
Solid-State Gas Sensors: Sensor System Challenges in the Civil Security Domain


Abstract
:1. Introduction

2. Target Substances
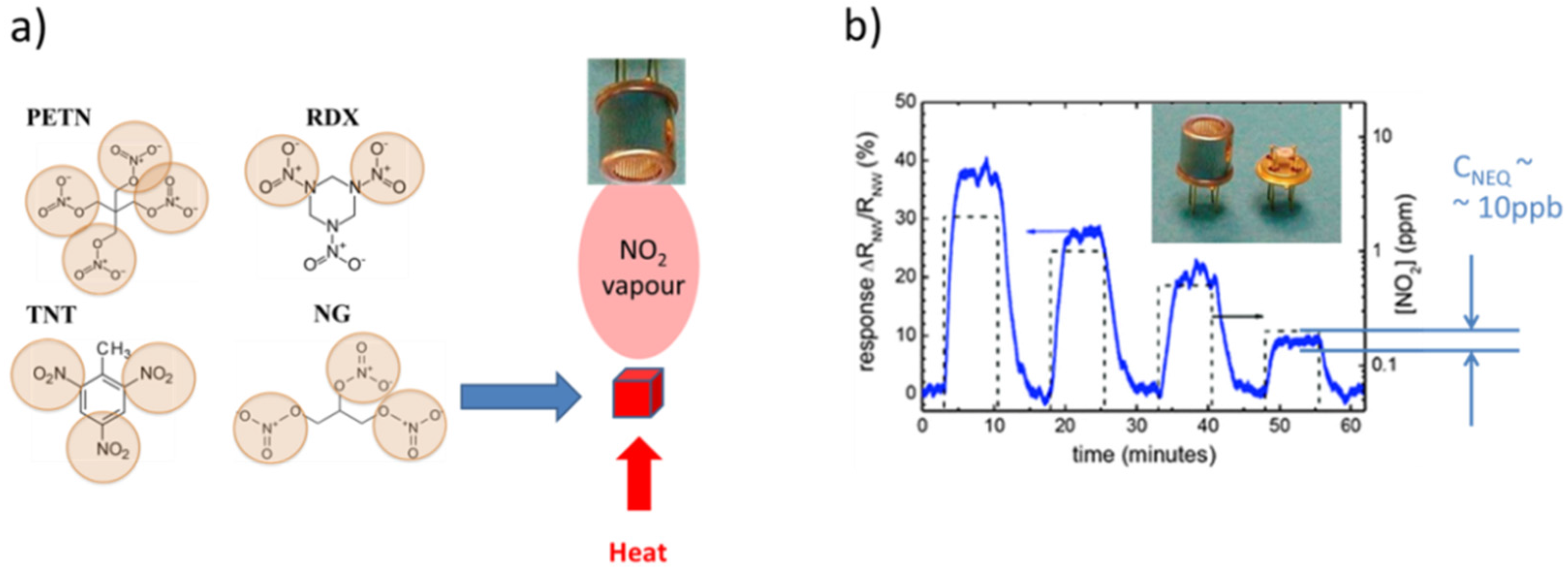
2.1. Explosives
- Military high explosives: trinitrotoluol (TNT), cyclotrimethylene-trinitramine (RDX), pentaerythritol tetranitrate (PETN);
- Improvised explosives: ammonium nitrate (AN), urea nitrate (UN).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Comment | Aggregate Form | Electron Affinity (eV) | Equil. Vapor Pressure at 25 °C (ppb) | Water Solubility (g/L) | Melting Point (°C) | Boiling Point (°C) | Product Gas |
|---|---|---|---|---|---|---|---|---|
| TNT [20] | Military high explosive | Solid | ~2.3 | 9.55 | 0.13 (20 °C) | 80 | 295 (igniting) | NO2 |
| RDX [21] | Military high explosive | Solid | ~1.2 | 0.006 | insoluble | 205 | 235 | NO2 |
| PETN [22] | Military high explosive | Solid | ~1.6 | 0.18 | 0.1 (50 °C) | 141 | 150 (de-com-poses) | NO2 |
| Ammonium nitrate (AN) [23] | Improvised explosive | Solid | 3.58 | 12.3 | 1500 (20 °C) | 170 | 210 | NO2 |
| Urea nitrate (UN) [24] | Improvised explosive | Solid | ~3.7 (NO3−) | 0.009 | 150 | 163 | unknown | NO2 |
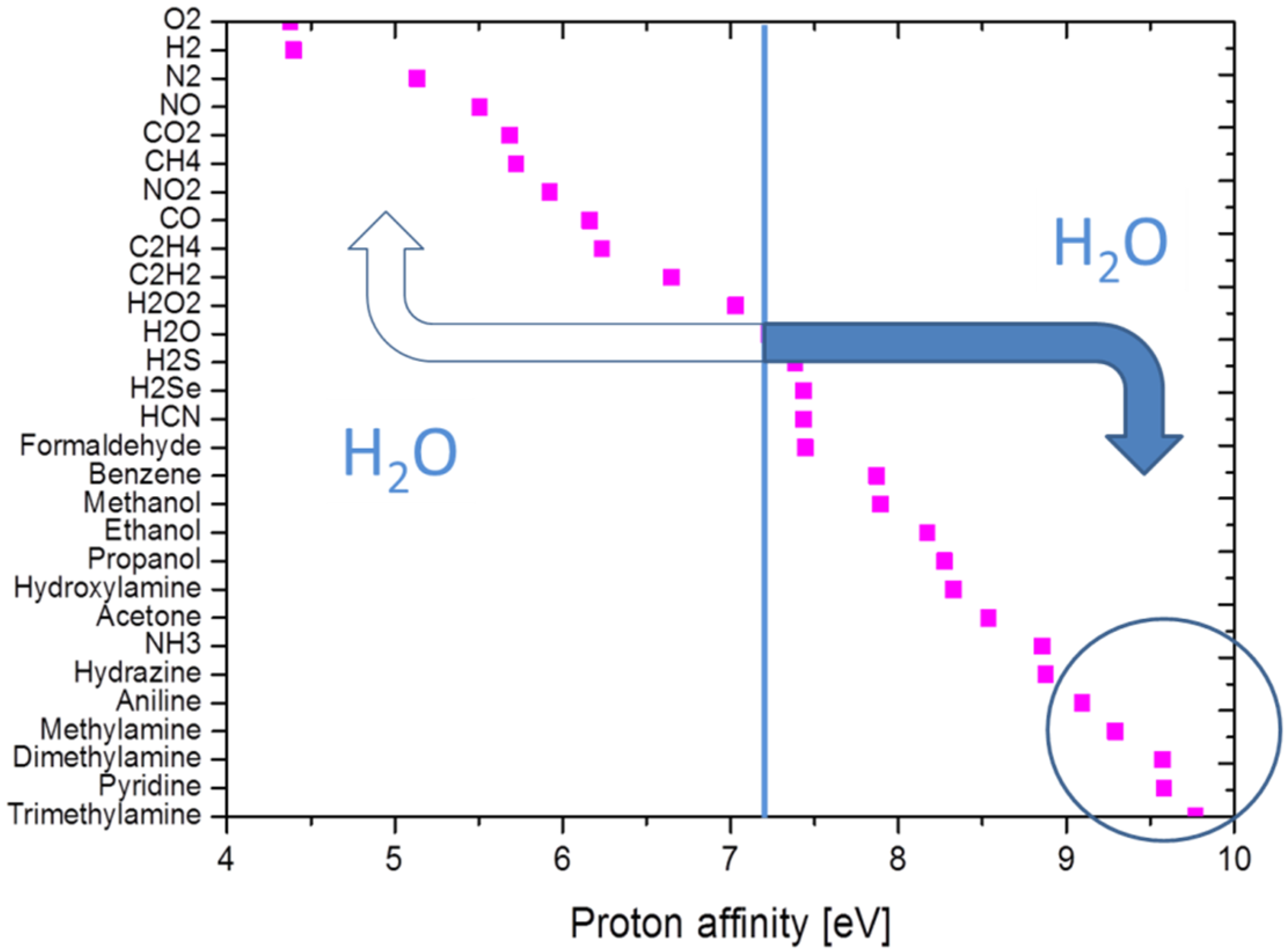
| Gas | Name | Electron Affinity (eV) |
|---|---|---|
| N2 | Nitrogen | −0.72 |
| O2 | Oxygen | 0.448 |
| H2O | Water vapor | negative |
| CO2 | Carbon dioxide | −0.6 |
| H2, HC | Hydrogen, most hydrocarbons | ≤0 |
| NO2 | Nitrogen dioxide | 2.273 |
| O3 | Ozone | 2.103 |
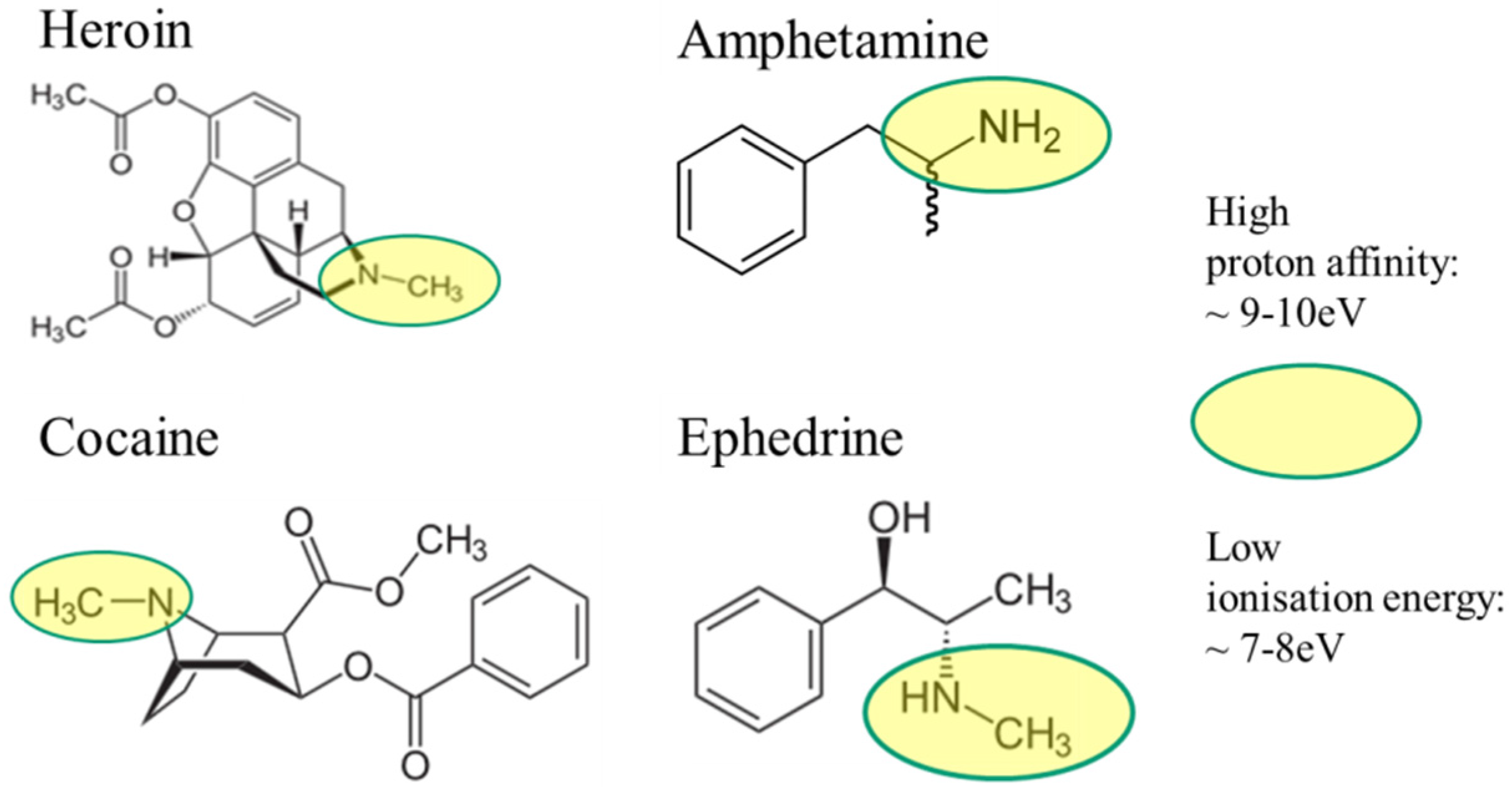
2.2. Illicit Drugs

| Name | Comment | Aggregate Form | Melting Point Free-Base/Salt (°C) | Boiling Point (°C) | Proton Affinity (eV) |
|---|---|---|---|---|---|
| Ecstasy [29] | Drug | solid | 152/11 (flash point) | 155 | 8 to 10 |
| Cocaine [30] | Drug | solid | 98/197 (decomposes) | 187 | 8 to 10 |
| Heroine [31] | Drug | solid | 171/250 (flash point) | 273 | 8 to 10 |
| Ephedrine [32] | Drug | solid | 40/220 | 225 | 8 to 10 |


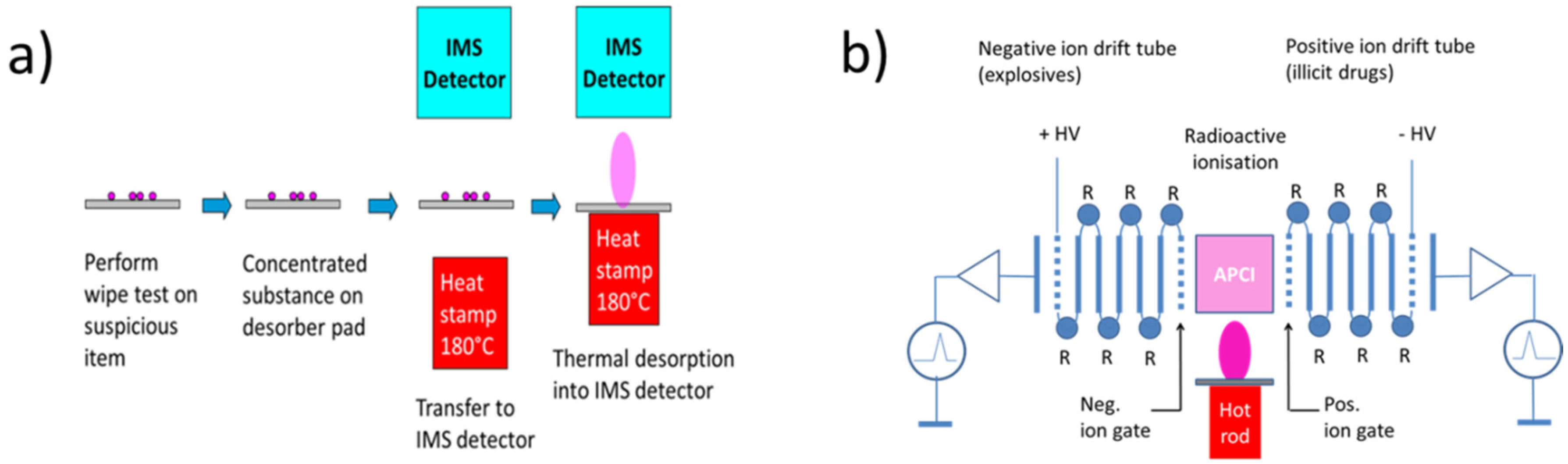
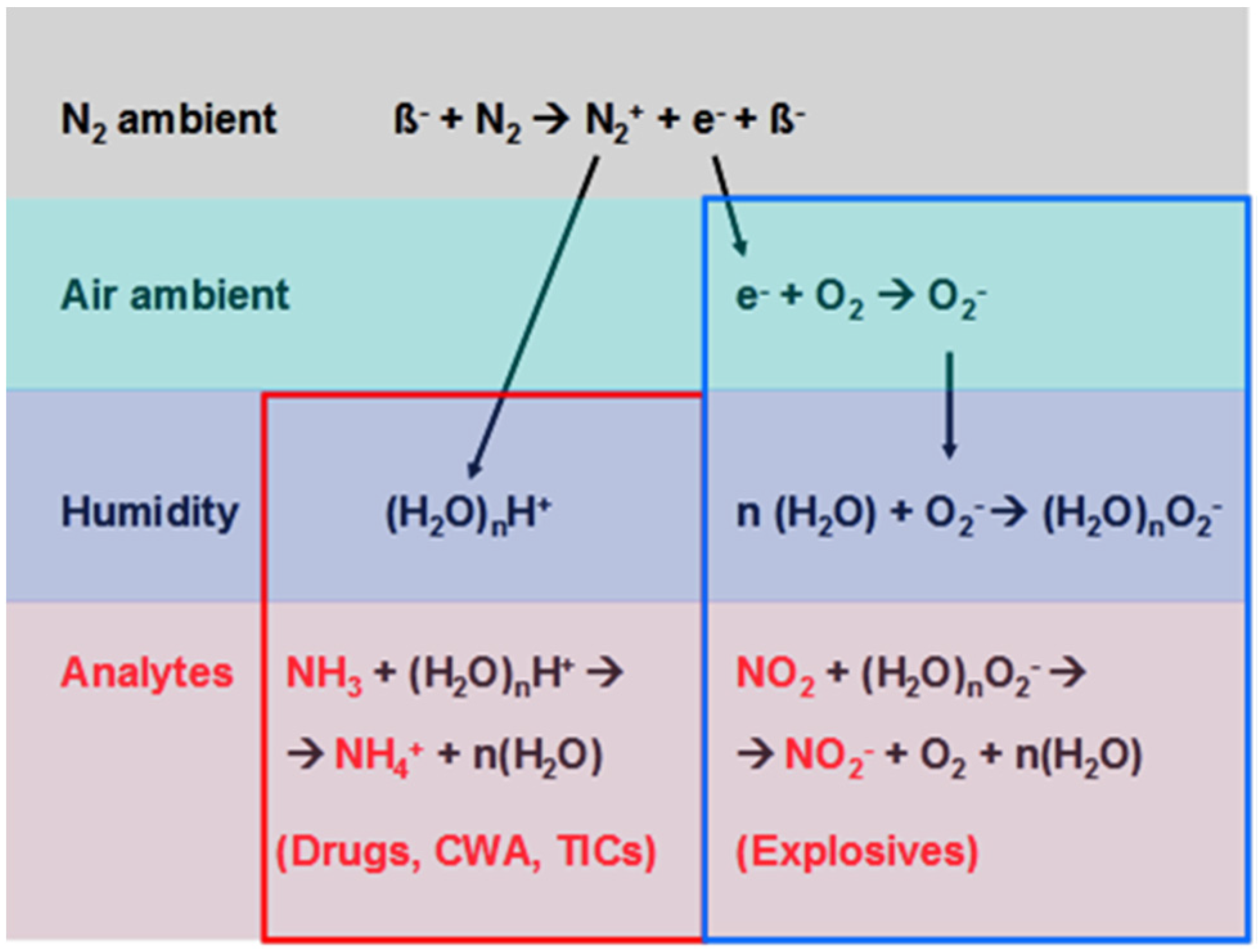
3. Security Screening: State of the Art (IMS)

| Detectable substances | Explosives, illicit drugs, chemical warfare agents (CWA), toxic industrial compounds (TICs) |
| Detection limits | Solids: 0.1 nanogram; gases, vapors (ppb) |
| Speed of response | Identification of sampled analyte within tens of seconds after flash evaporation |
| Time required for sampling | Dependent on suspect object and inspector |
| Volume/weight | Several liters/several kg |
| Price | ~40 to 50 thousand € |
| Downsides | Use of radioactive ionization only allowed by specially-trained personal in approved locations. |


4. Metal-Oxide-Based Security Sensor Systems
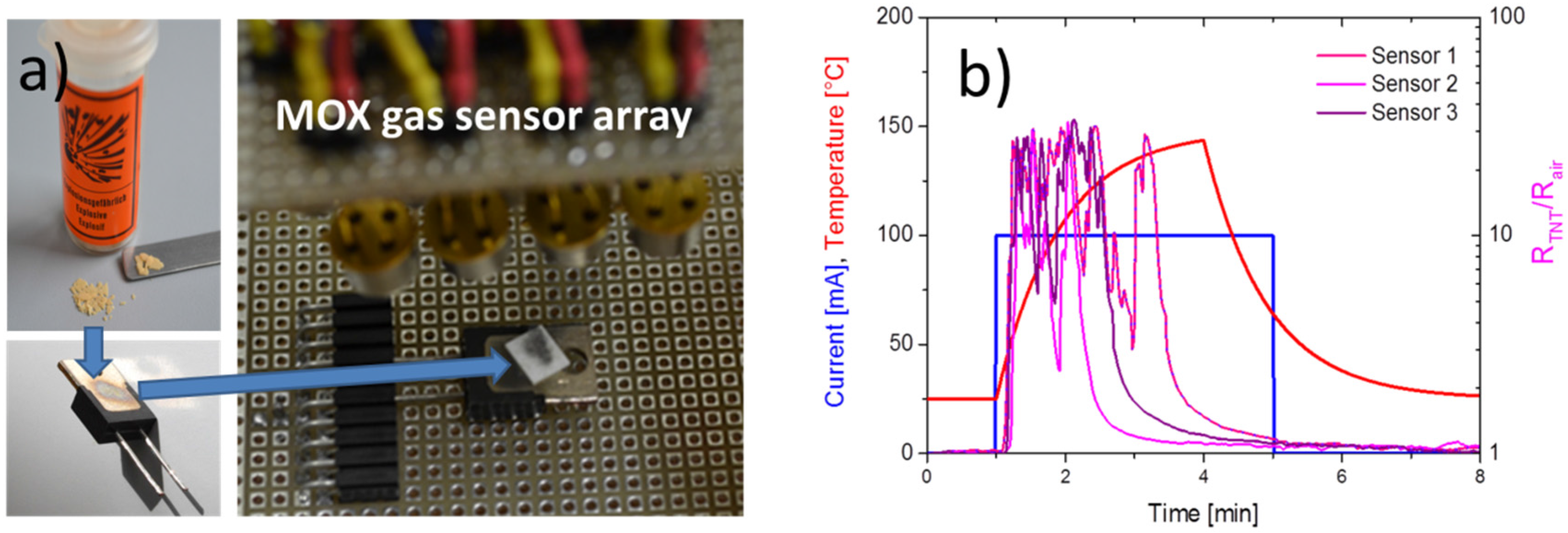
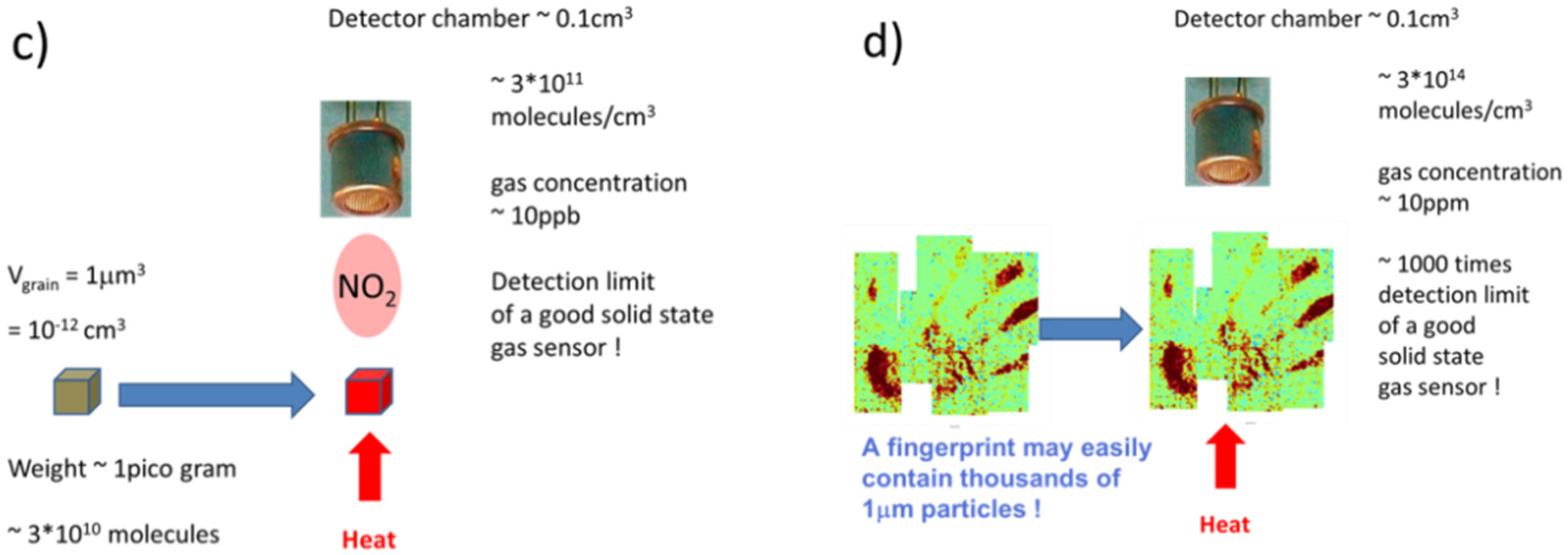
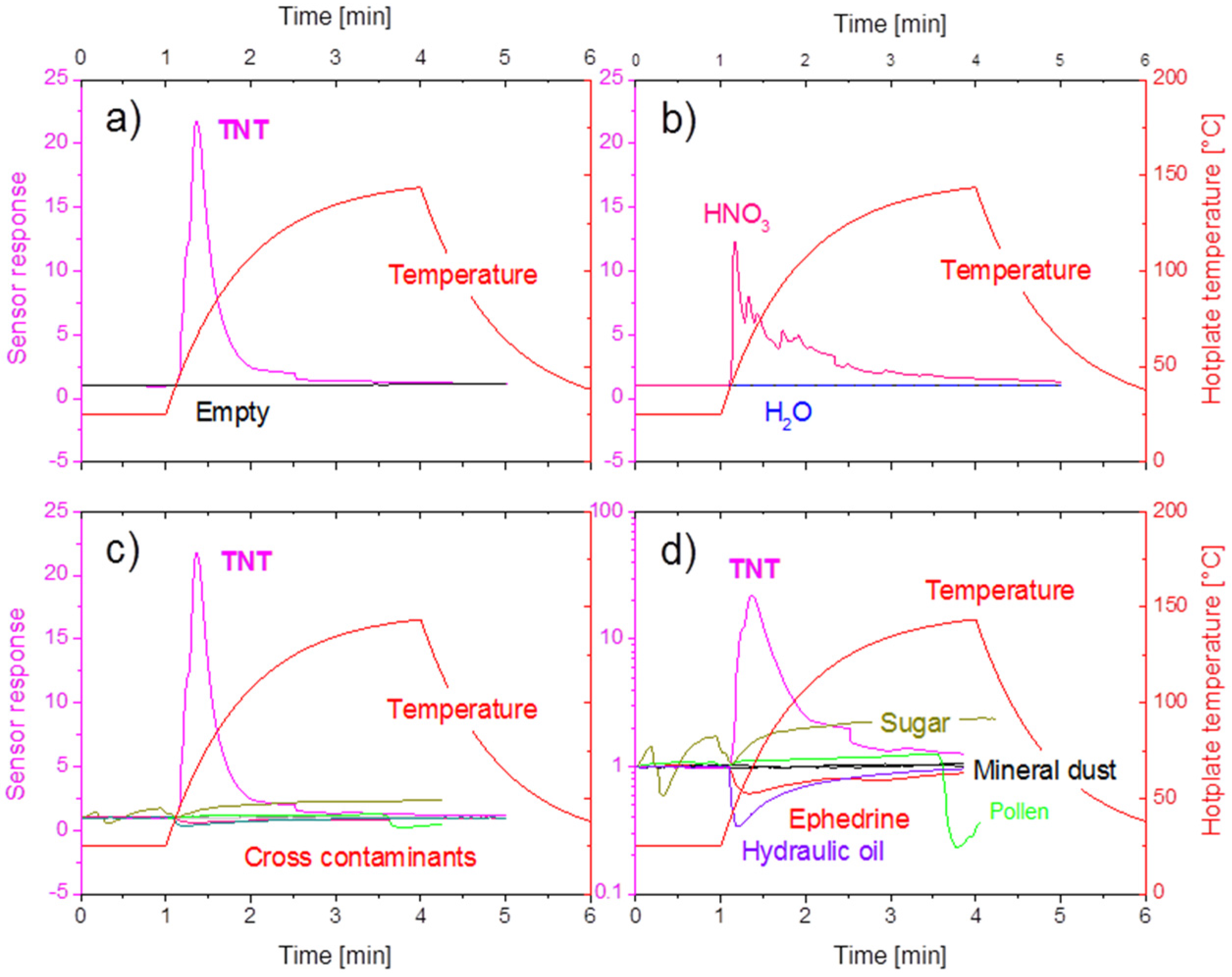
4.1. Feasibility of MOX Gas Sensors in the Field of Explosives Detection




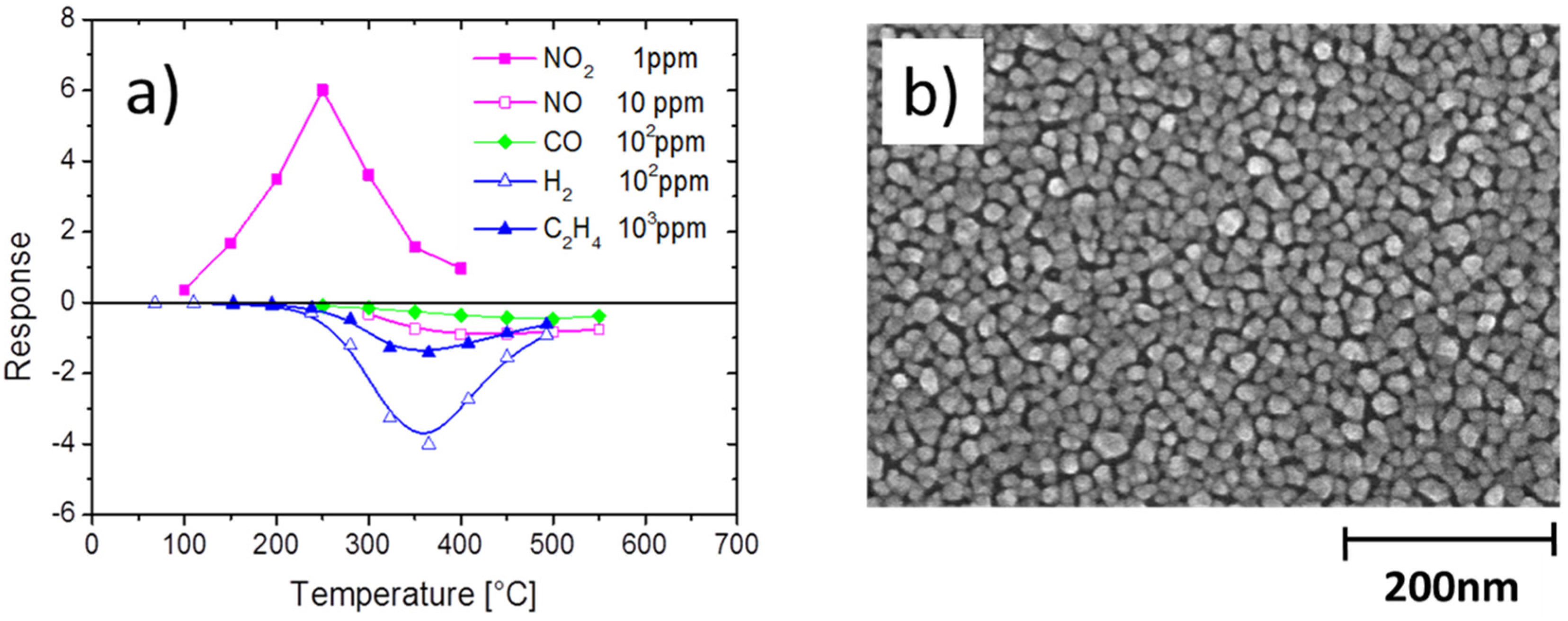

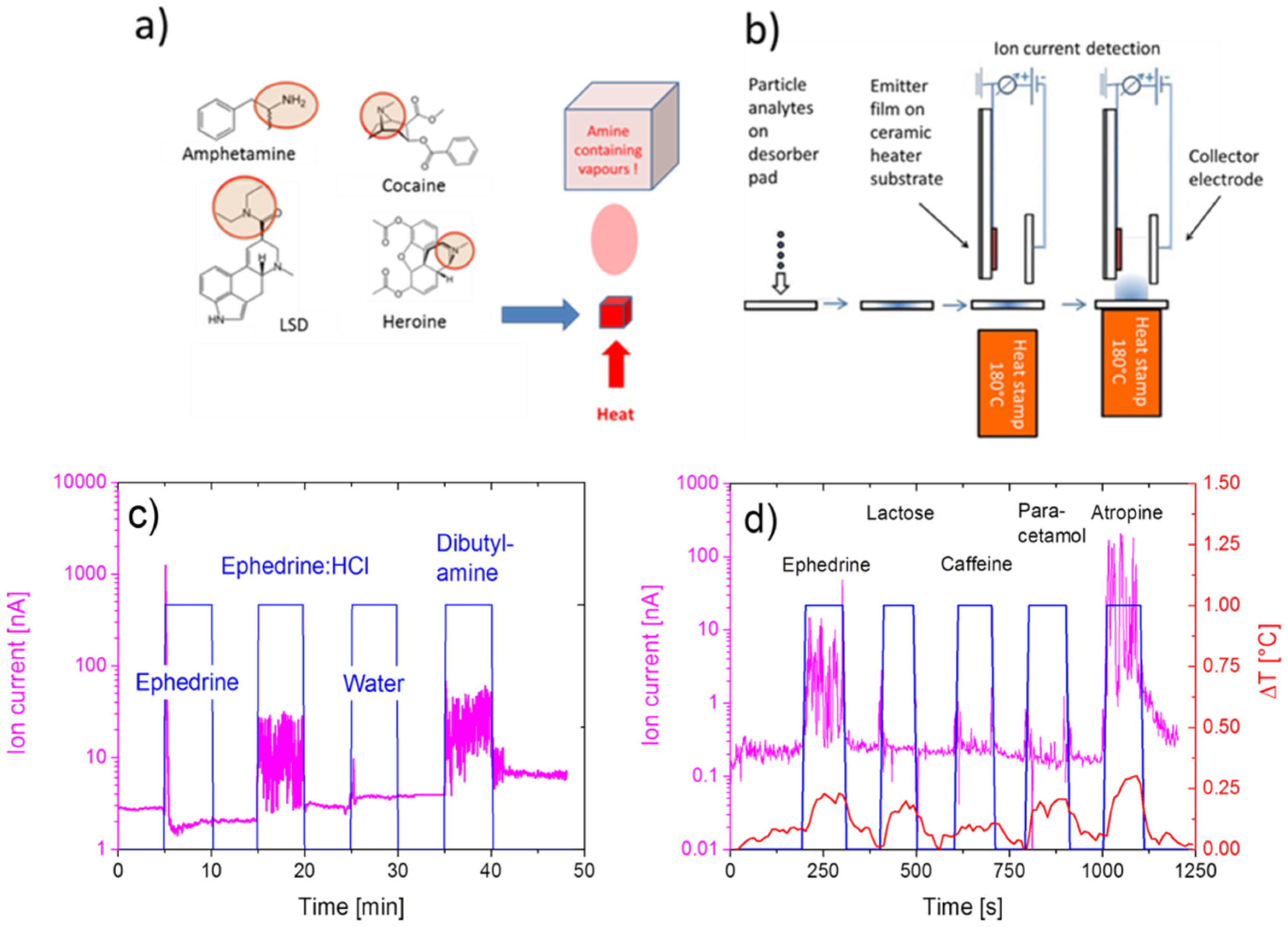
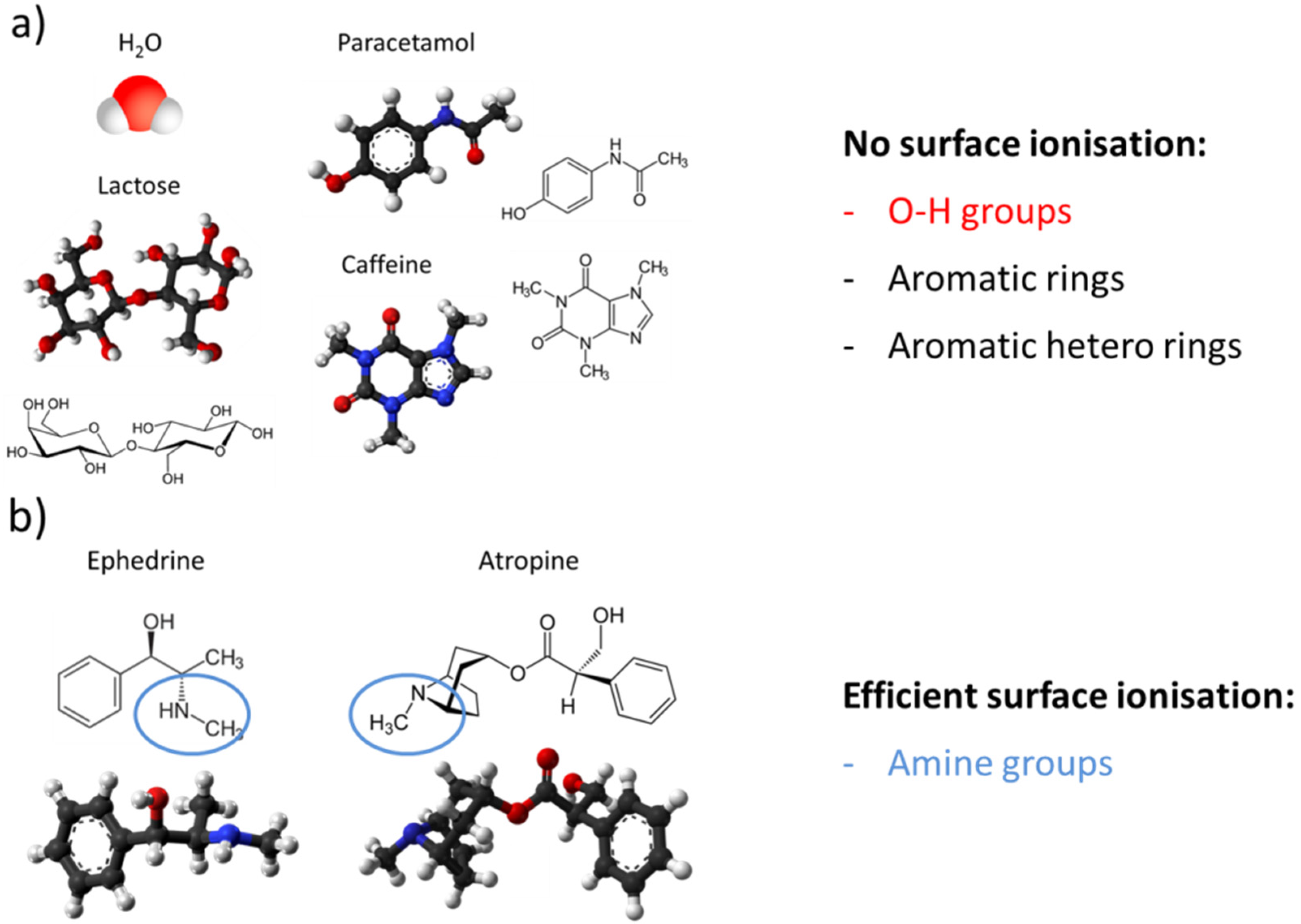
4.2. Feasibility of MOX Gas Sensors in the Field of Illicit Drug Detection





- Dissociative ionization: (M − H)+;
- Associative ionization: (M + H)+;
- Direct ionization: M+.

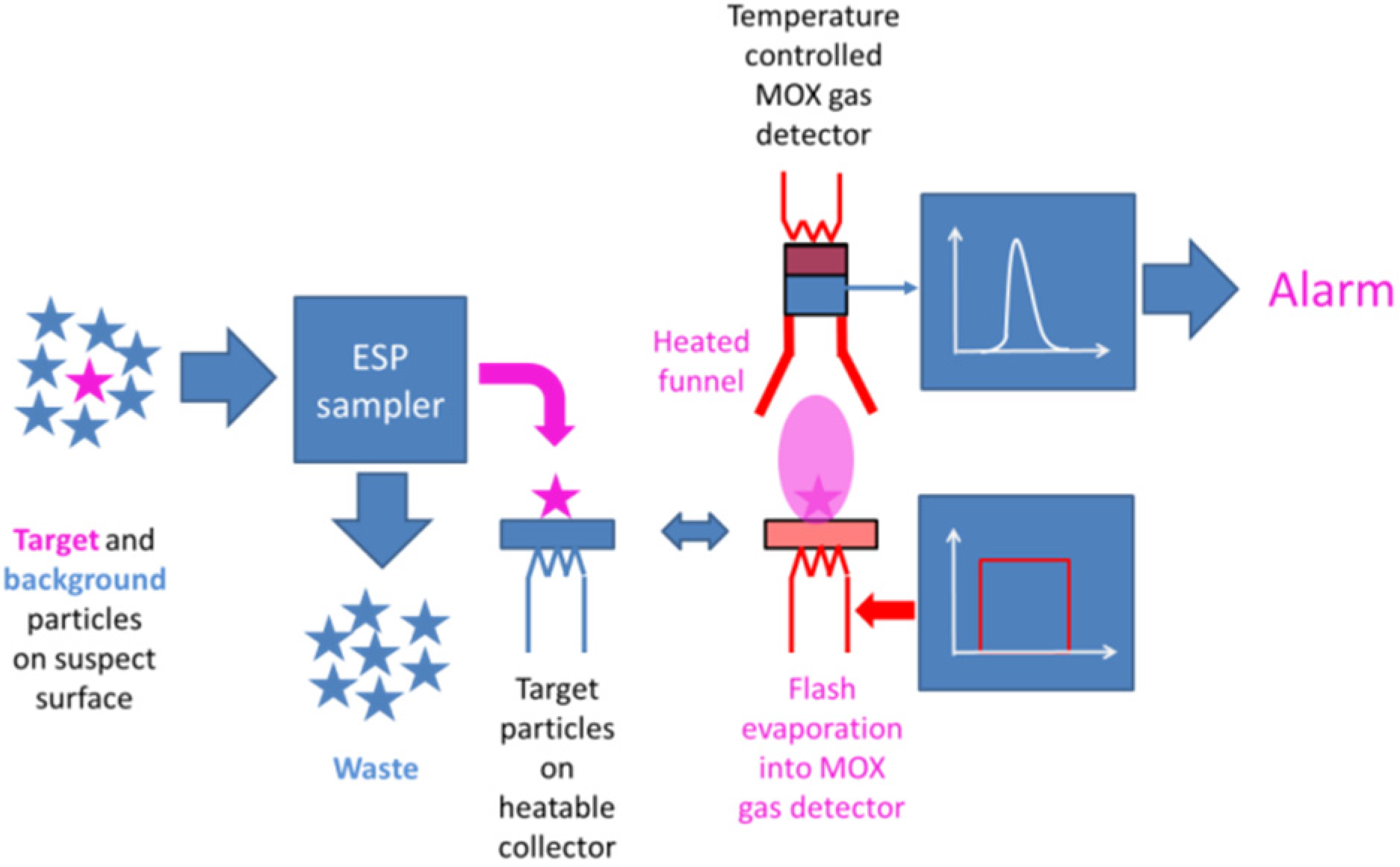
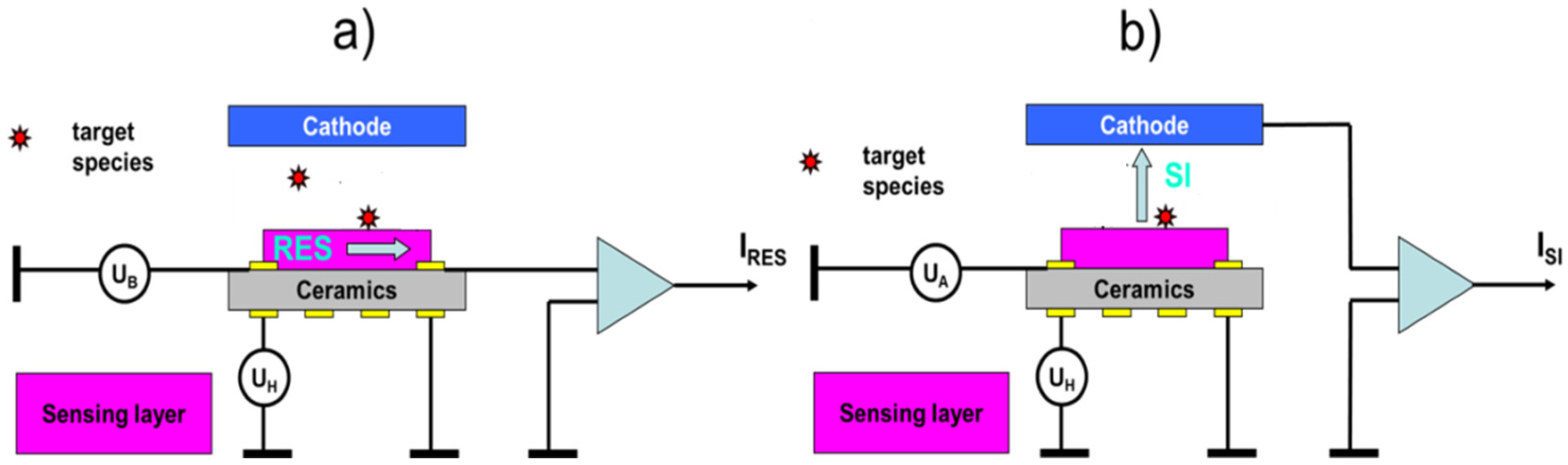
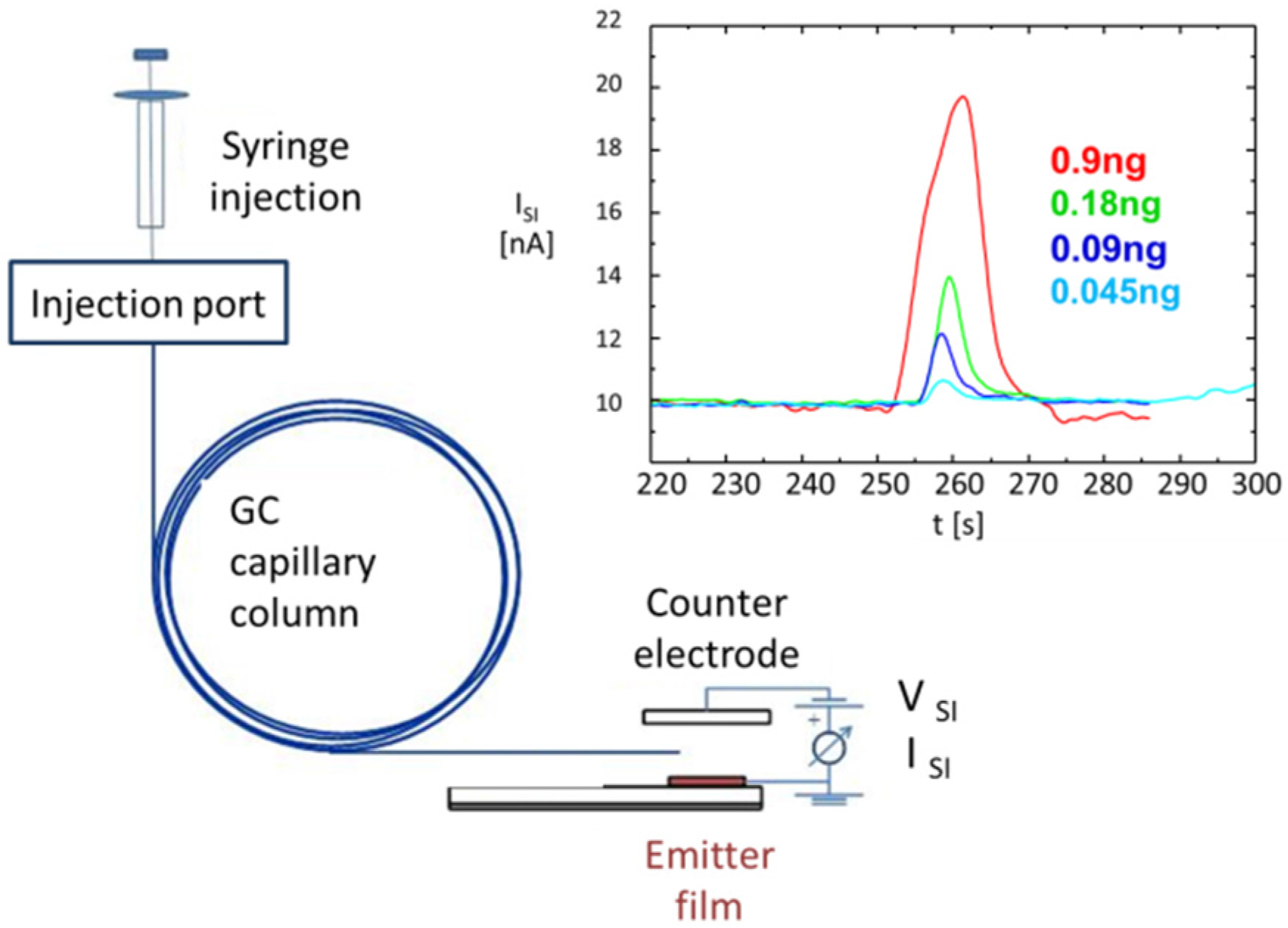
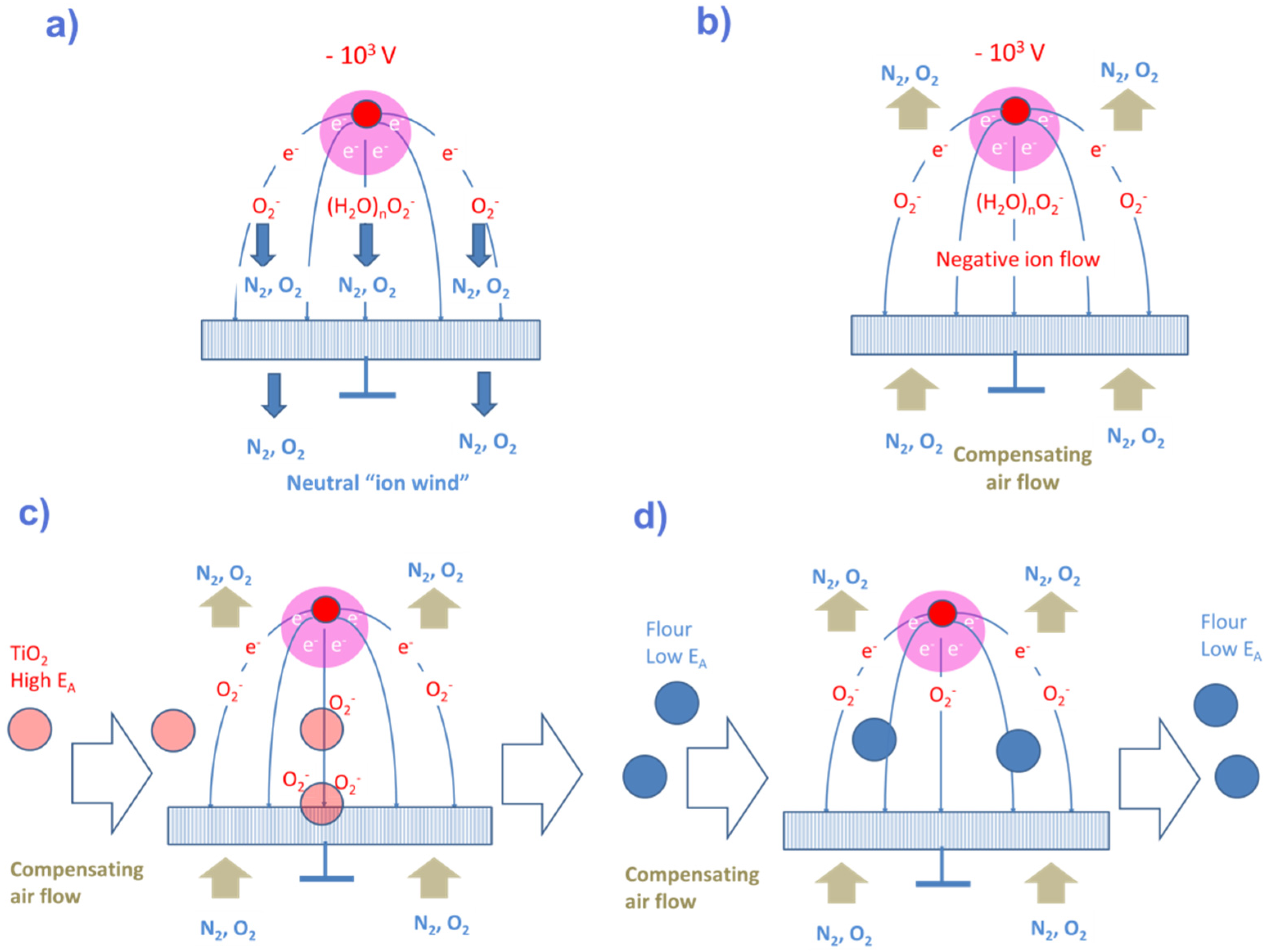
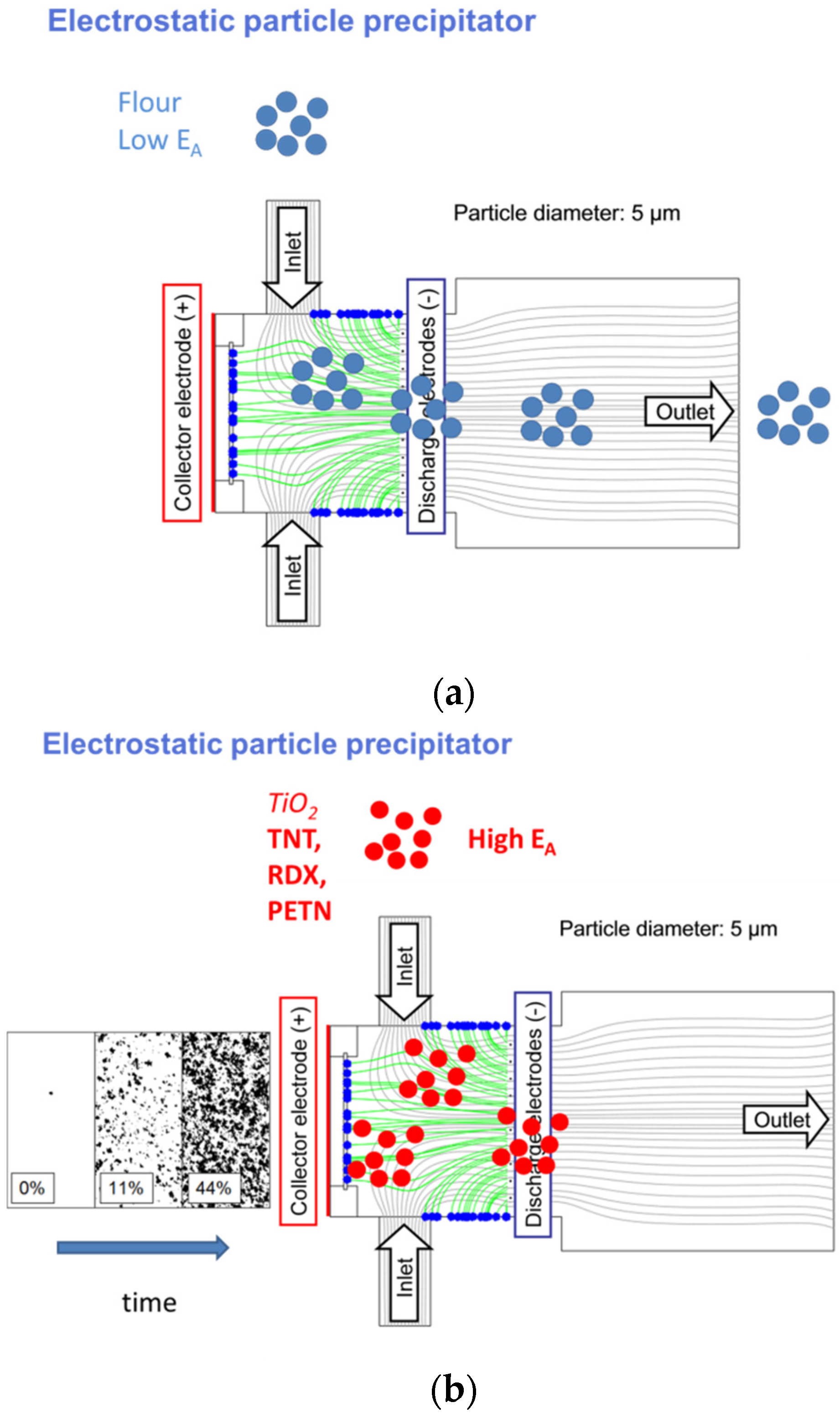
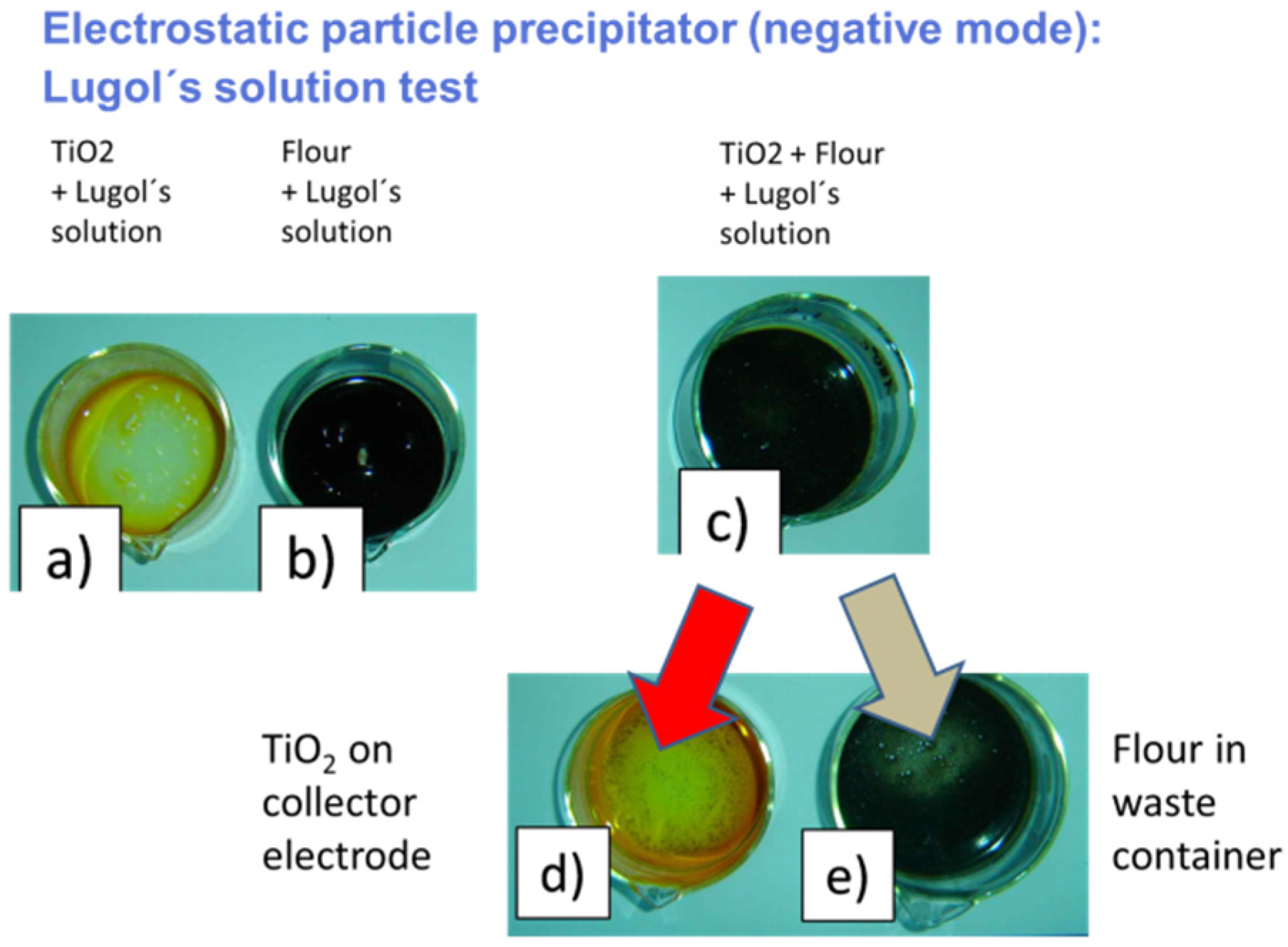
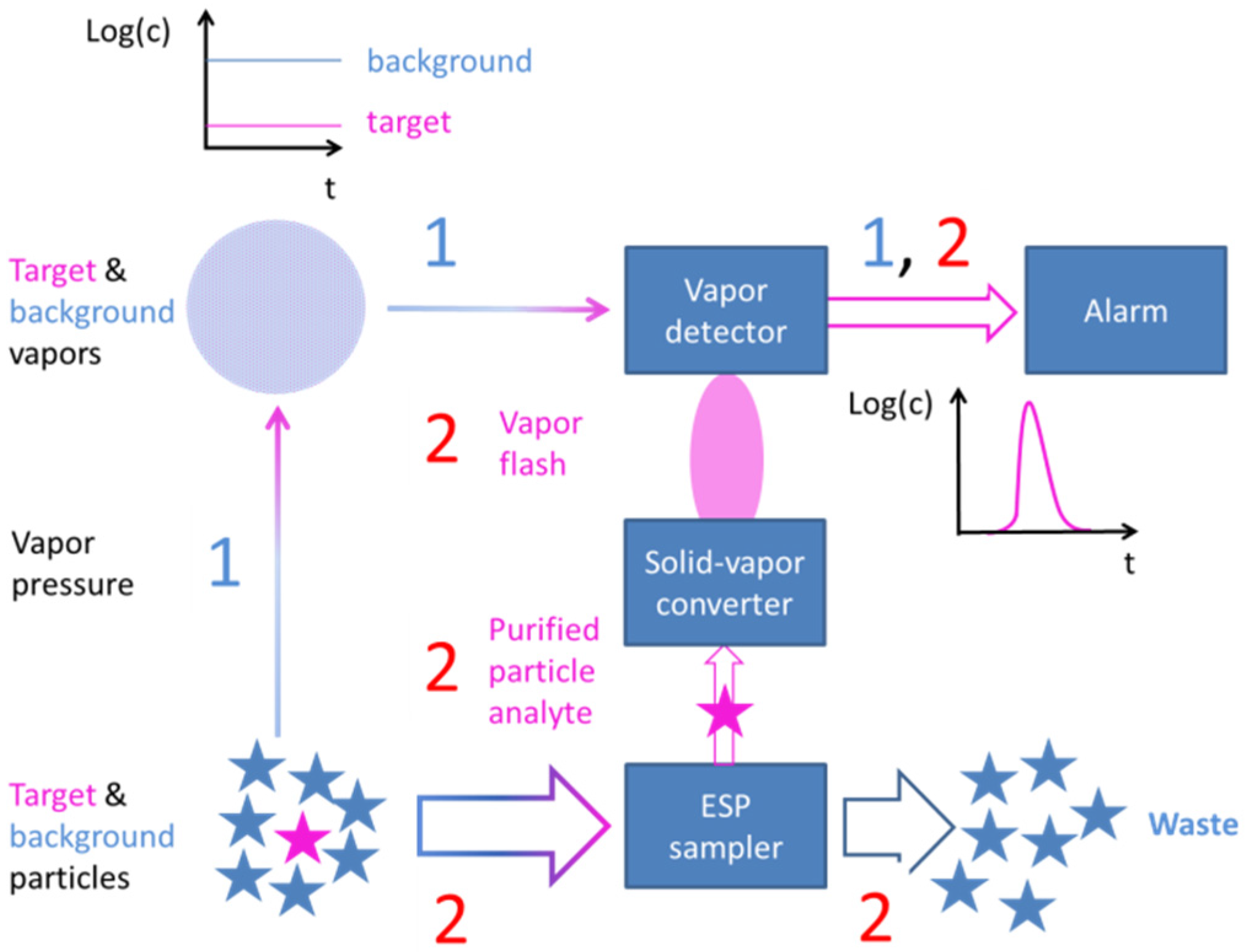
4.3. Selective Collection of Particle Residue Using Electrostatic Precipitation (ESP)




5. Achievements, Future Challenges and Outlook
5.1. Current Achievements
- Explosives and illicit drugs abound on suspect surfaces in the form of low vapor pressure solid particles. Both kinds of target substances are best detected by collecting particle residue from the suspect surfaces and by thermally converting it into detectable vapors.
- As particles can be rapidly collected from suspect surfaces and rapidly evaporated, particle collection and thermal vapor conversion ensure speed, which is an important requirement in many security screening scenarios. Further, as solid particle residue contains high densities of target molecules, flash evaporation can produce relatively intense bursts of target vapor with peak concentrations well above the lower limit of detection (LOD) of the backend vapor detectors. Particle collection and flash evaporation are therefore also key enablers for high sensitivity.
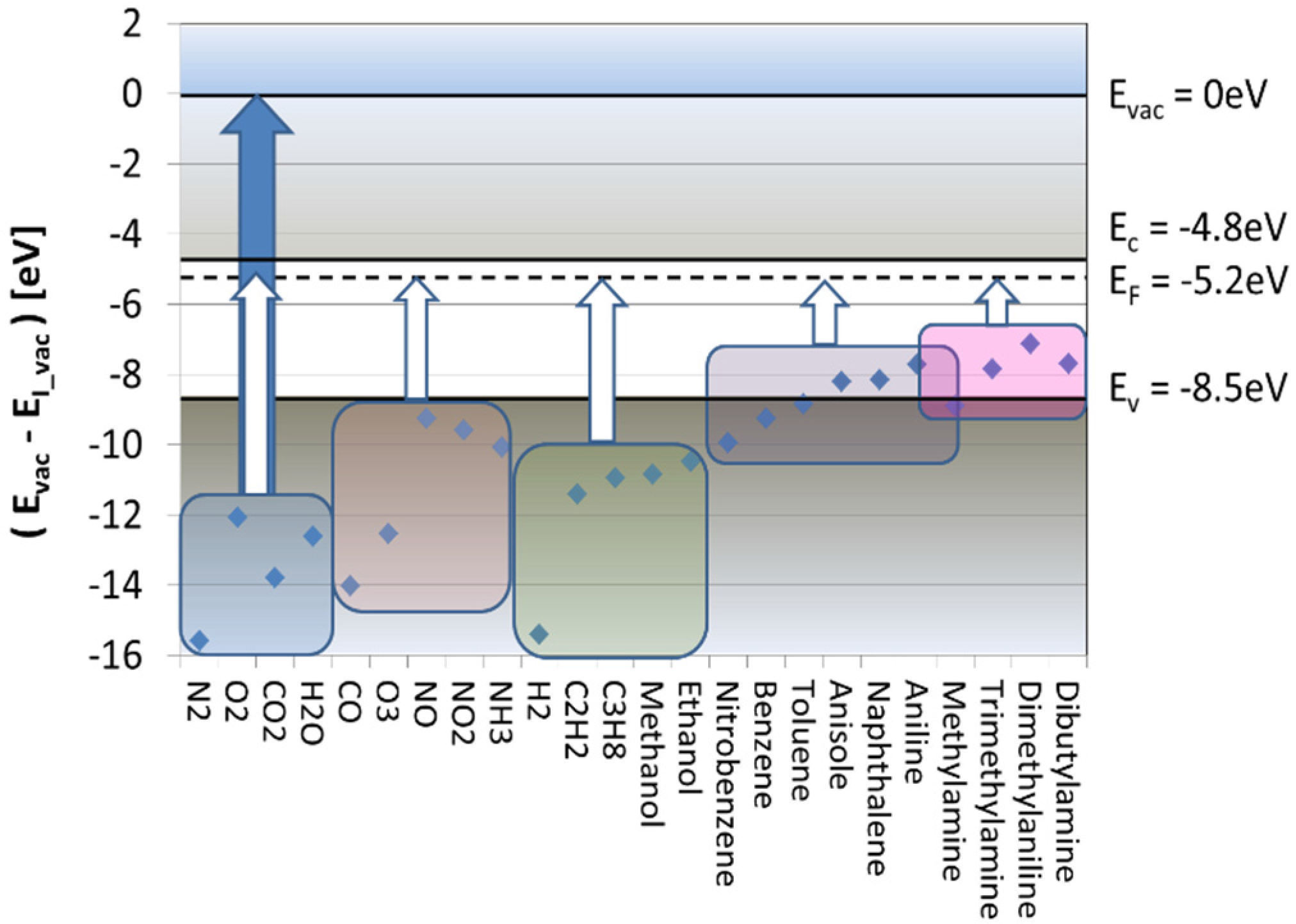
- Once vaporized and subjected to showers of electrons and protons, cascades of APCI processes selectively place the electron and proton charges on explosive and illicit drug molecules, as these are normally those components in the evaporated gas mix, which feature the highest electron (explosives) or proton (drugs) affinities. In this way, the target materials can be electrostatically separated from lower affinity background matter. APCI processes therefore are key enablers for attaining high selectivity towards explosives and illicit drugs.

5.2. Future Technology Enhancements
5.2.1. Electrostatic Particle Precipitators and Solid-Vapor Converters
5.2.2. Advanced Detector Technologies

Acknowledgments
- Snoopy “Sniffer for concealed people discovery”;
- Project Number: 313110, FP7-SEC-2012.3.4-4;
- Sniffer “A bio-mimicry enabled artificial sniffer”;
- Project Number: 285203, FP7-SEC-2011.3.4-2;
- Dirac “Portable system for rapid detection of illicit Drugs and key precursors by infrared Absorption spectroscopy and gas Chromatography”;
- Project Number: 242309, FP7-SEC-2009.1.3-2;
- S3 “Surface ionization and novel concepts in nano-MOX gas sensors with increased Selectivity, Sensitivity and Stability for detection of low concentrations of toxic and explosive agents”, Project Number: 247768, FP7-NMP-2009-1.2-3.
Author Contributions
Conflicts of Interest
References
- Chicago Project on Security and Terrorism, Suicide Attack Data Base. Available online: http://cpostdata.uchicago.edu/search_new.php (accessed on 21 August 2015).
- United Nations Office on Drugs and Crime (UNODC). Drug Trafficking. Available online: http://www.unodc.org/unodc/it/drug-trafficking/index.html (accessed on 21 August 2015).
- UNODC. World Drug Report 2010; United Nations Publications: New York, NY, USA, 2010. [Google Scholar]
- European Commission, Security. Available online: http://ec.europa.eu/programmes/horizon2020/en/area/security (accessed on 16 January 2016).
- European Commission, Community Research and Development Information Service. Available online: http://cordis.europa.eu/fp7/security/home_en.html (accessed on 16 January 2016).
- Practice. Preparedness and Resilience against CBRN Terrorism Using Integrated Concepts and Equipment Practice. Available online: https://www.cbrneltd.com/Downloads/PRACTICE_Project_Final_Report_CBRNE.pdf (accessed on 16 January 2016).
- CATO: CBRN Crisis Management, Architecture, Technologies and Operational Procedures. Available online: http://www.cato-project.eu/ (accessed on 16 January 2016).
- SNIFFER: A Bio-Mimicry Enabled Artificial Sniffer. Available online: http://www.sniffer-project.eu/ (accessed on 16 January 2016).
- DIRAC. Portable System for Rapid Detection of Illicit Drugs and Key Precursors by InfraRed Absorption Spectroscopy and Gas Chromatography. Available online: http://www.fp7-dirac.eu/ (accessed on 16 January 2016).
- BONAS BOmb Factory Detection by Networks of Advanced Sensors. Available online: http://cordis.europa.eu/project/rcn/98486_en.html (accessed on 16 January 2016).
- SNOOPY. Sniffer for Concealed People Discovery. Available online: http://www.snoopy-project.eu/ (accessed on 16 January 2016).
- Homeland Security Research. Available online: http://www2.epa.gov/homeland-security-research (accessed on 16 January 2016).
- Senesac, L.; Thundat, T.G. Nanosensors for trace explosive detection. Mater. Today 2008, 11, 28–36. [Google Scholar] [CrossRef]
- Camara, M.; Breuil, P.; Briand, D.; de Rooij, N.F.; Pijolat, C. A micro gas preconcentrator with improved performance for pollution monitoring and explosives detection. Anal. Chim. Acta 2011, 688, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Camara, M.; Rieu, M.; Breuil, P.; Pijolat, C.; Briand, D.; de Rooij, N.F. Gas preconcentrator made by rolling up a printed hotplate on foil. Procedia Eng. 2015, 120, 265–268. [Google Scholar] [CrossRef]
- Beer, S.; Müller, G.; Wöllenstein, J. Development and characterization of an electrostatic particle sampling system for the selective collection of trace explosives. Talanta 2012, 89, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Eiceman, G.A.; Karpas, Z. Ion Mobility Spectrometry, 2nd ed.; CRC Press Inc.: Boca Raton, FL, USA, 2005; ISBN 9780849322471. [Google Scholar]
- Richard, J.M.; Rowland, J.H., III; Harris, M.L.; Sapko, M.J. Behavior of Nitrogen Oxides in the Product Gases from Explosive Detonations. Available online: http://www.cdc.gov/niosh/mining/UserFiles/works/pdfs/bonoi.pdf (accessed on 16 January 2016).
- Cooper, J.K.; Grant, C.D.; Zhang, J.Z. Ab initio calculation of ionization potential and electron affinity of six common explosive compounds. Rep. Theor. Chem. 2012, 1, 11–19. [Google Scholar] [CrossRef]
- Trinitrotoluene. Available online: http://en.wikipedia.org/wiki/TNT_(explosive) (accessed on 24 August 2015).
- RDX. Available online: http://en.wikipedia.org/wiki/RDX (accessed on 24 August 2015).
- PET. Available online: http://en.wikipedia.org/wiki/PET (accessed on 24 August 2015).
- Ammonium Nitrate. Available online: http://en.wikipedia.org/wiki/Ammonium_nitrate (accessed on 24 August 2015).
- Urea Nitrate. Available online: http://en.wikipedia.org/wiki/Urea_nitrate (accessed on 24 August 2015).
- NIST. Chemistry WebBook. Available online: http://webbook.nist.gov/chemistry/ (accessed on 23 August 2015).
- Wu, T.Y. Electron Affinity of Boron, Carbon, Nitrogen, and Oxygen Atoms. Phys. Rev. 1955, 100, 1195–1196. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A. Compendium of Chemical Terminology: Iupac Recommendations: Gold Book; Blackwell Science: Hoboken, NJ, USA, 1997. [Google Scholar]
- Carroll, D.I.; Dzidic, I.; Stillwell, R.N.; Horning, M.G.; Horning, E.C. Subpicogram detection system for gas phase analysis based upon atmospheric pressure ionization (API) mass spectrometry. Anal. Chem. 1974, 46, 706–710. [Google Scholar] [CrossRef]
- Ecstasy. Available online: http://de.wikipedia.org/wiki/MDMA (accessed on 24 August 2015).
- Cocaine. Available online: http://en.wikipedia.org/wiki/Cocaine (accessed on 24 August 2015).
- Heroin. Available online: http://en.wikipedia.org/wiki/Heroin (accessed on 24 August 2015).
- Ephedrine. Available online: http://en.wikipedia.org/wiki/Ephedrine (accessed on 24 August 2015).
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; The “Gold Book”; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997; Available online: http://goldbook.iupac.org (accessed on 16 January 2016).
- Smiths Detection. Available online: http://www.smithsdetection.com/ (accessed on 24 August 2015).
- QS-H150 Handheld Explosives Trace Detector. Available online: http://www.implantsciences.com/products/handheld-explosives-trace-detector/ (accessed on 16 January 2016).
- Oberhüttinger, C.; Langmeier, A.; Oberpriller, H.; Kessler, M.; Göbel, J.; Müller, G. Hydrocarbon detection using laser ion mobility spectrometry. Int. J. Ion Mobil. Spectrom. 2009, 12, 23–32. [Google Scholar] [CrossRef]
- Sabo, M.; Páleník, J.; Kučera, M.; Han, H.; Wang, H.; Chu, Y.; Matejčík, Š. Atmospheric pressure corona discharge ionisation and ion mobility spectrometry/mass spectrometry study of the negative corona discharge in high purity oxygen and oxygen/nitrogen mixtures. Int. J. Mass Spectrom. 2010, 293, 23–27. [Google Scholar] [CrossRef]
- Ihokura, K.; Watson, J. The Stannic Oxide Gas Sensor Principles and Applications; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Korotcenkov, G.; Cho, B.K. Engineering approaches for the improvement of conductometric gas sensor parameters: Part 1. Improvement of sensor sensitivity and selectivity (short survey). Sens. Actuators B Chem. 2013, 188, 709–728. [Google Scholar] [CrossRef]
- Korotcenkov, G.; Cho, B.K. Engineering approaches to improvement of conductometric gas sensor parameters. Part 2: Decrease of dissipated (consumable) power and improvement stability and reliability. Sens. Actuators B Chem. 2014, 198, 316–341. [Google Scholar] [CrossRef]
- Comini, E.; Faglia, G.; Sberveglieri, G. Solid State Gas Sensing, 1st ed.; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Das, S.; Jayaraman, V. SnO2: A comprehensive review on structures and gas sensors. Prog. Mater. Sci. 2014, 66, 112–255. [Google Scholar] [CrossRef]
- Ruhland, B.; Becker, T.; Müller, G. Gas-kinetic interactions of nitrous oxides with SnO2 surfaces. Sens. Actuators B Chem. 1998, 50, 85–94. [Google Scholar] [CrossRef]
- Müller, G.; Friedberger, A.; Kreisl, P.; Ahlers, S.; Schulz, O.; Becker, T. A MEMS toolkit for metal-oxide-based gas sensing systems. Thin Solid Films 2003, 436, 34–45. [Google Scholar] [CrossRef]
- Pearce, T.C.; Schiffman, S.S.; Nagle, H.T.; Gardner, J.W. Handbook of Machine Olfaction: Electronic Nose Technology; Wiley-VCH: Hoboken, NJ, USA, 2003; p. 324. [Google Scholar]
- Maurer, S.; Makarov, R.; Holl, G.; Kaul, P. Heterogenes Sensorsystem Zum Nachweis von Explosivstoff-Typischen Merkmalen Durch Thermische Aktivierung. In Proceedings of the Dresdner Sensor Symposium, Dresden, Germany, 7–9 December 2015; Available online: https://www.researchgate.net/publication/286453345_Heterogenes_Sensorsystem_zum_Nachweis_von_Explosivstoff-typischen_Merkmalen_durch_thermische_Aktivierung (accessed on 16 January 2016).
- Oberhüttinger, C.; Hackner, A.; Müller, G.; Stutzmann, M. On the temperature dependence of the resistive and surface ionisation response of SnO2 gas sensing layers. Sens. Actuators B Chem. 2011, 156, 563–571. [Google Scholar] [CrossRef]
- Oberhüttinger, C.; Habauzit, A.; Hackner, A.; Müller, G. A rate equation approach towards surface ionisation gas detection. Sens. Actuators B Chem. 2011, 160, 981–990. [Google Scholar] [CrossRef]
- Hackner, A.; Beer, S.; Müller, G.; Fischer, T.; Mathur, S. Surface ionization detection of amphetamine-type illicit drugs. Sens. Actuators B Chem. 2012, 162, 209–215. [Google Scholar] [CrossRef]
- Hackner, A.; Legner, W.; Müller, G.; Biavardi, E.; Dalcanale, E.; Zampolli, S.; Elmi, I.; Cardinali, G.C. Surface ionization detection of amine containing drugs. Sens. Actuators B Chem. 2013, 185, 771–776. [Google Scholar] [CrossRef]
- Hackner, A.; Bouxin, B.; Müller, G. Surface ionisation gas detection: Vertical versus planar readout modes. Sens. Actuators B Chem. 2013, 188, 286–292. [Google Scholar] [CrossRef]
- Hellmich, W.; Müller, G.; Doll, T.; Eisele, I. Field-effect-induced gas sensitivity changes in metal oxides. Sens. Actuators B Chem. 1997, 43, 132–139. [Google Scholar] [CrossRef]
- Lactose. Available online: https://en.wikipedia.org/wiki/Lactose (accessed on 16 January 2016).
- Caffeine. Available online: https://en.wikipedia.org/wiki/Caffeine (accessed on 16 January 2016).
- Paracetamol. Available online: https://en.wikipedia.org/wiki/Paracetamol (accessed on 16 January 2016).
- Atropine. Available online: https://en.wikipedia.org/wiki/Atropine (accessed on 16 January 2016).
- Fujii, T.; Kitai, T. Surface ionization mass spectrometry of organic compounds. I. Nitrogen-containing aliphatic organic compounds. Int. J. Mass Specrtom. Ion Process. 1986, 71, 129–140. [Google Scholar] [CrossRef]
- Fujii, T.; Kakizaki, K.; Mitsutsuka, Y. Surface ionization mass spectrometry of organic compounds Part 4. Oxygen-containing organic compounds. Int. J. Mass Spectrom. Ion Process. 1991, 104, 129–136. [Google Scholar] [CrossRef]
- Loeb, L.B. Electrical Coronas: Their Basic Physical Mechanisms; University of California Press: Oakland, CA, USA, 1965. [Google Scholar]
- Parker, K.R. Applied Electrostatic Precipitation, 1st ed.; Springer Science & Business Media: Berlin, Germany; Heidelberg, Germany, 1997. [Google Scholar]
- Electrostatic precipitator. Available online: https://en.wikipedia.org/wiki/Electrostatic_precipitator (accessed on 16 January 2016).
- Beer, S. Development of a selective electrostatic sampling system for the detection of trace explosives and illicit drugs. Ph.D. Thesis, Microsystem Simulation, Design and Manufacture, IMTEK Freiburg, Der Andere Verlag, Uelvesbüll, Germany, 2013. [Google Scholar]
- Manai, R.; Scorsone, E.; Rousseau, L.; Ghassemi, F.; Possas Abreu, M.; Lissorgues, G.; Tremillon, N.; Ginisty, H.; Arnault, J.C.; Tuccori, E.; et al. Grafting odorant binding proteins on diamond bio-MEMS. Biosens. Bioelectron. 2014, 60, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Spannhake, J.; Helwig, A.; Schulz, O.; Müller, G. Micro-Fabrication of Gas Sensors. In Solid State Gas Sensing; Comini, E., Faglia, G., Sberveglieri, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 1–46. [Google Scholar]
- Hildenbrand, J.; Korvink, J.; Wollenstein, J.; Peter, C.; Kurzinger, A.; Naumann, F.; Ebert, M.; Lamprecht, F. Micromachined mid-infrared emitter for fast transient temperature operation for optical gas sensing systems. IEEE Sens. J. 2010, 10, 353–362. [Google Scholar] [CrossRef]
- Barros, T. Investigation of Corona Discharges for the Preconcentration of Trace Amounts of Explosives and Narcotics. Master’s Thesis, Munich University of Applied Sciences, Munich, Germany, 2013. [Google Scholar]
- Schedin, F.; Geim, A.K.; Morozov, S.V.; Hill, E.W.; Blake, P.; Katsnelson, M.I.; Novoselov, K.S. Detection of individual gas molecules adsorbed on graphene. Nat. Mater. 2007, 6, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.; Sudhölter, E.J.R.; de Smet, L.C.P.M. Silicon nanowire-based devices for gas-phase sensing. Sensors 2014, 14, 245–271. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, P.; Kimoto, T.; Ley, L.; Pensl, G. Silicon Carbide: Volume 2: Power Devices and Sensors; Wiley-VCH: Hoboken, NJ, USA, 2011. [Google Scholar]
- Bogue, R. Diamond sensors: The dawn of a new era? Sens. Rev. 2007, 27, 288–290. [Google Scholar]
- Ambacher, O.; Eickhoff, M.; Link, A.; Hermann, M.; Stutzmann, M.; Bernardini, F.; Fiorentini, V.; Smorchkova, Y.; Speck, J.; Mishra, U.; et al. Electronics and sensors based on pyroelectric AlGaN/GaN heterostructures. Phys. Status Solidi 2003, 0, 1878–1907. [Google Scholar] [CrossRef]
- Anderson, T.; Ren, F.; Pearton, S.; Kang, B.S.; Wang, H.T.; Chang, C.Y.; Lin, J. Advances in hydrogen, carbon dioxide, and hydrocarbon gas sensor technology using GaN and ZnO-based devices. Sensors 2009, 9, 4669–4694. [Google Scholar] [CrossRef] [PubMed]
- Vitushinsky, R.; Crego-Calama, M.; Brongersma, S.H.; Offermans, P. Enhanced detection of NO2 with recessed AlGaN/GaN open gate structures. Appl. Phys. Lett. 2013, 102, 172101. [Google Scholar] [CrossRef]
- Härtl, A.; Schmich, E.; Garrido, J.A.; Hernando, J.; Catharino, S.C.R.; Walter, S.; Feulner, P.; Kromka, A.; Steinmüller, D.; Stutzmann, M. Protein-modified nanocrystalline diamond thin films for biosensor applications. Nat. Mater. 2004, 3, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, G.; Hermann, M.; Schaff, W.J.; Eastman, L.F.; Stutzmann, M.; Eickhoff, M. pH response of GaN surfaces and its application for pH-sensitive field-effect transistors. Appl. Phys. Lett. 2003, 83, 177–179. [Google Scholar] [CrossRef]
- Stutzmann, M.; Garrido, J.A.; Eickhoff, M.; Brandt, M.S. Direct biofunctionalization of semiconductors: A survey. Phys. Status Solidi 2006, 203, 3424–3437. [Google Scholar] [CrossRef]
- Mitra, T.; Jelfs, K.E.; Schmidtmann, M.; Ahmed, A.; Chong, S.Y.; Adams, D.J.; Cooper, A.I. Molecular shape sorting using molecular organic cages. Nat. Chem. 2013, 5, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Younus, H.A.; Chughtai, A.H.; Verpoort, F. Metal-organic molecular cages: Applications of biochemical implications. Chem. Soc. Rev. 2015, 44, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Reiss, P.S.; Chong, S.Y.; Holden, D.; Jelfs, K.E.; Hasell, T.; Little, M.A.; Kewley, A.; Briggs, M.E.; Stephenson, A.; et al. Separation of rare gases and chiral molecules by selective binding in porous organic cages. Nat. Mater. 2014, 13, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Slater, A.G.; Cooper, A.I. Function-led design of new porous materials. Science 2015, 348. [Google Scholar] [CrossRef] [PubMed]
- Mulla, M.Y.; Tuccori, E.; Magliulo, M.; Lattanzi, G.; Palazzo, G.; Persaud, K.; Torsi, L. Capacitance-modulated transistor detects odorant binding protein chiral interactions. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Wang, Z.; Li, Y.; Liu, L. A microcalorimeter integrated with carbon nanotube interface layers for fast detection of trace energetic chemicals. J. Microelectromech. Syst. 2013, 22, 152–162. [Google Scholar] [CrossRef]
- Thundat, T.; Chen, G.Y.; Warmack, R.J.; Allison, D.P.; Wachter, E.A. Vapor detection using resonating microcantilevers. Anal. Chem. 1995, 67, 519–521. [Google Scholar] [CrossRef]
- Zuck, A.; Greenblatt, J.; Zifman, A.; Zaltsman, A.; Kendler, S.; Frishman, G.; Meltzer, S.; Fisher, I. Explosive detection by microthermal analysis. J. Energ. Mater. 2008, 26, 163–180. [Google Scholar] [CrossRef]
- Khan, M.F.; Kim, S.; Lee, D.; Schmid, S.; Boisen, A.; Thundat, T. Nanomechanical identification of liquid reagents in a microfluidic channel. Lab Chip 2014, 14, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Zampolli, S.; Elmi, I.; Mancarella, F.; Betti, P.; Dalcanale, E.; Cardinali, G.C.; Severi, M. Real-time monitoring of sub-ppb concentrations of aromatic volatiles with a MEMS-enabled miniaturized gas-chromatograph. Sens. Actuators B Chem. 2009, 141, 322–328. [Google Scholar] [CrossRef]
- Garg, A.; Akbar, M.; Vejerano, E.; Narayanan, S.; Nazhandali, L.; Marr, L.C.; Agah, M. Zebra GC: A mini gas chromatography system for trace-level determination of hazardous air pollutants. Sens. Actuators B Chem. 2015, 212, 145–154. [Google Scholar] [CrossRef]
- Akbar, M.; Shakeel, H.; Agah, M. GC-on-chip: Integrated column and photoionization detector. Lab Chip 2015, 15, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Rice, G.; Agah, M. A micro-discharge photoionization detector for micro-gas chromatography. Microchim. Acta 2013, 181, 493–499. [Google Scholar] [CrossRef]
- Rogers, B.; Malekos, S.; Deal, L.; Whitten, R.; Adams, J. Combined, solid-state molecular property and gamma spectrometers for CBRN & E detection. In Proceedings of the 2013 IEEE International Conference on Technologies for Homeland Security (HST), Waltham, MA, USA, 12–14 November 2013; pp. 607–612.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, G.; Hackner, A.; Beer, S.; Göbel, J. Solid-State Gas Sensors: Sensor System Challenges in the Civil Security Domain. Materials 2016, 9, 65. https://doi.org/10.3390/ma9010065
Müller G, Hackner A, Beer S, Göbel J. Solid-State Gas Sensors: Sensor System Challenges in the Civil Security Domain. Materials. 2016; 9(1):65. https://doi.org/10.3390/ma9010065
Chicago/Turabian StyleMüller, Gerhard, Angelika Hackner, Sebastian Beer, and Johann Göbel. 2016. "Solid-State Gas Sensors: Sensor System Challenges in the Civil Security Domain" Materials 9, no. 1: 65. https://doi.org/10.3390/ma9010065
APA StyleMüller, G., Hackner, A., Beer, S., & Göbel, J. (2016). Solid-State Gas Sensors: Sensor System Challenges in the Civil Security Domain. Materials, 9(1), 65. https://doi.org/10.3390/ma9010065