoy, R.N.; Alonso, M.J.; Fernandez-Megia, E.; Novoa-Carballal, R.; Quinoa, E.; Riguera, R.; Sargon, M.F.; Celik, H.H.; Demir, A.S.; Hincal, A.A.; Dalkara, T.; Capan, Y.; Couvreur, P. Development and brain delivery of chitosan-PEG nanoparticles functionalized with the monoclonal antibody OX26. Bioconjug. Chem. 2005, 16, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Oyarzun-Ampuero, F.A.; Brea, J.; Loza, M.I.; Torres, D.; Alonso, M.J. Chitosan-hyaluronic acid nanoparticles loaded with heparin for the treatment of asthma. Int. J. Pharm. 2009, 381, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Bae, K.H.; Moon, C.W.; Lee, Y.; Park, T.G. Intracellular delivery of heparin complexed with chitosan-g-poly(ethylene glycol) for inducing apoptosis. Pharm. Res. 2009, 26, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wong, H.; Lin, K.; Chen, H.; Wey, S.; Sonaje, K.; Lin, Y.; Chu, C.; Sung, H. The characteristics, biodistribution and bioavailability of a chitosan-based nanoparticulate system for the oral delivery of heparin. Biomaterials 2009, 30, 6629–6637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.; Wu, Z.; Wang, Z.; Niu, H.; Li, C. Nasal absorption enhancement of insulin using PEG-grafted chitosan nanoparticles. Eur. J. Pharm. Biopharm. 2008, 68, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, C.; Wu, Z.; Teng, D.; Zhang, X.; Wang, Z.; Li, C. Chitosan-NAC nanoparticles as a vehicle for nasal absorption enhancement of insulin. J. Biomed. Mater. Res. Part B 2009, 88, 150–161. [Google Scholar] [CrossRef]
- Yin, L.; Ding, J.; He, C.; Cui, L.; Tang, C.; Yin, C. Drug permeability and mucoadhesion properties of thiolated trimethyl chitosan nanoparticles in oral insulin delivery. Biomaterials 2009, 30, 5691–5700. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, Y.S.; Park, K.; Kang, E.; Lee, S.; Nam, H.Y.; Kim, K.; Park, J.H.; Chi, D.Y.; Park, R.W.; Kim, I.S.; Choi, K.; Chan Kwon, I. Self-assembled glycol chitosan nanoparticles for the sustained and prolonged delivery of antiangiogenic small peptide drugs in cancer therapy. Biomaterials 2008, 29, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Progress in human tumour immunology and immunotherapy. Nature 2001, 2, 380–384. [Google Scholar] [CrossRef]
- Sallusto, F.; Cella, M.; Danieli, C.; Lanzavecchia, A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products. J. Exp. Med. 1995, 182, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Elamanchili, P.; Diwan, M.; Cao, M.; Samuel, J. Characterization of poly(d,l-lactic-co-glycolic acid) based nanoparticulate system for enhanced delivery of antigens to dendritic cells. Vaccine 2004, 22, 2406–2412. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 2006, 112, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Lutsiak, M.E.; Robinson, D.R.; Coester, C.; Kwon, G.S.; Samuel, J. Analysis of poly(d,l-lactic-co-glycolic acid) nanosphere uptake by human dendritic cells and macrophages in vitro. Pharm. Res. 2002, 19, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Thiele, L.; Merkle, H.P.; Walter, E. Phagocytosis and phagosomal fate of surface-modified microparticles in dendritic cells and macrophages. Pharm. Res. 2003, 20, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Avrameas, A.; McIlroy, D.; Hosmalin, A.; Autran, B.; Debre, P.; Monsigny, M.; Roche, A.C.; Midoux, P. Expression of a mannose/fucose membrane lectin on human dendritic cells. Eur. J. Immunol. 1996, 26, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Murthy, N.; Campbell, J.; Fausto, N.; Hoffman, A.S.; Stayton, P.S. Bioinspired pH-responsive polymers for the intracellular delivery of biomolecular drugs. Biochim. Biophys. Acta 2003, 14, 412–419. [Google Scholar]
- Randolph, G.J.; Angeli, V.; Swartz, M.A. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 2005, 5, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A. The physiology of the lymphatic system. Adv. Drug Delivery Rev. 2001, 50, 3–20. [Google Scholar] [CrossRef]
- Sayin, B.; Somavarapu, S.; Li, X.W.; Thanou, M.; Sesardic, D.; Alpar, H.O.; Senel, S. Mono-N-carboxymethyl chitosan (MCC) and N-trimethyl chitosan (TMC) nanoparticles for non-invasive vaccine delivery. Int. J. Pharm. 2008, 363, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Minigo, G.; Scholzen, A.; Tang, C.K.; Hanley, J.C.; Kalkanidis, M.; Pietersz, G.A.; Apostolopoulos, V.; Plebanski, M. Poly-L-lysine-coated nanoparticles: A potent delivery system to enhance DNA vaccine efficacy. Vaccine 2007, 25, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Pandit, S.; Bramwell, V.W.; Alpar, H.O. Diphtheria toxoid loaded poly-(ε-caprolactone) nanoparticles as mucosal vaccine delivery systems. Methods 2006, 38, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Bharali, D.J.; Pradhan, V.; Elkin, G.; Qi, W.; Hutson, A.; Mousa, S.A.; Thanavala, Y. Novel nanoparticles for the delivery of recombinant hepatitis B vaccine. Nanomedicine 2008, 4, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Yoshii, H.; Ishikawa, T.; Akagi, T.; Akashi, M.; Takahashi, M.; Yamanishi, K.; Mori, Y. Single dose of inactivated Japanese encephalitis vaccine with poly(γ-glutamic acid) nanoparticles provides effective protection from Japanese encephalitis virus. Vaccine 2008, 26, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Yoshii, H.; Akagi, T.; Akashi, M.; Ishikawa, T.; Okuno, Y.; Takahashi, M.; Yamanishi, K.; Mori, Y. Influenza hemagglutinin vaccine with poly(γ-glutamic acid) nanoparticles enhances the protection against in uenza virus infection through both humoral and cell-mediated immunity. Vaccine 2007, 25, 8270–8278. [Google Scholar] [CrossRef] [PubMed]
- Bivas-Benita, M.; Lin, M.Y.; Bal, S.M.; van Meijgaarden, K.E.; Franken, K.L.; Friggen, A.H.; Junginger, H.E.; Borchard, G.; Klein, M.R.; Ottenhoff, T.H. Pulmonary delivery of DNA encoding Mycobacterium tuberculosis latency antigen Rv1733c associated to PLGA-PEI nanoparticles enhances T cell responses in a DNA prime/protein boost vaccination regimen in mice. Vaccine 2009, 27, 4010–4017. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, Z.; Liao, B.; Zhong, N. Induction of Th1-type Immune response by chitosan nanoparticles containing plasmid DNA encoding house dust mite allergen Der p 2 for oral vaccination in mice. Cel. Mol. Immunol. 2009, 6, 45–50. [Google Scholar] [CrossRef]
- Khatri, K.; Goyal, A.K.; Gupta, P.N.; Mishra, N.; Vyas, S.P. Plasmid DNA loaded chitosan nanoparticles for nasal mucosal immunization against hepatitis B. Int. J. Pharm. 2008, 354, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Bivas-Benita, M.; van Meijgaarden, K.E.; Franken, K.L.; Junginger, H.E.; Borchard, G.; Ottenhoff, T.H.; Geluk, A. Pulmonary delivery of chitosan-DNA nanoparticles enhances the immunogenicity of a DNA vaccine encoding HLA-A*0201-restricted T-cell epitopes of Mycobacterium tuberculosis. Vaccine 2004, 22, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Shen, Y.; Jiang, Z.; Wang, Y.; Chu, Y.; Xiong, S. Intranasal delivery of chitosan-DNA vaccine generates mucosal SIgA and anti-CVB3 protection. Vaccine 2004, 22, 3603–3612. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yuan, X.; Cai, D.; Wang, S.; Zong, L. Low molecular weight chitosan in DNA vaccine delivery via mucosa. Int. J. Pharm. 2009, 375, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Middaugh, C.R.; Wiethoff, C.M. Barriers to nonviral gene delivery. J. Pharm. Sci. 2003, 92, 203–217. [Google Scholar] [PubMed]
- Kim, S.H.; Mok, H.; Jeong, J.H.; Kim, S.W.; Park, T.G. Comparative evaluation of target-specific GFP gene silencing efficiencies for antisense ODN, synthetic siRNA, and siRNA plasmid complexed with PEI-PEG-FOL conjugate. Bioconjugate Chem. 2006, 17, 241–244. [Google Scholar] [CrossRef]
- Dang, J.M.; Leong, K.W. Natural polymers for gene delivery and tissue engineering. Adv. Drug Delivery Rev. 2006, 58, 487–499. [Google Scholar] [CrossRef]
- Park, T.G.; Jeong, J.H.; Kim, S.W. Current status of polymeric gene delivery systems. Adv. Drug Delivery Rev. 2006, 58, 467–486. [Google Scholar] [CrossRef]
- Sato, T.; Ishii, T.; Okahata, Y. In vitro gene delivery mediated by chitosan. Effect of pH, serum, and molecular mass of chitosan on the transfection efficiency. Biomaterials 2001, 22, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Fong, C.W.; Khor, E.; Lim, L.Y. Transfection efficiency of chitosan vectors: Effect of polymer molecular weight and degree of deacetylation. J. Control. Release 2005, 106, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Liu, L.R.; Jiang, Q.; Zhang, Q.Q. Self-aggregated nanoparticles of cholesterol-modified chitosan conjugate as a novel carrier of epirubicin. Eur. Polym. J. 2007, 43, 43–51. [Google Scholar] [CrossRef]
- Kim, T.H.; Park, I.K.; Nah, J.W.; Choi, Y.J.; Cho, C.S. Galactosylated chitosan/DNA nanoparticles prepared using water-soluble chitosan as a gene carrier. Biomaterials 2004, 25, 3783–3792. [Google Scholar] [CrossRef] [PubMed]
- Weecharangsan, W.; Opanasopit, P.; Ngawhirunpat, T.; Apirakaramwong, A.; Rojanarata, T.; Ruktanonchai, U.; Lee, R.J. Evaluation of chitosan salts as non-viral gene vectors in CHO-K1 cells. Int. J. Pharm. 2008, 348, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Corsi, K.; Chellat, F.; Fernandes, J.C. Mesenchymal stem cells, MG63 and HEK293 transfection using chitosan-DNA nanoparticles. Biomaterials 2003, 24, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.R.; Remant Bahadur, K.C.; Aryal, S.; Bhattarai, N.; Kim, S.Y.; Yi, H.K.; Hwang, P.H.; Kim, H.Y. Hydrophobically modified chitosan/gold nanoparticles for DNA delivery. J. Nanopart. Res. 2008, 10, 151–162. [Google Scholar] [CrossRef]
- Koping-Hoggard, M.; Tubulekas, I.; Guan, H.; Edwards, K.; Nilsson, M.; Varum, K.M.; Artursson, P. Chitosan as a nonviral gene delivery system. Structure-property relationships and characteristics compared with polyethylenimine in vitro and after lung administration in vivo. Gene Ther. 2001, 8, 1108–1121. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Lezoualc'h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Pasumarthy, M.; Cooper, M.J. Biological properties of poly-L-lysine-DNA complexes generated by cooperative binding of the polycation. J. Biol. Chem. 2001, 276, 34379–34387. [Google Scholar] [CrossRef] [PubMed]
- Petersen, H.; Fechner, P.M.; Martin, A.L.; Kunath, K.; Stolnik, S.; Roberts, C.J.; Fischer, D.; Davies, M.C.; Kissel, T.; Kingdom, U. Polyethylenimine-graft-poly(ethylene glycol) copolymers: Influence of copolymer block structure on DNA complexation and biological activities as gene delivery system. Bioconjugate Chem. 2002, 13, 845–854. [Google Scholar] [CrossRef]
- Choi, Y.H.; Liu, F.; Kim, J.-S.; Choi, Y.K.; Park, J.S.; Kim, S.W. Polyethylene glycol-grafted poly-L-lysine as polymeric gene carrier. J. Control. Release 1998, 54, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Zhong, Z.; Lok, M.C.; Jiang, X.; Hennink, W.E.; Feijen, J.; Engbersen, J.F.J. Linear poly(amido amine)s with secondary and tertiary amino groups and variable amounts of disulfide linkages: Synthesis and in vitro gene transfer properties. J. Control. Release 2006, 116, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Oupicky, D.; Parker, A.L.; Seymour, L.W. Laterally stabilized complexes of DNA with linear reducible polycations: Strategy for triggered intracellular activation of DNA delivery vectors. J. Am. Chem. Soc. 2002, 124, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Engbersen, J.F.J. Effect of chemical functionalities in poly(amido amine)s for non-viral gene transfection. J. Control. Release 2008, 132, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Wolfert, M.; Seymour, L. Chloroquine and amphipathic peptide helices show synergistic transfection in vitro. Gene Ther. 1998, 5, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, Z.; Jiang, J.; Gao, Y.; Gu, W.; Chen, L.; Tang, X.; Li, Y. Poly(imidazole/DMAEA)phosphazene/DNA self-assembled nanoparticles for gene delivery: Synthesis and in vitro transfection. J. Control. Release 2008, 127, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jeong, J.H.; Lee, S.H.; Kim, S.W.; Park, T.G. Local and systemic delivery of VEGF siRNA using polyelectrolyte complex micelles for effective treatment of cancer. J. Control. Release 2008, 129, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Foger, F.; Noonpakdee, W.; Loretz, B.; Joojuntr, S.; Salvenmoser, W.; Thaler, M.; Bernkop-Schnurch, A. Inhibition of malarial topoisomerase II in plasmodium falciparum by antisense nanoparticles. Int. J. Pharm. 2006, 319, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Cai, Z.; Song, X.; Yu, B.; Bi, Y.; Chen, Q.; Zhao, D.; Xu, J.; Hou, S. Receptor mediated gene delivery by folate conjugated N-trimethyl chitosan in vitro. Int. J. Pharm. 2009, 382, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Cuie, Y.; Winnik, F.; Qin, S.; Lavigne, P.; Benderdour, M.; Beaumont, E.; Fernandes, J.C. Characterization of folate-chitosan-DNA nanoparticles for gene therapy. Biomaterials 2006, 27, 2060–2065. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kakimoto, S.; Kano, H.; Nakatani, M.; Shinkai, S.; Nagasaki, T. In vitro gene delivery to HepG2 cells using galactosylated 6-amino-6-deoxychitosan as a DNA carrier. Carbohydr. Res. 2007, 342, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Fuente, M.D.L.; Seij, B.N.; Alonso, M.J. Novel hyaluronic acid-chitosan nanoparticles for ocular gene therapy. Invest. Ophthalmol. 2008, 49, 2016–2024. [Google Scholar] [CrossRef]
- Hashimoto, M.; Morimoto, M.; Saimoto, H.; Shigemasa, Y.; Yanagie, H.; Eriguchi, M.; Sato, T. Gene transfer by DNA/mannosylated chitosan complexes into mouse peritoneal macrophages. Biotechnol. Lett. 2006, 28, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.; Wang, H.; Jreyssaty, C.; Benderdour, M.; Lavigne, P.; Qiu, X.; Winnik, F.M.; Zhang, X.; Dai, K.; Shi, Q. Bone-protective effects of nonviral gene therapy with folate-chitosan DNA nanoparticle containing interleukin-1 receptor antagonist gene in rats with adjuvant-induced arthritis. Mol. Ther. 2008, 16, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.-Q.; Roy, K.; Troung-le, V.L.; Janes, K.A.; Lin, K.Y.; Wang, Y.; August, J.T.; Leong, K.W. Chitosan-DNA nanoparticles as gene carriers: Synthesis, characterization and transfection efficiency. J. Control. Release 2001, 70, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Okahata, Y.; Sato, T. Mechanism of cell transfection with plasmid/chitosan complexes. Biochim. Biophys. Acta 2001, 1514, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, W.-L.; Li, G.; Qian, J.; Xue, J.-L.; Fu, S.-K.; Lu, D.-R. Transfection of mEpo gene to intestinal epithelium in vivo mediated by oral delivery of chitosan-DNA nanoparticles. World J. Gastroenterol. 2004, 10, 112–116. [Google Scholar] [PubMed]
- Lee, D.; Zhang, W.; Shirley, S.A.; Kong, X.; Hellermann, G.R.; Lockey, R.F.; Mohapatra, S.S. Thiolated chitosan/DNA nanocomplexes exhibit enhanced and sustained gene delivery. Pharm. Res. 2007, 24, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Shi, X.-W.; Yang, G.-F.; Gong, L.-L.; Yuan, H.-Y.; Cui, Y.-J.; Wang, Y.; Du, Y.-M.; Li, Y. Chitosan nanoparticle as gene therapy vector via gastrointestinal mucosa administration: Results of an in vitro and in vivo study. Life Sci. 2007, 80, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Z.; Chen, L.; Gu, W.; Li, Y. Synthesis of 6-N,N,N-trimethyltriazole chitosan via "click chemistry" and evaluation for gene delivery. Biomacromolecules 2009, 10, 2175–2182. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wan, X.; Zhang, W.; Wang, Z.; Peng, R.; Tao, F.; Cai, L.; Li, Y.; Jiang, Q.; Gao, R. Synthesis of biodegradable polycationic methoxy poly(ethylene glycol)-polyethylenimine-chitosan and its potential as gene carrier. Carbohydr. Polym. 2009, 78, 46–53. [Google Scholar] [CrossRef]
- Peng, S.; Yang, M.; Su, C.; Chen, H.; Lee, P.; Wei, M.; Sung, H. Effects of incorporation of poly(γ-glutamic acid) in chitosan/DNA complex nanoparticles on cellular uptake and transfection efficiency. Biomaterials 2009, 30, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lee, I.L.; Lim, W.S.; Chia, S.M.; Yu, H.; Leong, K.W.; Mao, H.Q. Evaluation of collagen and methylated collagen as gene carriers. Int. J. Pharm. 2004, 279, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Kushibiki, T.; Nagata-nakajima, N.; Sugai, M.; Shimizu, A.; Tabata, Y. Delivery of plasmid DNA expressing small interference RNA for TGF-b type II receptor by cationized gelatin to prevent interstitial renal fibrosis. J. Control. Release 2005, 105, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Kommareddy, S.; Amiji, M. Poly(ethylene glycol)-modified thiolated gelatin nanoparticles for glutathione-responsive intracellular DNA delivery. Nanomedicine 2007, 3, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Acute and chronic in vivo therapeutic resistance. Biochem. Pharmacol. 2009, 77, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Patil, Y.B.; Swaminathan, S.K.; Sadhukha, T.; Panyam, J. The use of nanoparticle-mediated targeted gene silencing and drug delivery to overcome tumor drug resistance. Biomaterials 2010, 31, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Wiradharma, N.; Tong, Y.W.; Yang, Y.-Y. Self-assembled oligopeptide nanostructures for co-delivery of drug and gene with synergistic therapeutic effect. Biomaterials 2009, 30, 3100–3109. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.-S.; Goh, S.-H.; Yang, Y.-Y. Synthesis and characterization of cationic micelles self-assembled from a biodegradable copolymer for gene delivery. Biomacromolecules 2007, 8, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Martimprey, H.D.; Bertrand, J.-R.; Fusco, A.; Santoro, M.; Couvreur, P.; Vauthier, C.; Malvy, C. siRNA nanoformulation against the Ret/PTC1 junction oncogene is efficient in an in vivo model of papillary thyroid carcinoma. Nucleic Acids Res. 2008, 36, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Howard, K.A.; Paludan, S.R.; Behlke, M.A.; Besenbacher, F.; Deleuran, B.; Kjems, J. Chitosan/siRNA nanoparticle-mediated TNF-alpha knockdown in peritoneal macrophages for anti-inflammatory treatment in a murine arthritis model. Mol. Ther. 2009, 17, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Elfinger, M.; Mauckscha, C.; Rudolph, C. Characterization of lactoferrin as a targeting ligand for nonviral gene delivery to airway epithelial cells. Biomaterials 2007, 28, 3448–3455. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Yockman, J.W.; Lee, M.; Jeong, J.H.; Kim, Y.-H.; Kim, S.W. Soluble Flt-1 gene delivery using PEI-g-PEG-RGD conjugate for anti-angiogenesis. J. Control. Release 2005, 106, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jeong, J.H.; Lee, S.H.; Kim, S.W.; Park, T.G. LHRH receptor-mediated delivery of sirna using polyelectrolyte complex micelles self-assembled from siRNA-PEG-LHRH conjugate and PEI. Bioconjugate Chem. 2008, 19, 2156–2162. [Google Scholar] [CrossRef]
- Blessing, T.; Kursa, M.; Holzhauser, R.; Kircheis, R.; Wagner, E. Different strategies for formation of PEGylated EGF-conjugated PEI/DNA complexes for targeted gene delivery. Bioconjugate Chem. 2001, 12, 529–537. [Google Scholar] [CrossRef]
- Zenga, J.; Wanga, X.; Wang, S. Self-assembled ternary complexes of plasmid DNA, low molecular weight polyethylenimine and targeting peptide for nonviral gene delivery into neurons. Biomaterials 2007, 28, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Elfinger, M.; Geiger, J.; Hasenpusch, G.; Uzgun, S.; Sieverling, N.; Aneja, M.K.; Maucksch, C.; Rudolph, C. Targeting of the β-adrenoceptor increases nonviral gene delivery to pulmonary epithelial cells in vitro and lungs in vivo. J. Control. Release 2009, 135, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Chul Cho, K.; Hoon Jeong, J.; Jung Chung, H.; Joe, C.O.; Wan Kim, S.; Gwan Park, T. Folate receptor-mediated intracellular delivery of recombinant caspase-3 for inducing apoptosis. J. Control. Release 2005, 108, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Lee, M.; Kim, W.J.; Yockman, J.W.; Park, T.G.; Kim, Y.H.; Kim, S.W. Anti-GAD antibody targeted non-viral gene delivery to islet beta cells. J. Control. Release 2005, 107, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.; Ueno, N.T.; Lee, R.J. Tumor-targeted gene delivery via anti-HER2 antibody (trastuzumab, HerceptinR) conjugated polyethylenimine. J. Control. Release 2004, 97, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Cotten, M. Mannose polyethylenimine conjugates for targeted DNA Delivery into dendritic cells. J. Biol. Chem. 1999, 274, 19087–19094. [Google Scholar] [CrossRef] [PubMed]
- Fewell, J.G.; Matar, M.; Slobodkin, G.; Han, S.O.; Rice, J.; Hovanes, B.; Lewis, D.H.; Anwer, K. Synthesis and application of a non-viral gene delivery system for immunogene therapy of cancer. J. Control. Release 2005, 109, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.; Jong, C. Improved stability of polycationic vector by dextran-grafted branched polyethylenimine. Biomacromolecules 2003, 4, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Park, J.H.; Lee, M.; Park, T.G.; Kim, S.W. Polyethylenimine with acid-labile linkages as a biodegradable gene carrier. J. Control. Release 2005, 103, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Quick, G.K.; Yeo, Y. Gene delivery through the use of a hyaluronate-associated intracellularly degradable crosslinked polyethyleneimine. Biomaterials 2009, 30, 5834–5843. [Google Scholar] [CrossRef] [PubMed]
- Hashida, M.; Takemura, S.; Nishikawa, M.; Takakura, Y. Targeted delivery of plasmid DNA complexed with galactosylated poly(L-lysine). J. Control. Release 1998, 53, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Nagasaki, Y.; Itaka, K.; Nishiyama, N.; Kataoka, K. Lactosylated poly(ethylene glycol)-siRNA conjugate through acid-labile -thiopropionate linkage to construct pH-sensitive polyion complex micelles achieving enhanced gene silencing in hepatoma cells. J. Am. Chem. Soc. 2005, 127, 1624–1625. [Google Scholar] [CrossRef] [PubMed]
- Nah, J.-W.; Yu, L.; Han, S.-O.; Ahn, C.-H.; Kim, S.W. Artery wall binding peptide-poly(ethylene glycol)-grafted-poly(l-lysine)-based gene delivery to artery wall cells. J. Control. Release 2002, 78, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Suh, W.; Chung, J.K.; Park, S.H.; Kim, S.W. Anti-JL1 antibody-conjugated poly (l-lysine) for targeted gene delivery to leukemia T cells. J. Control. Release 2001, 72, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Oba, M.; Aoyagi, K.; Miyata, K.; Matsumoto, Y.; Itaka, K.; Nishiyama, N.; Yamasaki, Y.; Koyama, H.; Kataoka, K. Polyplex micelles with cyclic RGD peptide ligands and disulfide cross-links directing to the enhanced transfection via controlled intracellular trafficking. Mol. Pharm. 2008, 5, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Zhang, W.; Lockey, R.F.; Auais, A.; Piedimonte, G.; Mohapatra, S.S. Respiratory syncytial virus infection in Fischer 344 rats is attenuated by short interfering RNA against the RSV-NS1 gene. Genet. Vaccines Ther. 2007, 5, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, W.; Mohapatra, S.; Kong, X.; Li, X.; Lockey, R.F.; Mohapatra, S.S. Prevention of airway inflammation with topical cream containing imiquimod and small interfering RNA for natriuretic peptide receptor. Genet. Vaccines Ther. 2008, 6, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.S.; Lee, M.; Koh, J.J.; Kim, S.W. Combined administration of plasmids encoding IL-4 and IL-10 prevents the development of autoimmune diabetes in nonobese diabetic mice. Mol. Ther. 2001, 4, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Putnam, D.; Langer, R. Poly(4-hydroxy-L-proline ester): Low-temperature polycondensation and plasmid DNA complexation. Macromolecules 1999, 32, 3658–3662. [Google Scholar] [CrossRef]
- Lin, C.; Zhong, Z.; Lok, M.C.; Jiang, X.; Hennink, W.E.; Feijen, J.; Engbersen, J.F.J. Novel bioreducible poly(amido amine)s for highly efficient gene delivery. Bioconjugate Chem. 2007, 18, 138–145. [Google Scholar] [CrossRef]
- Ferruti, P.; Franchini, J.; Bencini, M.; Ranucci, E.; Zara, G.P.; Serpe, L.; Primo, L.; Cavalli, R. Prevailingly cationic agmatine-based amphoteric polyamidoamine as a nontoxic, nonhemolytic, and "stealthlike" DNA complexing agent and transfection promoter. Biomacromolecules 2007, 8, 1498–1504. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-F.; Chin, W.-K.; Cherng, J.-Y. Synthesis of novel biodegradable cationic polymer: N,N-Diethylethylenediamine polyurethane as a gene carrier. Biomacromolecules 2004, 5, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gonçalves, C.; Pereira, P.; Gama, M. Self-Assembled Hydrogel Nanoparticles for Drug Delivery Applications. Materials 2010, 3, 1420-1460. https://doi.org/10.3390/ma3021420
Gonçalves C, Pereira P, Gama M. Self-Assembled Hydrogel Nanoparticles for Drug Delivery Applications. Materials. 2010; 3(2):1420-1460. https://doi.org/10.3390/ma3021420
Chicago/Turabian StyleGonçalves, Catarina, Paula Pereira, and Miguel Gama. 2010. "Self-Assembled Hydrogel Nanoparticles for Drug Delivery Applications" Materials 3, no. 2: 1420-1460. https://doi.org/10.3390/ma3021420
APA StyleGonçalves, C., Pereira, P., & Gama, M. (2010). Self-Assembled Hydrogel Nanoparticles for Drug Delivery Applications. Materials, 3(2), 1420-1460. https://doi.org/10.3390/ma3021420