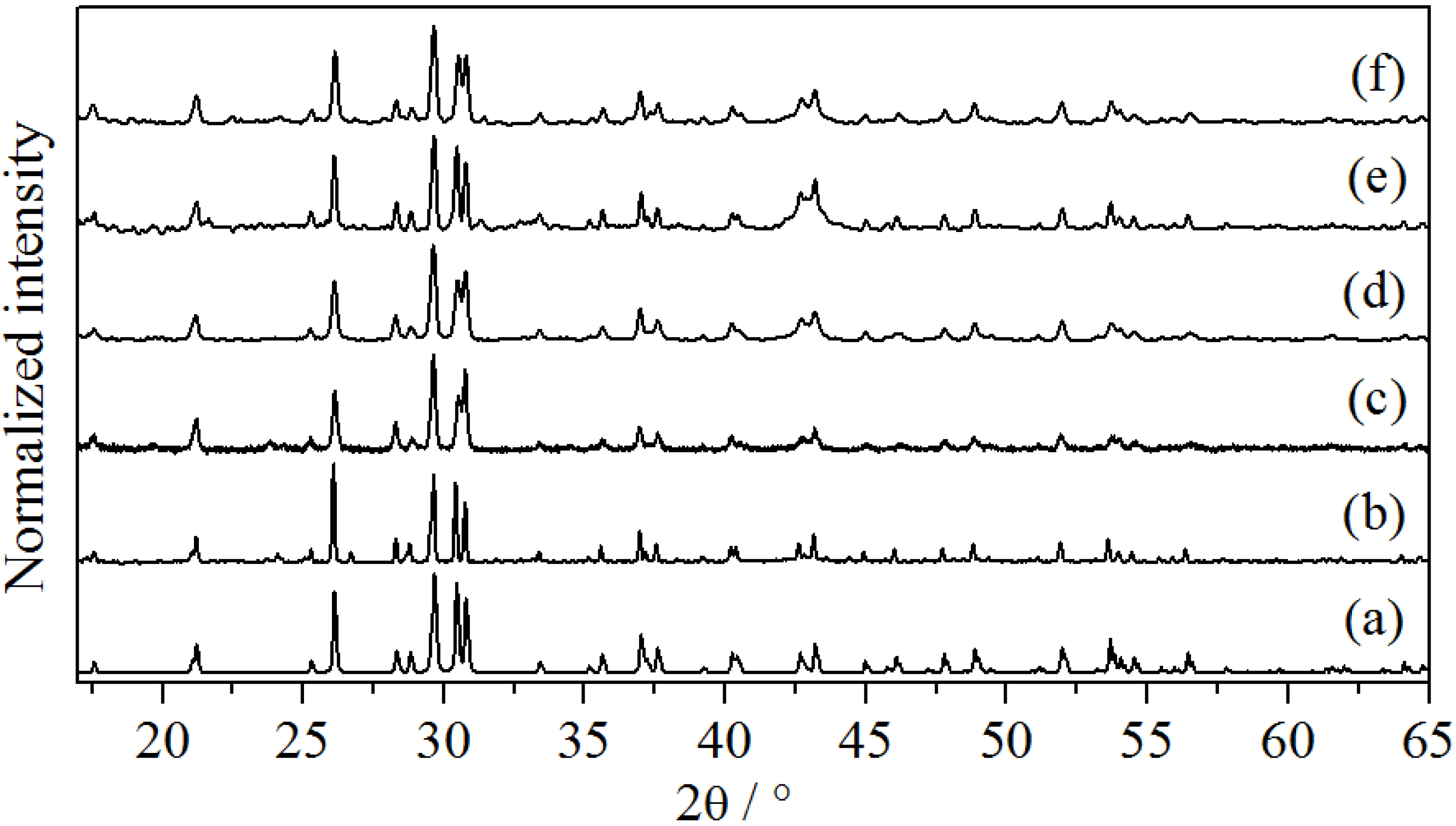
2.3.1. Ba2SiO4:Eu2+/3+
Excitation spectra of Ba
2SiO
4:Eu
2+ bulk and nanophosphors are both broadly distributed over the UV spectral range between 20,000 and 42,000 cm
−1 (
Figure 5). The broader bands of the bulk material and the intensity increase around 32,000 cm
−1 in contrast to the formation of two discrete absorption bands of the NPs may be explained by four different hypotheses: (i) the presence of an impurity phase in the bulk material, which cannot be detected by XRD; (ii) the occupation of two different Ba
2+-sites by the Eu
2+ ions; (iii) the crystal field splitting of the 4f
65d
1 electronic configuration due to the low-symmetry coordination of the Eu
2+ ions and (iv) a saturation effect due to the high doping concentration of 1% [
17]. In order to investigate the origin of the two discrete bands on the excitation spectra, several emission measurements were performed, which are depicted in the
Figure 6a,b.
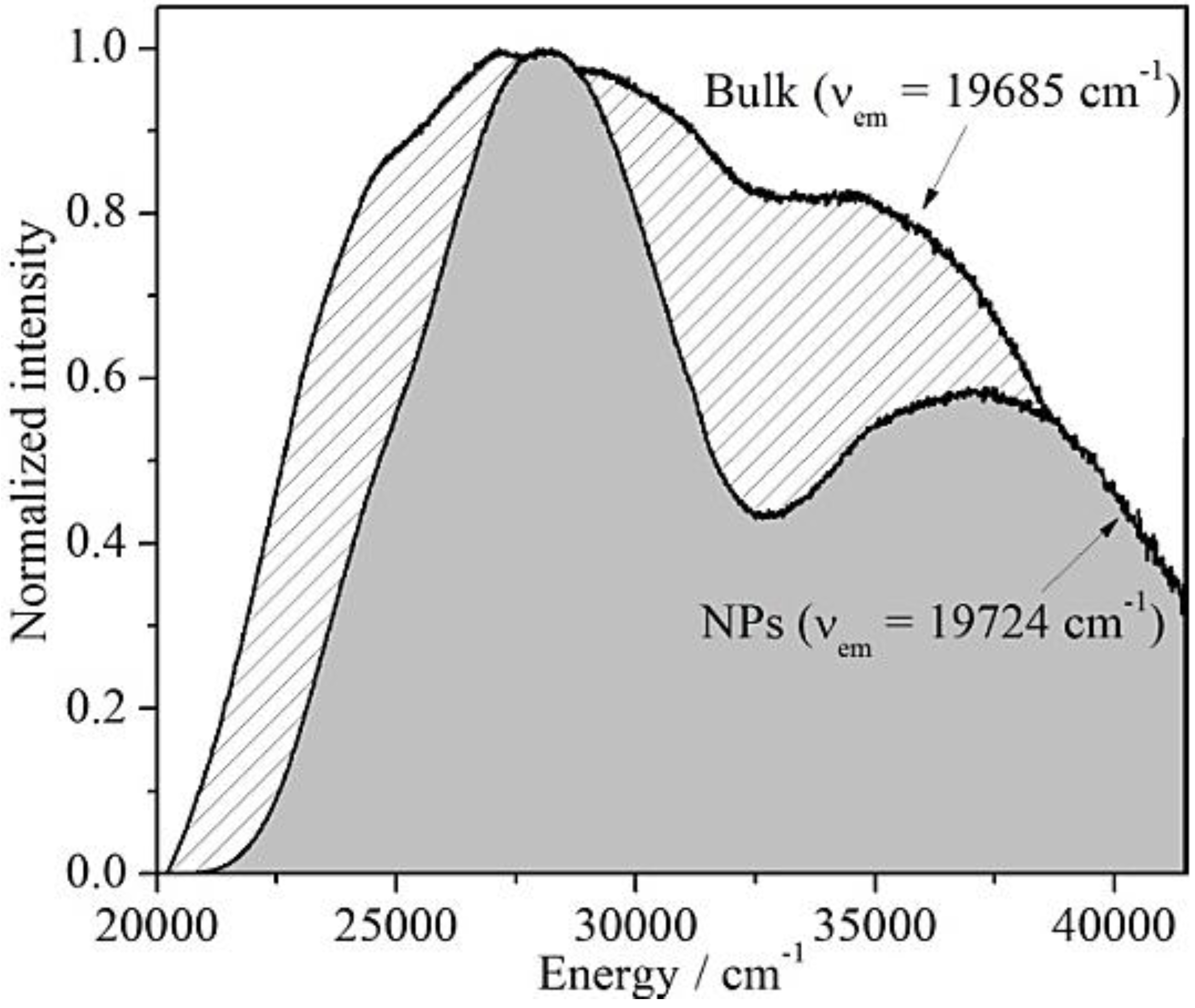
Figure 5.
Excitation spectra of Ba2SiO4:Eu2+ microcrystals (νem = 19,685 cm−1) and nanoparticles (νem = 19,724 cm−1).
Figure 5.
Excitation spectra of Ba2SiO4:Eu2+ microcrystals (νem = 19,685 cm−1) and nanoparticles (νem = 19,724 cm−1).
In general, the emission spectrum of Ba
2SiO
4:Eu
2+ consists of a broad band peaking at 19,706 cm
−1 (
Figure 6a–c), resulting from the 4f
65d
1 → 4f
7 electronic transition of Eu
2+ in this lattice. As the optical properties of divalent europium strongly depend on the environmental conditions, the presence of an impurity phase would result in a change on the emission spectrum. Therefore, if the two discrete excitation bands in the
Figure 5 would belong to two different structures, excitations of the sample at 27,855 cm
−1 and 37,037 cm
−1 would generate deviations of the respective emission spectra. However, the emission spectra recorded for these two excitation energies are both centered at 19,706 cm
−1 and present similar values of full width at half maximum (FWHM) of 2403 cm
−1 and 2413 cm
−1 for the spectra excited at, respectively, 27,855 cm
−1 and 37,037 cm
−1 (
Figure 6a). The maximum of the emission spectrum at 19,706 cm
−1 is comparable to the value of 19,531 cm
−1, described in the literature for Ba
2SiO
4:Eu
2+ particles [
8]. In this case, the minor deviation of approximately 200 cm
−1 may be explained by the different doping concentrations [
4] of 1% in our work and of 3% in the work of Han
et al. [
8].
As previously described in the literature [
1,
2,
3], the barium ions occupy two different sites in the crystal structure of Ba
2SiO
4: one nine-fold and another ten-fold coordinated site. The occupancy of Eu
2+ of both Ba
2+-sites causes a slight asymmetry of the Gaussian shape of the Ba
2SiO
4:Eu
2+ emission band. This is assigned to the overlap of two emission bands centered at approximately 19,200 cm
−1 and 19,800 cm
−1 [
1,
3].
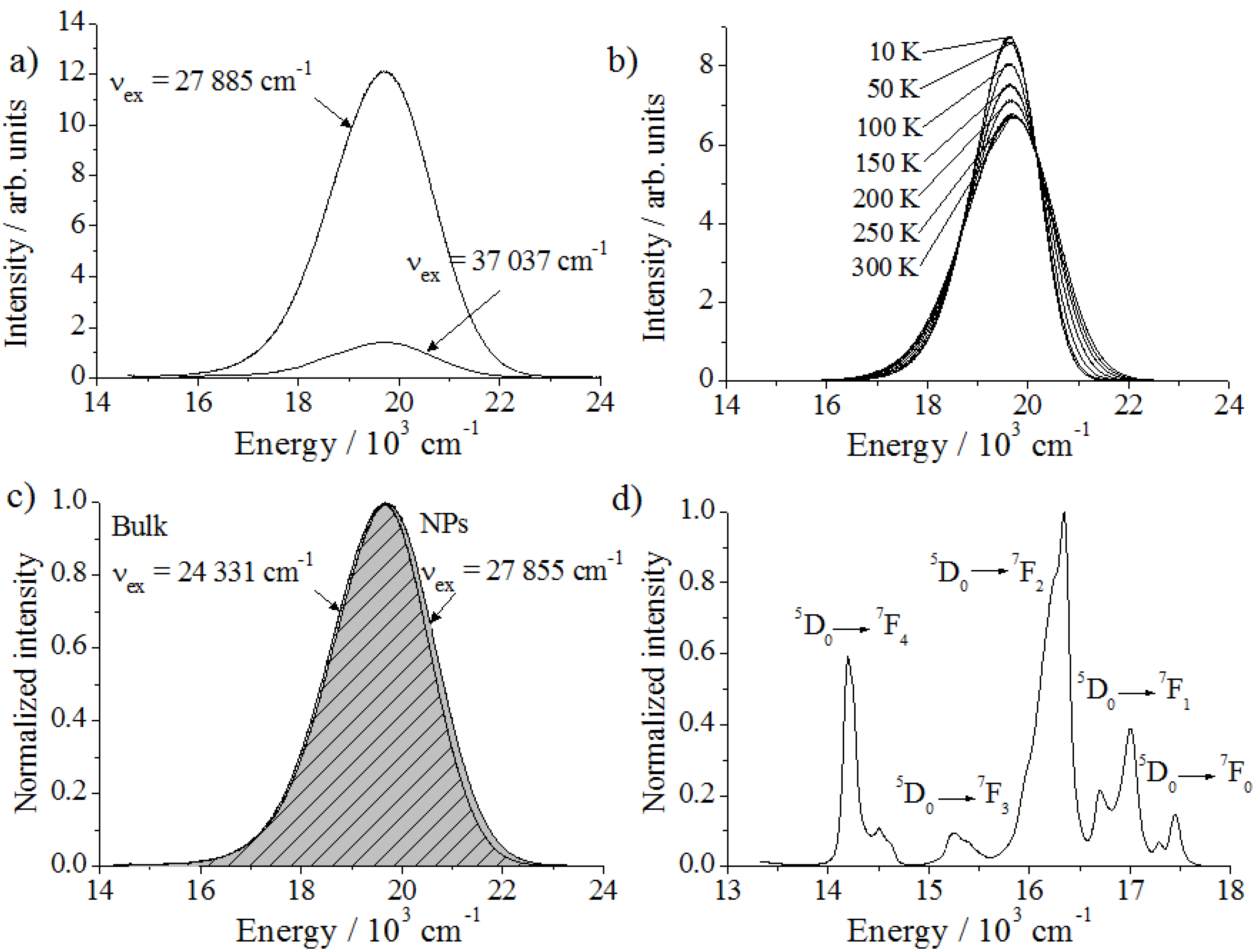
Figure 6.
Emission spectra. (a) Ba2SiO4:Eu2+ NPs for different excitation energies and (b) temperature-dependent measurement of Ba2SiO4:Eu2+ bulk phosphor (νex = 24,510 cm−1). (c) Comparison between Ba2SiO4:Eu2+ in bulk and in nano form and (d) Ba2SiO4:Eu3+ nanophosphor (νex = 25,316 cm−1).
Figure 6.
Emission spectra. (a) Ba2SiO4:Eu2+ NPs for different excitation energies and (b) temperature-dependent measurement of Ba2SiO4:Eu2+ bulk phosphor (νex = 24,510 cm−1). (c) Comparison between Ba2SiO4:Eu2+ in bulk and in nano form and (d) Ba2SiO4:Eu3+ nanophosphor (νex = 25,316 cm−1).
Trying to improve the resolution of these two overlapped emission bands, the luminescence spectra of Ba
2SiO
4:Eu
2+ were measured at temperatures varying between 10 and 300 K (
Figure 6b). At 10 K, the maximum of the emission intensity is located at 19,627 cm
−1 with a fwhm of 1552 cm
−1. As expected, after cooling down to 10 K, an increase in the emission intensity and a decrease on the band width were observed. However, the emission band is not split. Presumably, the energetic position of the excited states related to Eu
2+ on both Ba
2+-sites are very similar and cannot be detected spectroscopically for the doping concentration of 1%. Furthermore, it agrees with the fact that the excitation spectra recorded for different emission energies between 16,000 cm
−1 and 22,000 cm
−1 remain practically invariable. Another explanation, which does not necessarily exclude the previously mentioned one, is the possible energy transfer between Eu
2+ ions occupying two different sites, resulting in one single emission band. An enhanced interaction between Eu
2+ ions is expected because of the applied high doping concentrations. Due to these reasons, the two absorption bands presented in the
Figure 5 are also not related to the occupation of the different Ba
2+ sites by the activator ion and are presumably associated to the crystal field splitting. A saturation effect in the bulk material owing to the high doping concentration of Eu
2+ ions, however, could also be an explanation, which had already been discussed in literature [
17]. Even though the doping concentration is equal for the nano and bulk material, the saturation effect is expected to be more pronounced manifested in the bulk Ba
2SiO
4:Eu
2+ phosphor, causing the broadening on the excitation spectrum observed on
Figure 5. In nanoparticles, the single Eu
2+ ions are mostly separated by the particle boundaries, reducing the interaction with each other.
Besides understanding the origin of the emission and excitation energies, it is also important to investigate possible changes on the photoluminescence of miniaturized phosphors. A decrease on the crystal size can influence the band gap energy and, consequently, the optical properties of nanophosphors [
6]. As depicted in
Figure 6c, the difference between the emission spectrum of nano and bulk Ba
2SiO
4:Eu
2+ is negligible. Therefore, it was possible to reduce the crystal size of the Eu
2+-activated barium orthosilicate down to the nano-scale avoiding drastic changes on the emission and excitation energies.
In addition to Ba
2SiO
4:Eu
2+, undoped Ba
2SiO
4 as well as Ba
2SiO
4:Eu
3+ and Ba
2SiO
4:Sr
2+ nanoparticles were spectroscopically characterized. The undoped Ba
2SiO
4 NPs do not show any luminescence, while the nanophosphors doped with Eu
2+, Eu
3+ and Sr
2+ emit in the green, red and blue range, respectively (
Figure 7 and
Figure 8a).
As expected, the emission spectrum of the Ba
2SiO
4:Eu
3+ NPs consists of several peaks distributed over the red spectral range, which are assigned to the
5D
0 →
7F
J (
J = 0–4) transitions of the trivalent europium ions (
Figure 6d). The distortions and the defects in the nanoparticles also due to the large surface cause slightly different coordination spheres of the Eu
3+ ions. Therefore, the transitions are relatively broad, and no crystal field splitting could be detected, which is generally the case for nanoparticles doped with trivalent lanthanides. It is remarkable that the transition
5D
0 →
7F
0 shows some intensity, which can be understood because the site symmetry is expected to be rather low. Also the
5D
0 →
7F
4 transition is relatively strong compared to the
5D
0 →
7F
2 one.
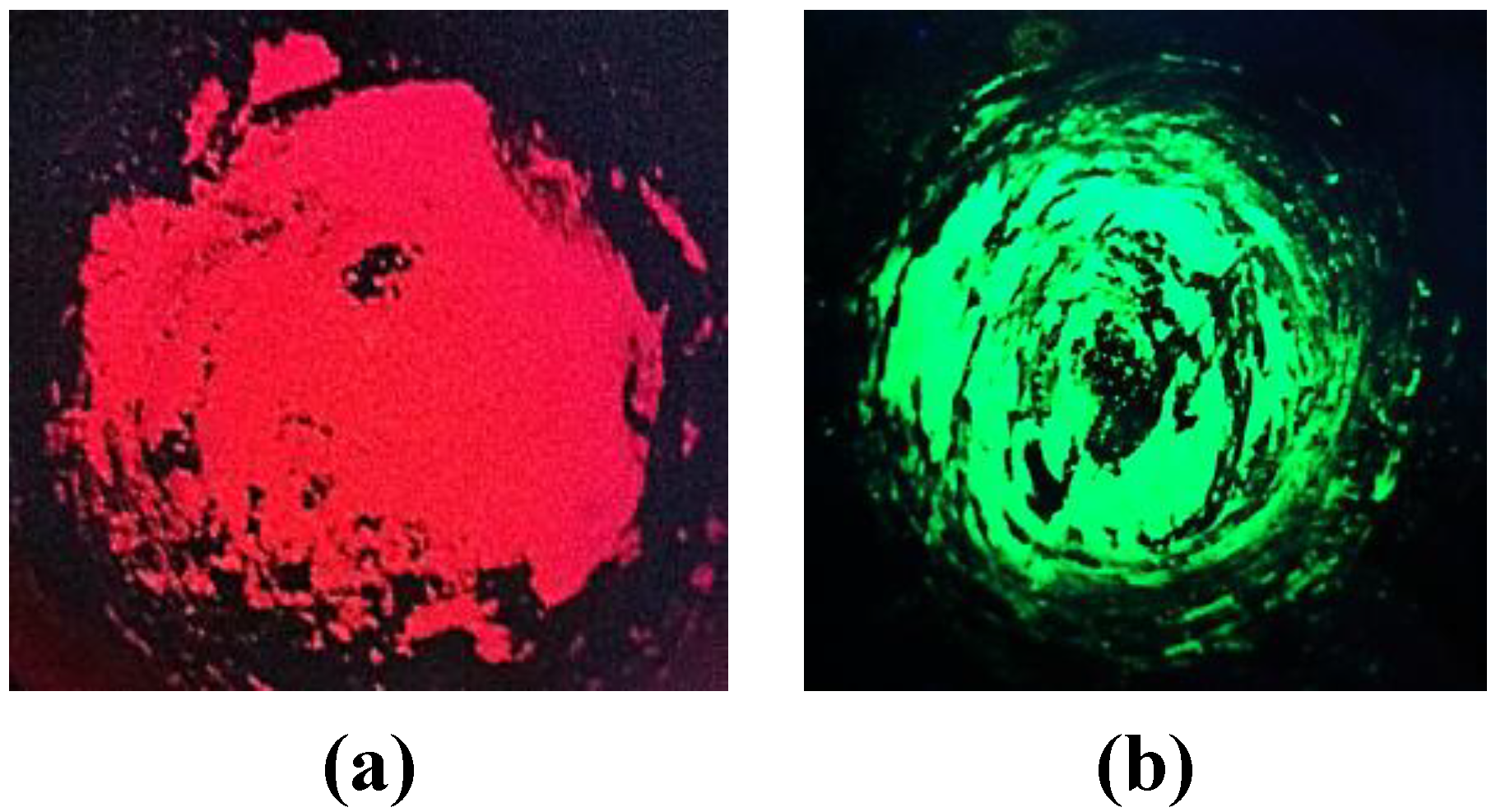
Figure 7.
Barium orthosilicate nanoparticles doped with Eu3+ (a) and Eu2+ (b), irradiated with UV light.
Figure 7.
Barium orthosilicate nanoparticles doped with Eu3+ (a) and Eu2+ (b), irradiated with UV light.
2.3.2. Ba2SiO4:Sr2+
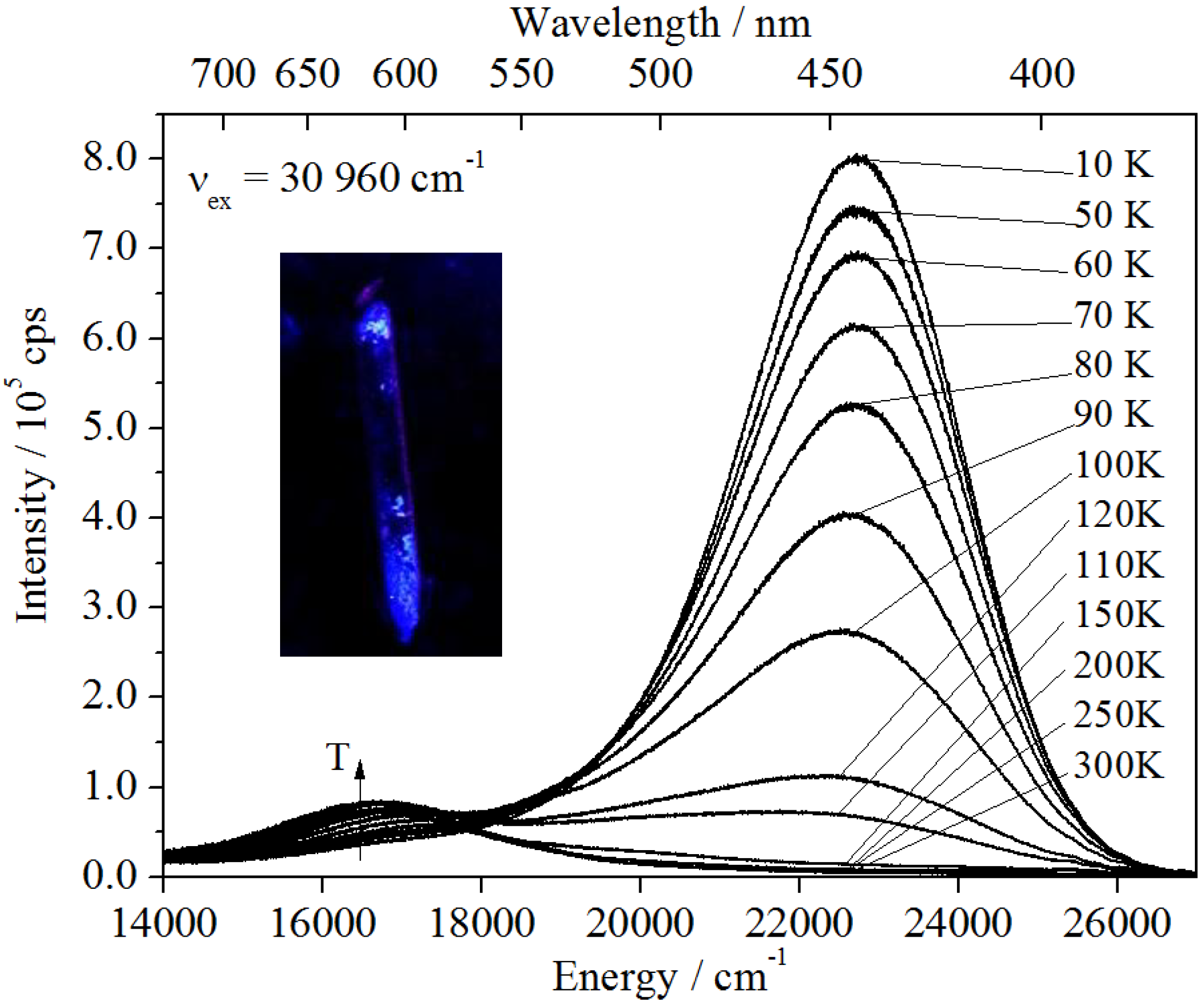
In the case of Ba
2SiO
4:Sr
2+, two emission bands can be observed (see
Figure 8b), while undoped Ba
2SiO
4 show no luminescence. The blue-shifted band is located at 22,321 cm
−1 (FWHM = 3286 cm
−1) giving rise to the strong blue luminescence of the nanoparticles at low temperatures (
Figure 8a). It is assigned to the presence of self-trapped excitons (STEs) in the nanoparticles (see below). However, it is totally quenched at temperatures above 120–130 K. At that temperature, another emission band located at 16,643 cm
−1 arises, which increases in intensity with increasing temperature. Thus, a correlation between these two bands is assumed, as will be discussed in more detail below.
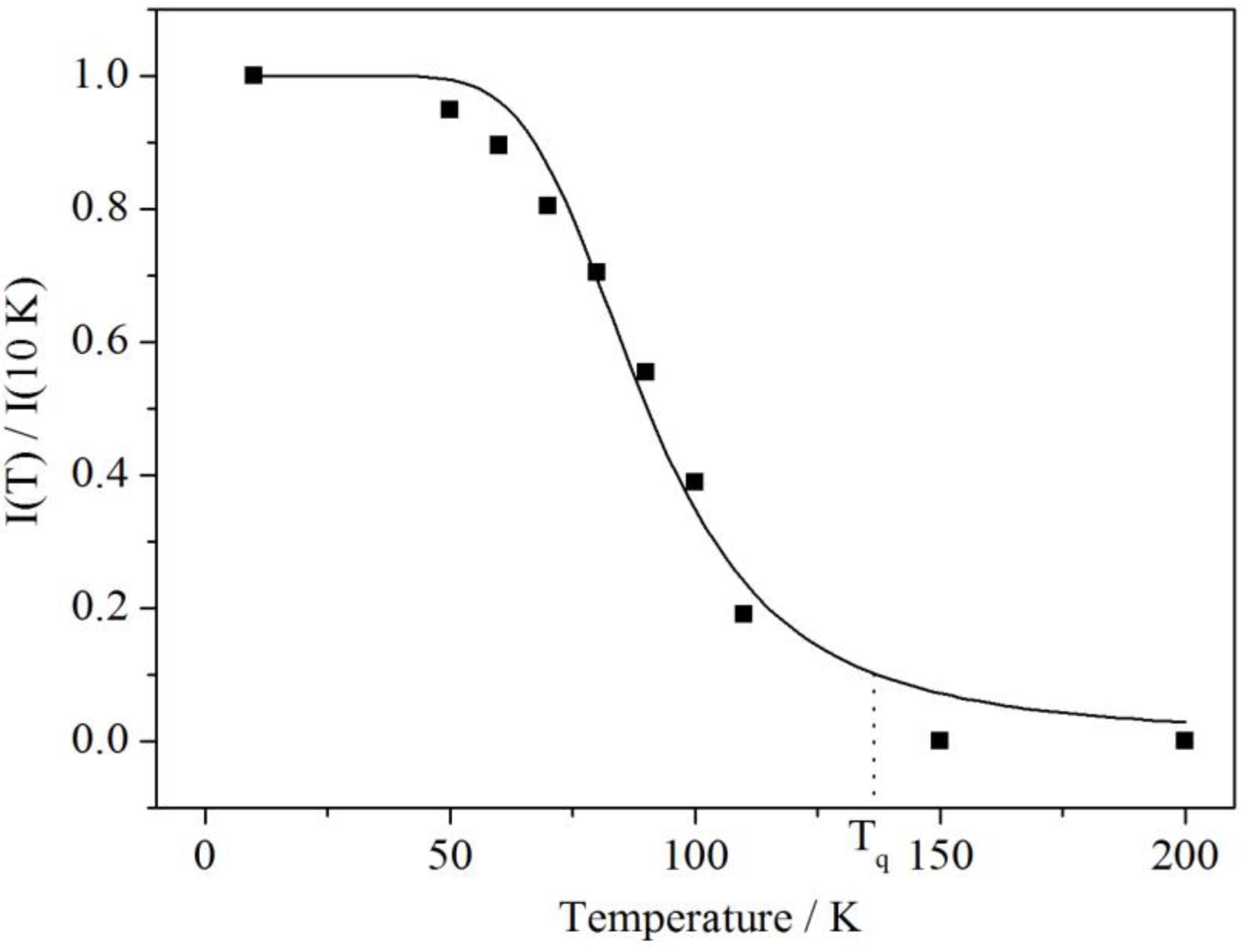
Figure 9 shows the temperature dependence of the integrated intensity of the blue emission band. It can be fitted to the well-known Mott Equation (1) [
18]
where
C is the ratio between the thermal quenching rate and the radiative decay rate (
C =
p/
krad) and
Ea is the activation barrier for the non-radiative process.
Figure 8.
Temperature dependent emission of Ba2SiO4:Sr2+ NPs: decreasing maximum at 22,760 cm−1 and increasing maximum at 16,690 cm−1 for higher temperatures. Inset: Ba2SiO4:Sr2+ NPs under UV light, cooled down with liquid N2.
Figure 8.
Temperature dependent emission of Ba2SiO4:Sr2+ NPs: decreasing maximum at 22,760 cm−1 and increasing maximum at 16,690 cm−1 for higher temperatures. Inset: Ba2SiO4:Sr2+ NPs under UV light, cooled down with liquid N2.
Figure 9.
Temperature dependence of the normalized integrated luminescence intensity of the blue-shifted emission in Ba2SiO4:Sr2+. The black curve was fitted according to Equation (1) with C = 616 and Ea = 50 meV or 400 cm−1.
Figure 9.
Temperature dependence of the normalized integrated luminescence intensity of the blue-shifted emission in Ba2SiO4:Sr2+. The black curve was fitted according to Equation (1) with C = 616 and Ea = 50 meV or 400 cm−1.
From the fit of the temperature dependent measurements the values
C = 616 and
Ea= 50 meV or 400 cm
−1 are obtained, respectively. The large value of C indicates a more efficient quenching rate, which is in agreement with the relatively low temperature of
Tq(
I(
Tq)/
I0 = 0.1) ≈ 120–130 K. At higher temperatures the high-energy band is quenched very efficiently. On the other hand, the activation energy is in a similar range to the energies of the vibrational modes of the SiO
44− tetrahedron, which have been reported by Handke and Urban for Ba
2SiO
4 to be roughly 350 cm
−1 for the E bending mode and 490–520 cm
−1 for the T
2 bending mode [
19]. Furthermore, as studied for SiO
2, the structural nature of the STE in silicates is solely based on the SiO
44− tetrahedron and consists of a peroxy linkage (Si–O–O) with two holes in a σ
u bond of an O
2 molecule and two electrons trapped in an oxygen vacancy (
) [
20]. Thus, a thermally induced activation of electrons via coupling to the vibrational modes of the silicate anion is expected. Moreover, this interpretation makes our assignment of the blue-shifted band clearly reasonable since the spectral position coincides with results found for SiO
2, which was reported to show an emission at 2.8 eV (22,582 cm
−1) [
20,
21]. A similar temperature behavior was also found for that band totally quenching in the range between 110 and 120 K [
22]. However, in these nanoparticles, the stabilization of the excitons arises from the presence of the Sr
2+ ions, which induce distortions large enough to trap the excitons. This also explains why no such emission is observed in the case of undoped Ba
2SiO
4.
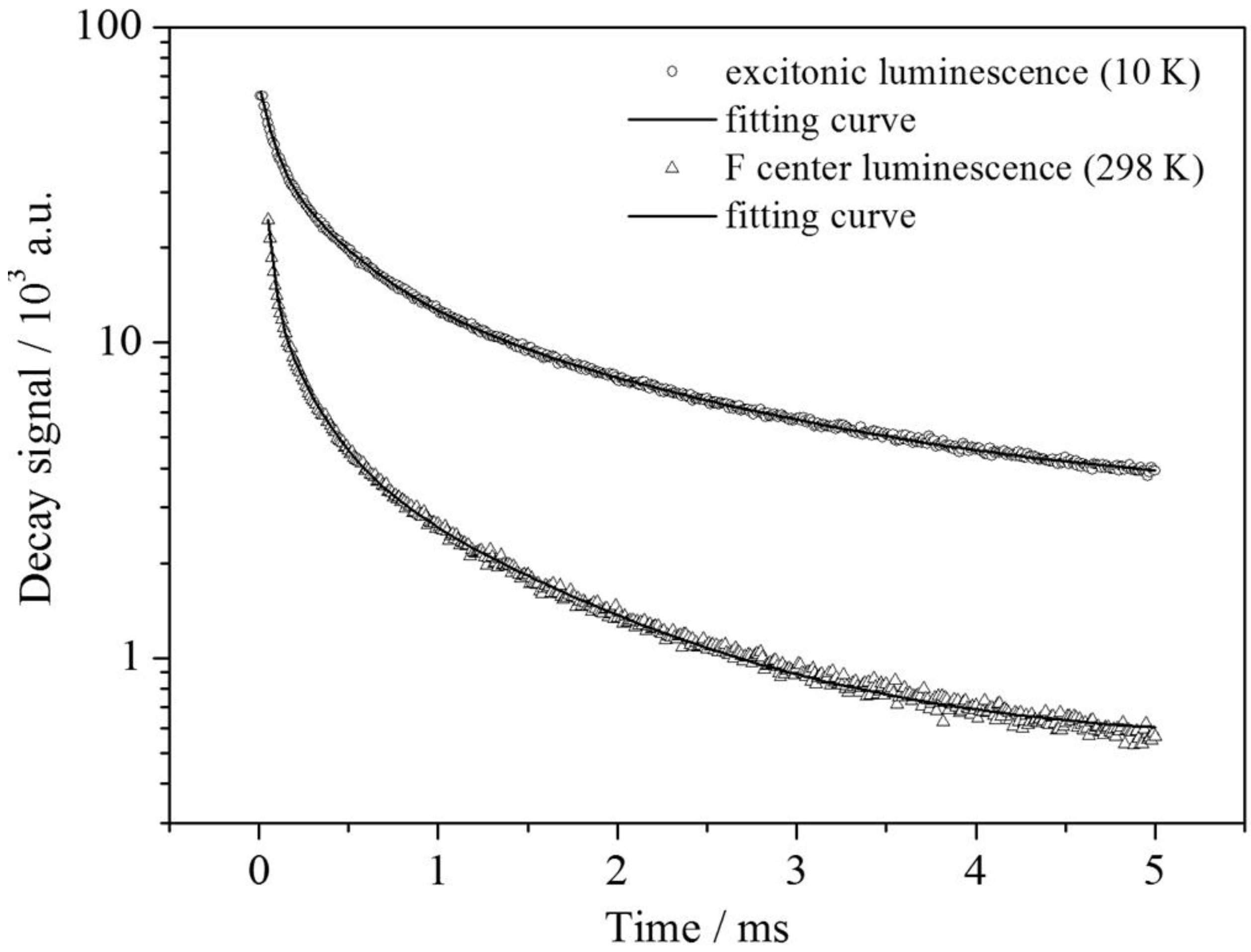
In order to investigate other intrinsic characteristics of the excitonic emission, which are also useful for the interpretation, lifetime measurements at 10 K and room temperature have been performed. The luminescence decay of the blue emission shows a tri-exponential behavior (see
Figure 10) with the lifetimes
τ1 = 1.76 ms (22%),
τ2 = 0.36 ms (36%) and
τ3 = 0.07 ms (41%). The first value is interpreted as the radiative decay of the STE and in good agreement with the value found for self-trapped excitons in SiO
2 (~1 ms) [
23,
24,
25]. The other two lifetimes already indicate trapping processes at the Sr
2+ sites inducing a distortion that is obviously necessary to stabilize the excitons.
Figure 10.
Semi-log plot of the luminescence decays of the observed emission bands in Ba2SiO4:Sr2+. The black curves correspond to tri-exponential fits, respectively. For the values: see text.
Figure 10.
Semi-log plot of the luminescence decays of the observed emission bands in Ba2SiO4:Sr2+. The black curves correspond to tri-exponential fits, respectively. For the values: see text.
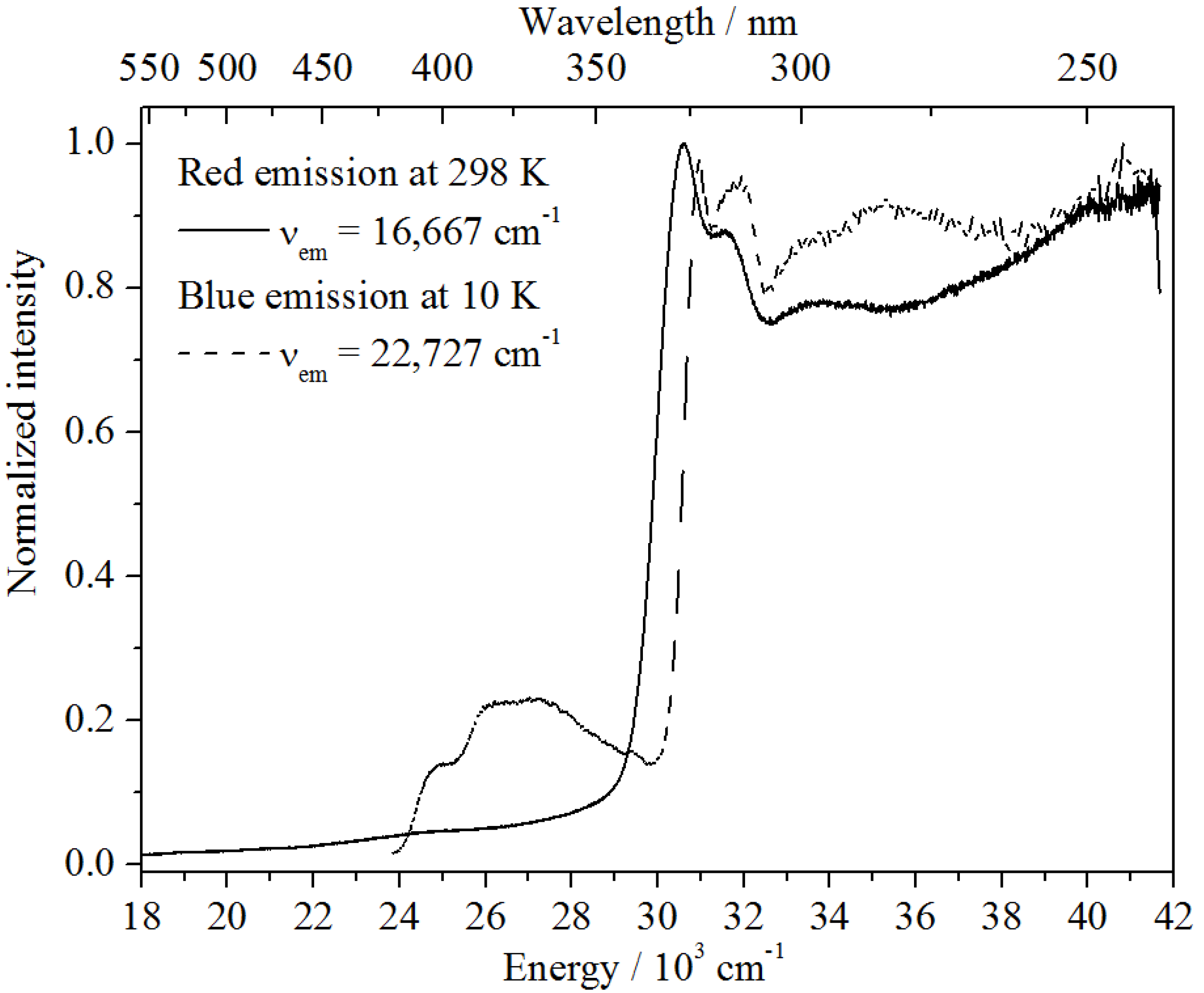
For an understanding of the origin of the red-shifted band at 16,643 cm
−1, one has to include both the temperature behavior as well as the lifetime measurements. Since that emission shows an increase just once the blue-shifted emission has been thermally quenched, a deep correlation between these two centers is supposed. This also becomes clear from the excitation spectra depicted in
Figure 11. Upon detection of the red emission at room temperature, a very similar excitation spectrum to the excitonic emission at 10 K is obtained. The slight redshift can be explained by the temperature difference. Moreover, the luminescence decay of the color center detected at room temperature is very similar to the decay of the excitonic emission at 10 K (see
Figure 10). As in the upper case, a tri-exponential fit can be performed affording the lifetimes,
τ1 = 1.15 ms (8%),
τ2 = 0.21 ms (18%) and
τ3 = 0.03 ms (74%). The deviations can be explained by the shortening of the lifetimes due to non-radiative relaxation.
Figure 11.
Excitation spectra of the red emission band at 10 K (bold curve) and of the blue emission band at 298 K (dashed curve).
Figure 11.
Excitation spectra of the red emission band at 10 K (bold curve) and of the blue emission band at 298 K (dashed curve).
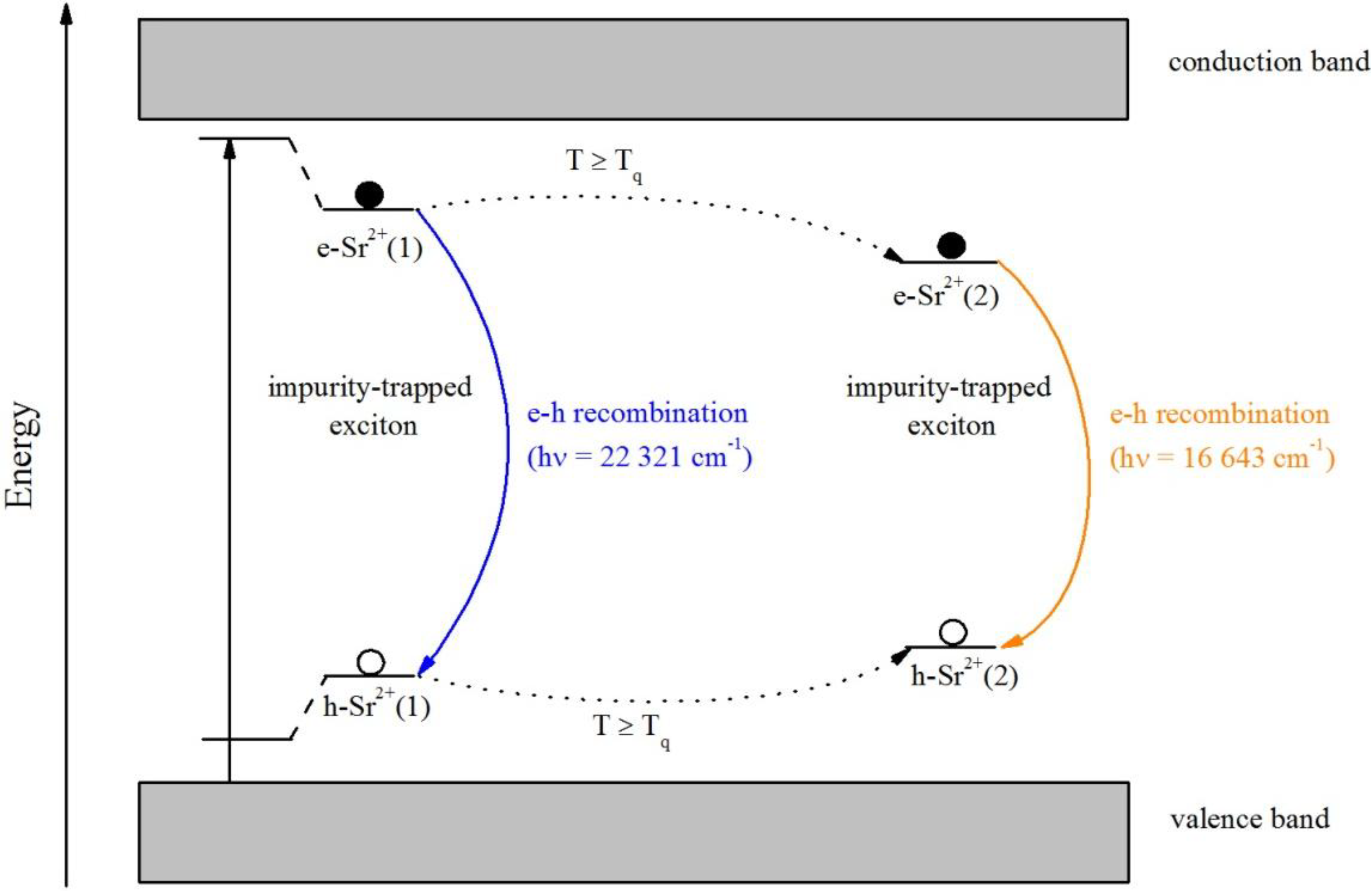
Thus, we assume that after heating at temperatures higher than
Tq, the excitons located at the Sr
2+ impurities become mobile and move within the lattice until they become trapped at another Sr
2+ site due to the induced distortion. A pictorial representation of the model is depicted in
Figure 12. During the movement, they lose energy non-radiatively, which gives rise to a larger Stokes shift and therefore a red-shift of the emission. Therefore,
Tq can be interpreted as the temperature at which the excitons become mobile. After trapping at another Sr
2+ site, the intensity decreases with increasing temperature via multiphonon relaxation. The similarity of the two centers is indicated by the similarity of the excitation spectra and the decay curves. Photoconductivity and thermoluminescence measurements could still clarify whether an involvement of the conduction band in the mechanism is the case.
Figure 12.
Proposed model for the electron transfer between the valence and conduction band in Sr2+-doped Ba2SiO4 NPs.
Figure 12.
Proposed model for the electron transfer between the valence and conduction band in Sr2+-doped Ba2SiO4 NPs.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











