
Photophysical Study of Polymer-Based Solar Cells with an Organo-Boron Molecule in the Active Layer


 ,
, 
Abstract
:1. Introduction
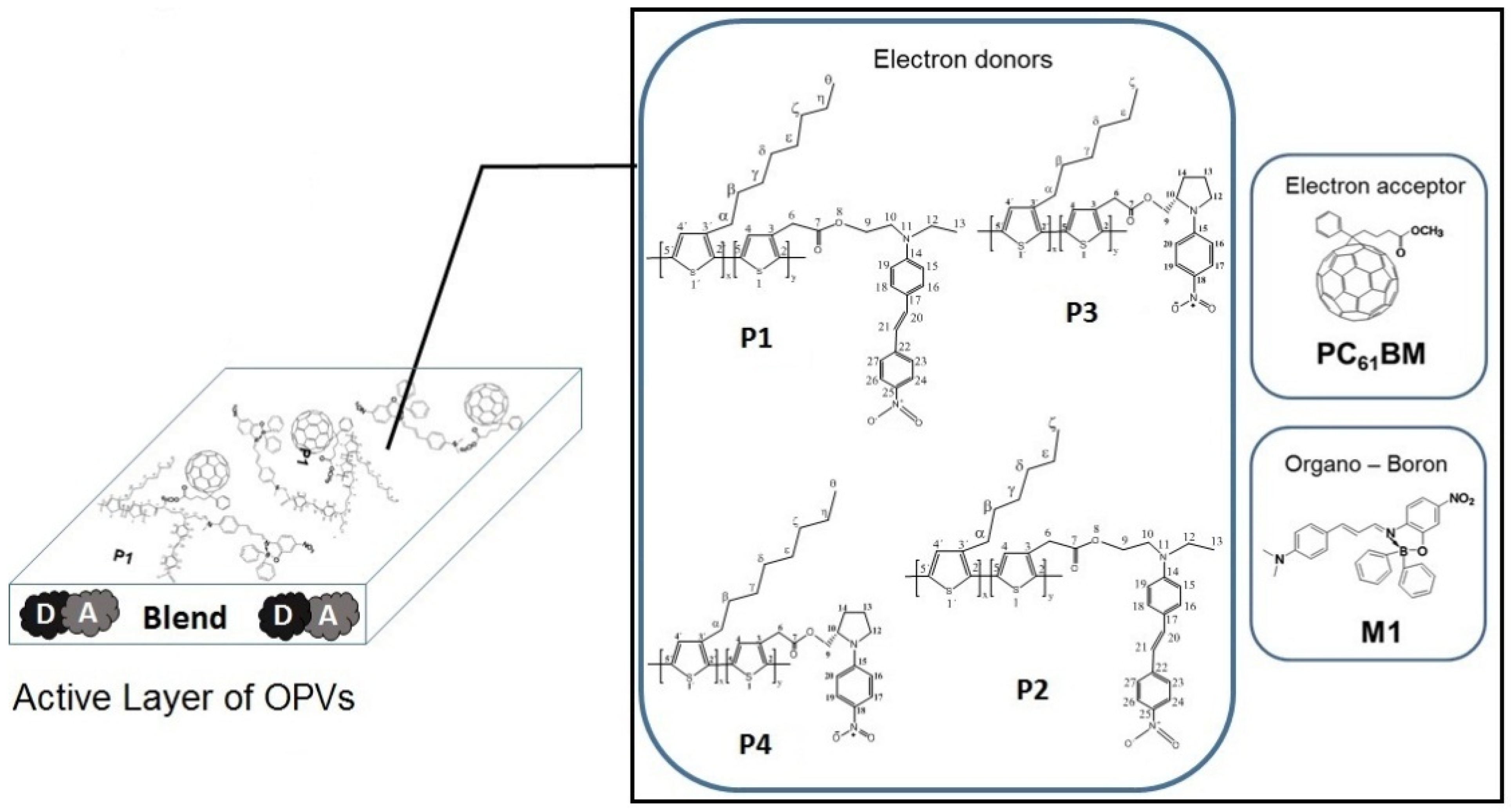
2. Results and Discussion
2.1. Characterization of Solar Cells Devices

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Soluble yield (%) | Monomer ratio a | Molecular weight | Configuration | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diads (%) | Triads (%) | |||||||||
| (g/mol) | HT | HH | HT-HT | TT-HT | HT-HH | TT-HH | ||||
| P1 | 79 | 1.4/98.6 | 12,600 | 80,000 | 76 | 24 | 58 | 14 | 21 | 8 |
| P2 | 63 | 5/95 | 24,600 | 236,000 | 67 | 33 | 43 | 18 | 20 | 19 |
| P3 | 32 | 26/74 | 16,800 | 106,000 | 64 | 36 | 43 | 18 | 19 | 20 |
| P4 | 15 | 12/88 | 11,000 | 117,000 | 68 | 32 | 50 | 15 | 18 | 17 |

| Components (weight ration) | η (%) |
|---|---|
| P1:PC61BM (1:2) | 0.17 |
| P1:PC61BM:M1 (1:2:1) | 0.39 |
| P2:PC61BM (1:2) | 0.15 |
| P2:PC61BM:M1 (1:2:1) | 0.41 |
| P3:PC61BM (1:2) | 0.14 |
| P3:PC61BM:M1 (1:2:1) | 0.18 |
| P4:PC61BM (1:2) | 0.02 |
| P4:PC61BM:M1 (1:2:1) | 0.04 |
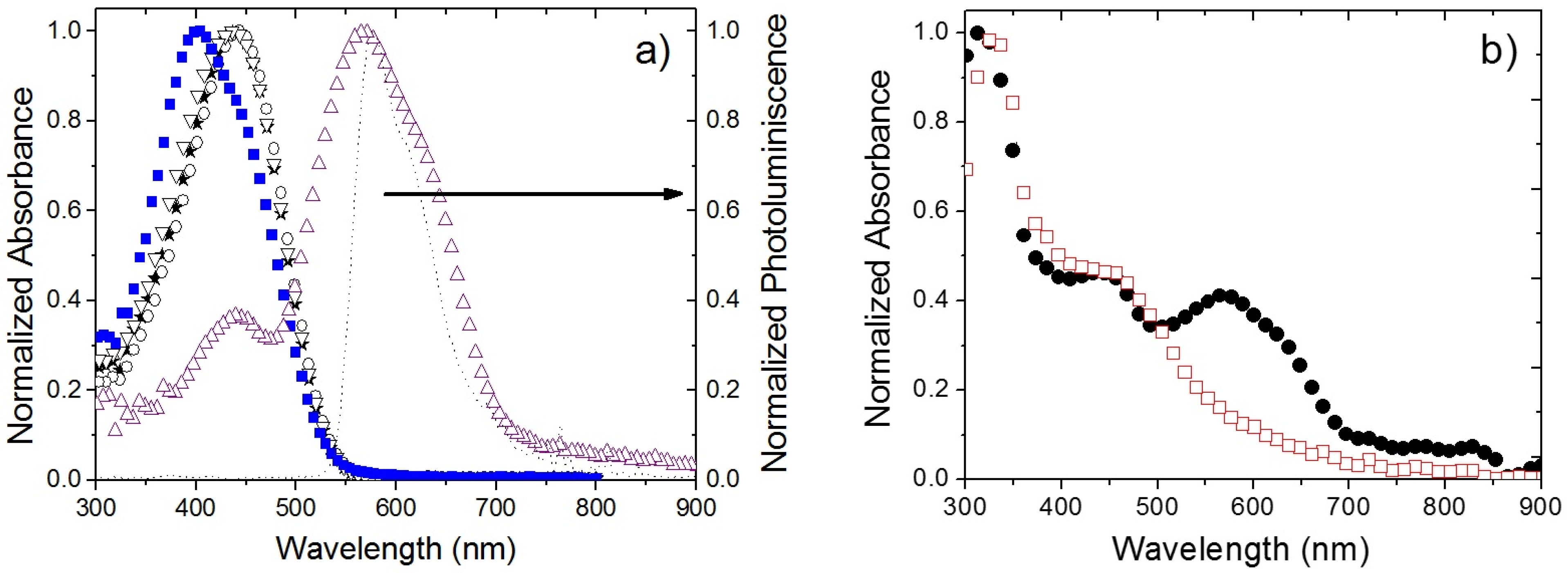
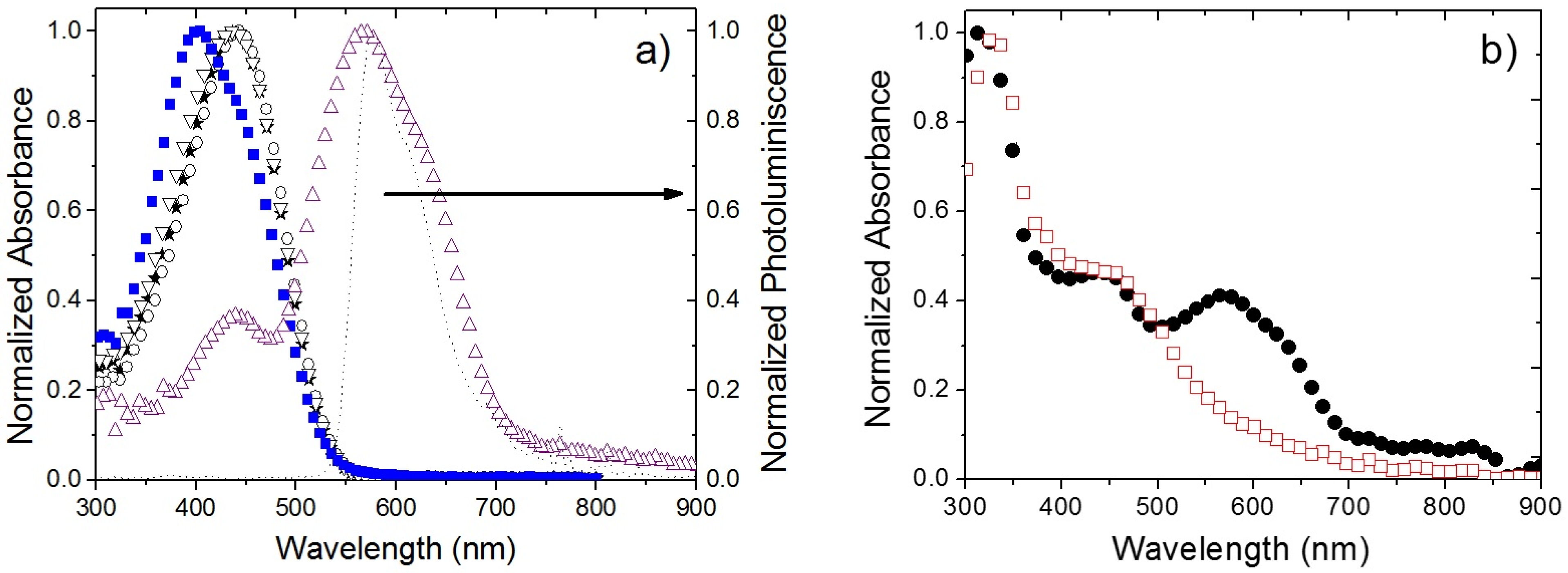
2.2. Photophysical Studies of OPVs’ Active Layers

| Electrochemical | P1 | M1 | PC61BM |
|---|---|---|---|
| LUMO (eV) | −3.54 | −3.66 | −3.7 |
| HOMO (eV) | −5.55 | −5.22 | −6.1 |
| GAP (eV) | 2.01 | 1.56 | 2.4 |

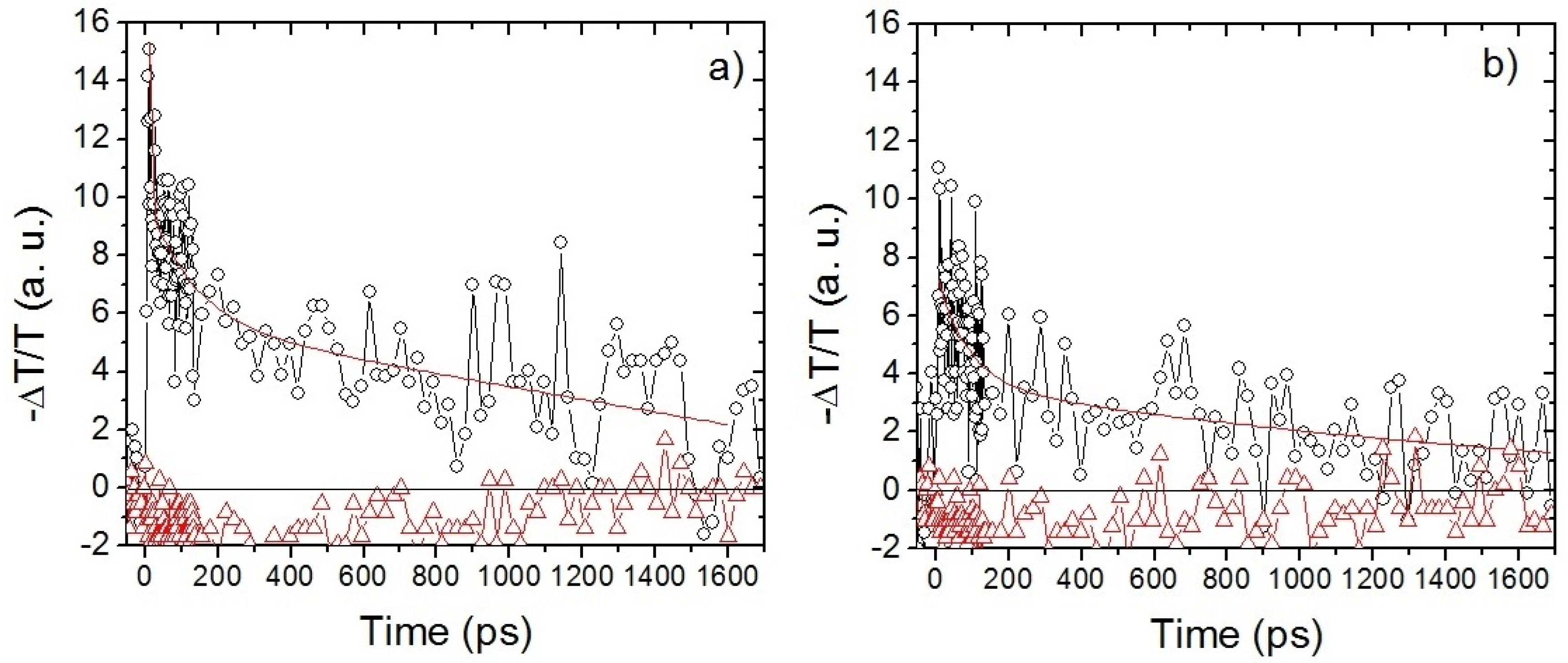
| Probe Wavelength (nm) | Time Constants of biexpontial decay | |||
|---|---|---|---|---|
| P1:PC61BM | P1:PC61BM:M1 | |||
| Short τ1 (ps) | Long τ2 (ns) | Short τ1 (ps) | Long τ2 (ns) | |
| 650 | 17 | 0.36 | 47 | 2.31 |
| 735 | 12 | 0.45 | 86 | 2.26 |
| 900 | 4 | 1.25 | 0.92 | 0.212 |
3. Experimental Section
3.1. Polythiophene Derivatives
3.2. Sample Preparation
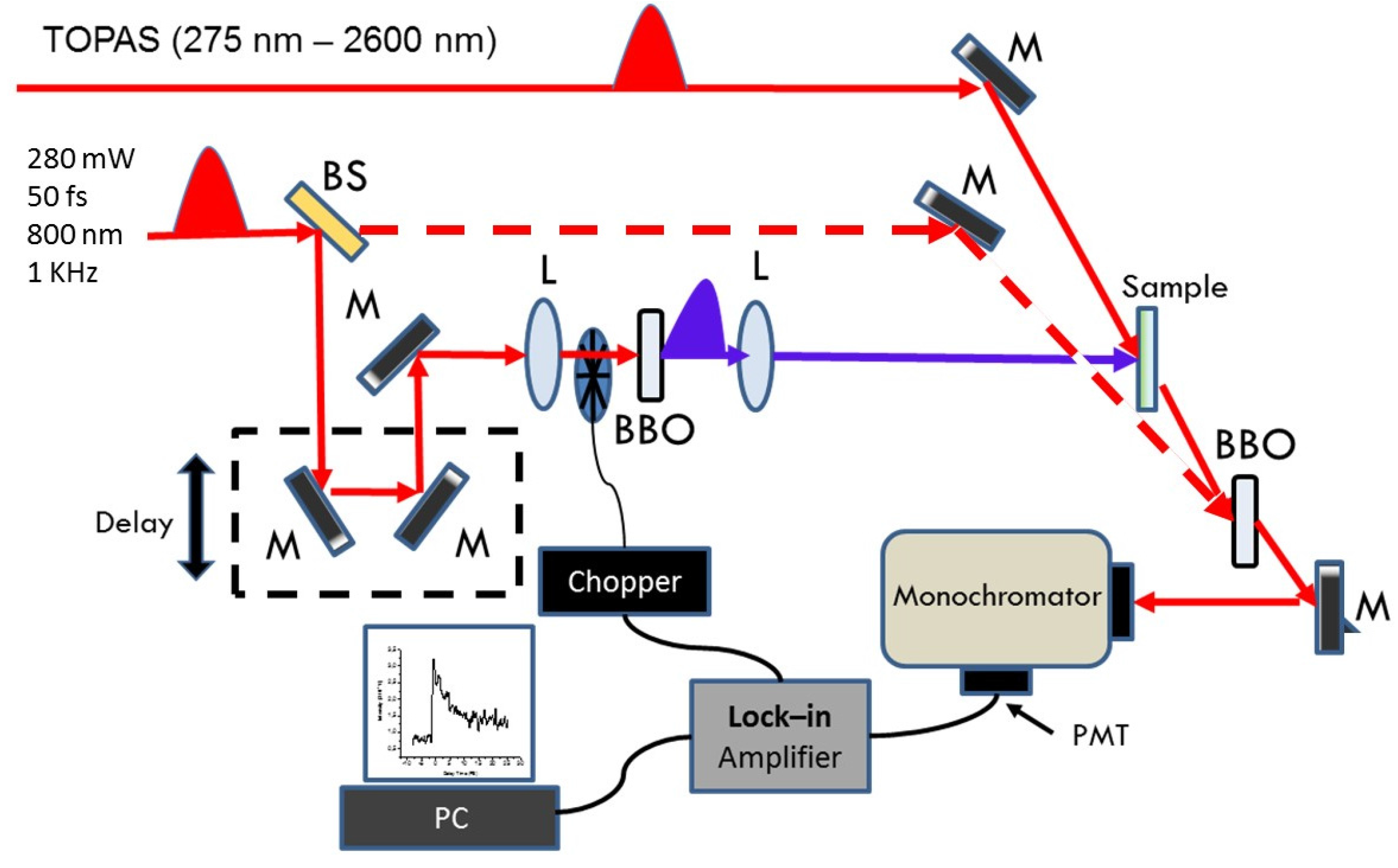
3.3. Steady-State and Transient Absorption Experiments

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoppe, H.; Serdar Sariciftci, N. Organic solar cells: An overview. J. Mater. Res. 2004, 19, 1924–1945. [Google Scholar] [CrossRef]
- Tang, C.W.; Albrecht, A.C. Photovoltaic effects of metal-chlorophyll-a-metal sandwich cells. J. Chem. Phys. 1975, 62, 2139–2149. [Google Scholar] [CrossRef]
- Tang, C.W. Two-layer organic photovoltaic cell. Appl. Phys. Lett. 1986, 48, 183–185. [Google Scholar] [CrossRef]
- Ohori, Y.; Hoashi, T.; Yanagi, Y.; Okukawa, T.; Fujii, S.; Kataura, H.; Nishioka, Y. Organic solar cells based on ternary blend active layer of two donors PTB7, P3HT and acceptor PC61BM. J. Photopolym. Sci. Technol. 2014, 27, 569–575. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, S.H.; Lee, H.-H.; Lee, K.; Ma, W.; Gong, X.; Heeger, A.J. New architecture for high efficiency polymer photovoltaic cells using solution-based titanium oxide as an optical spacer. Adv. Mater. 2006, 18, 572–576. [Google Scholar] [CrossRef]
- Park, S.H.; Roy, A.; Beaupré, S.; Cho, S.; Coates, N.; Moon, J.S.; Moses, D.; Leclerc, M.; Lee, K.; Heeger, A.J. Bulk heterojunction solar cell with internal quantum efficiency approaching 100%. Nat. Photonics 2009, 3, 297–302. [Google Scholar] [CrossRef]
- Li, G.; Zhu, R.; Yang, Y. Polymer solar cells. Nat. Photonics 2012, 6, 153–161. [Google Scholar]
- Sun, Y.; Welch, G.C.; Leong, W.L.; Takacs, C.J.; Bazan, G.C.; Heeger, A.J. Solution-processed small-molecule solar cells with 6.7% efficiency. Nat. Mater. 2012, 11, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yang, L.; You, W. Rational design of high performance conjugated polymers for organic solar cells. Macromolecules 2012, 45, 607–632. [Google Scholar] [CrossRef]
- Wong, W. Challenges in organometallic research–great opportunity for solar cells and oleds. J. Organomet. Chem. 2009, 694, 2644–2647. [Google Scholar]
- Qiao, F.; Liu, A.; Zhou, Y.; Xiao, Y.; Yang, P.O. Bulk heterojunction organic solar cell based on a novel fluorescent fluorine-boron complex. J. Mater. Sci. 2009, 44, 1283–1286. [Google Scholar]
- Pandey, R.; Zou, Y.; Holmes, R.J. Efficient bulk heterojunction organic photo-voltaic cells based on boron subphathalocyanine chloride-C70. Appl. Phys. Lett. 2012, 101. [Google Scholar] [CrossRef]
- Jakle, F. Advances in the synthesis of organoborone polymers for optical, electrical and sensory applications. Chem. Rev. 2010, 110, 3985–4022. [Google Scholar]
- Qin, Y.; Kiburu, I.; Shah, S.; Jakle, F. Luminescence tuning of organoboron quinolates through substituent variation at the 5-position of the quinolato moiety. Org. Lett. 2006, 8, 5227–5230. [Google Scholar]
- Cataldo, S.; Fabiano, S.; Ferrante, F.; Preveti, F.; Patane, S.; Pignataro, B. Organoboron polymers for photovoltaic bulk heterojunctions. Macromol. Rapid Commun. 2010, 31, 1281–1286. [Google Scholar]
- Wu, Q.; Esteghamatian, M.; Hu, N.; Popovic, Z.; Enright, G.; Tao, Y.; D’lorio, M.; Wang, S. Synthesis, structure, and electroluminescence of BR2q(R = Et, Ph, 2-Naphthyland q = 8-Hydroxyquinolto). Chem. Mater. 2000, 12, 79–83. [Google Scholar]
- Li, D.; Wang, K.; Huang, S.; Qu, S.; Liu, X.; Zhu, Q.; Zhang, H.; Wang, Y. Brightly fluorescent red organic solids bearing boron-bridged π-conjugated skeletons. J. Mater. Chem. 2011, 21, 15298–15304. [Google Scholar]
- Jakle, F. Lewis acidic organoboron Polymers. Coord. Chem. Rev. 2006, 250, 1107–1121. [Google Scholar]
- Rodriguez, M.; Maldonado, J.L.; Ramos-Ortiz, G.J.; Lamere, F.; Lacroix, P.G.; Farfan, N.; Ochoa, M.E.; Santillan, R.; Meneses-Nava, M.A.; Barbosa-Garcia, O.; et al. Syntehsis and non-linear optical characterization of novel borinate derivatives of cinnamaldehyde. New J. Chem. 2009, 33, 1693–1702. [Google Scholar]
- Clarke, T.; Durrant, J. Charge photogeneration in organic solar cells. Chem. Rev. 2010, 110, 6736–6767. [Google Scholar]
- Cheng, Y.; Yang, C.; Hsu, C. Synthesis of conjugated polymers for organic solar cells applications. Chem. Rev. 2009, 109, 5868–5923. [Google Scholar]
- Chasteen, S.V.; Sholin, V.; Carter, S.A.; Rumbles, G. Towards optimization of device performance in conjugated polymer photovoltaics: Charge generation, transfer and transport in poly(p-phenylene-vinylene) polymer heterojunctions. Sol. Energy Mater. Sol. Cells 2008, 92, 651–659. [Google Scholar] [CrossRef]
- Agina, E.V.; Ponomarenko, S.A.; Muzafrov, A.M. Macromolecular systems with the p-type conductivity. Russ. Chem. Bull. 2010, 59, 1080–1098. [Google Scholar]
- Del-Oso, J.A.; Maldonado, J.L.; Ramos-Ortiz, G.; Rodriguez, M.; Guizado-Rodriguez, M.; Escalante, J.; Frontana-Uribe, B.A.; Perez-Gutierrez, E.; Santillan, R. New polythiopene derivatives and enhanced photovoltaic effect by a boron compound blended with them in OPVs cells. Synth. Met. 2014, 196, 83–91. [Google Scholar] [CrossRef]
- Salinas, J.F.; Maldonado, J.L.; Ramos-Ortíz, G.; Rodríguez, M.; Meneses-Nava, M.A.; Barbosa-García, O.; Santillan, R.; Farfán, N. On the use of woods metal for fabricating and testing polymeric organic solar cells: An easy and fast method. Sol. Energy Mater. Sol. Cells 2011, 95, 595–601. [Google Scholar] [CrossRef]
- Yu, J.; Zheng, Y.; Huang, J. Towards high performance organic photovoltaic cells: A review of recent development in organic photovoltaics. Polymers 2014, 6, 2473–2509. [Google Scholar] [CrossRef]
- Kaur, N.; Singh, M.; Pathak, D.; Wagner, T.; Nunzi, J.M. Organic materials for photovoltaic applications: Review and mechanism. Synth. Met. 2014, 190, 20–26. [Google Scholar] [CrossRef]
- Yan, J.; Saunders, B. Third-generation solar cells: A review and comparison of polymer: Fullerene, hybrid polymer and perovskite solar cells. RSC Adv. 2014, 4, 43286–43314. [Google Scholar] [CrossRef]
- Huang, Y.; Kramer, E.J.; Heeger, A.J.; Bazan, G.C. Bulk heterojunction solar cells: Morphology and performance relathionships. Chem. Rev. 2014, 114, 7006–7043. [Google Scholar] [CrossRef] [PubMed]
- Kandada, R.S.; Grancini, G.; Perozza, A.; Perissinotto, S.; Fazzi, D.; Raavi, S.S.K.; Lanzani, G. Ultrafast energy transfer in ultrathin organic donor/acceptor blend. Sci. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Ohkita, H.; Benten, H.; Ito, S. Near-IR femtosecond transient absorption spectroscopy of ultrafast polaron and triplet exciton formation in polythiophene films with different regioregularities. J. Am. Chem Soc. 2009, 131, 16869–16880. [Google Scholar] [CrossRef] [PubMed]
- Ohkita, H.; Cook, S.; Astuti, Y.; Duffy, W.; Tierney, S.; Zhang, W.; Heeney, M.; McCulloch, I.; Nelson, J.; Bradley, D.D.C.; et al. Charge carrier formation in polythiophene/fullerene blend films studied by transient absorption spectroscopy. J. Am. Chem. Soc. 2008, 130, 3030–3042. [Google Scholar] [CrossRef] [PubMed]
- Roncali, J. Conjugated poly(thiophenes): Synthesis, functionalization and applications. Chem. Rev. 1992, 92, 711–738. [Google Scholar] [CrossRef]
- Piris, J.; Dykstra, T.E.; Bakulin, A.A.; van Loosdrecht, P.H.M.; Knulst, W.; Trinh, M.T.; Schins, J.M.; Siebbeles, L.D.A. Photogeneration and ultrafast dynamics of excitons and charges in P3HT/PCBM blends. J. Phys. Chem. C. 2009, 113, 14500–14506. [Google Scholar] [CrossRef]
- Cook, S.; Katoh, R.; Furube, A. Ultrafast studies of charge generation in PCBM:P3HT blend films following excitation of the fullerene PCBM. J. Phys. Chem. C 2009, 113, 2547–2552. [Google Scholar] [CrossRef]
- Albota, M.A.; Xu, C.; Webb, W.W. Two-photon fluorescence excitation cross sections of biomolecular probes from 690 to 960 nm. Appl. Opt. 1998, 37, 7352–7356. [Google Scholar] [CrossRef]
- Martini, B.; Smith, A.D.; Schwartz, B.J. Exciton-exciton annihilation and the production of interchain species in conjugated polymer films: Comparing the ultrafast stimulated emission and photoluminescence dynamics of MEH-PPV. Phys. Rev. B 2004, 69. [Google Scholar] [CrossRef]
- Hodgkiss, M.; Albert-Seifried, S.; Rao, A.; Barker, A.J.; Campbell, A.R.; Marsh, R.A.; Friend, R.H. Exciton-charge annihilation in organic semiconductor films. Adv. Funct. Mater. 2012, 22, 1567–1577. [Google Scholar] [CrossRef]
- Benson-Smith, J.; Goris, L.; Vandewal, K.; Haenen, K.; Manca, J.V.; Vanderzande, D.; Bradley, D.D.C.; Nelson, J. Formation of a ground-state charge-tranfer complex in polyfluorene/[6,6]-phenyl-C61 Butyric Acid Methyl Ester (PCBM) blend films and its role in the function of polymer/PCBM solar cells. Adv. Funct. Mater. 2007, 17, 451–457. [Google Scholar] [CrossRef]
- Scheblykin, G.; Yartsev, A.; Pullerits, T.; Gulbinas, V.; Sundstrom, V. Excited state and charge photogeneration dynamics in conjugated polymers. J. Phys. Chem. B 2007, 111, 6303–6321. [Google Scholar] [CrossRef] [PubMed]
- Vandewal, A.; Gadisa, W.D.; Oosterbaan, S.; Bertho, F.; Banishoeib, I.V.; Severen, L.; Lutsen, T.; Cleij, J.; Vanderzande, D.; Manca, J.V. The relation between open-circuit voltage and the onset of photocurrent generation by charge-transfer absorption in polymer: Fullerene bulk heterojunction solar cells. Adv. Funct. Mater. 2008, 18, 2064–2070. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Servin, S.; Villa, M.D.A.; Carriles, R.; Ramos-Ortíz, G.; Maldonado, J.-L.; Rodríguez, M.; Güizado-Rodríguez, M. Photophysical Study of Polymer-Based Solar Cells with an Organo-Boron Molecule in the Active Layer. Materials 2015, 8, 4258-4272. https://doi.org/10.3390/ma8074258
Romero-Servin S, Villa MDA, Carriles R, Ramos-Ortíz G, Maldonado J-L, Rodríguez M, Güizado-Rodríguez M. Photophysical Study of Polymer-Based Solar Cells with an Organo-Boron Molecule in the Active Layer. Materials. 2015; 8(7):4258-4272. https://doi.org/10.3390/ma8074258
Chicago/Turabian StyleRomero-Servin, Sergio, Manuel De Anda Villa, R. Carriles, Gabriel Ramos-Ortíz, José-Luis Maldonado, Mario Rodríguez, and M. Güizado-Rodríguez. 2015. "Photophysical Study of Polymer-Based Solar Cells with an Organo-Boron Molecule in the Active Layer" Materials 8, no. 7: 4258-4272. https://doi.org/10.3390/ma8074258
APA StyleRomero-Servin, S., Villa, M. D. A., Carriles, R., Ramos-Ortíz, G., Maldonado, J.-L., Rodríguez, M., & Güizado-Rodríguez, M. (2015). Photophysical Study of Polymer-Based Solar Cells with an Organo-Boron Molecule in the Active Layer. Materials, 8(7), 4258-4272. https://doi.org/10.3390/ma8074258