
A Review on Grafting of Biofibers for Biocomposites

Abstract
:1. Introduction
2. Biofibers
2.1. Lignocellulosic Biofibers
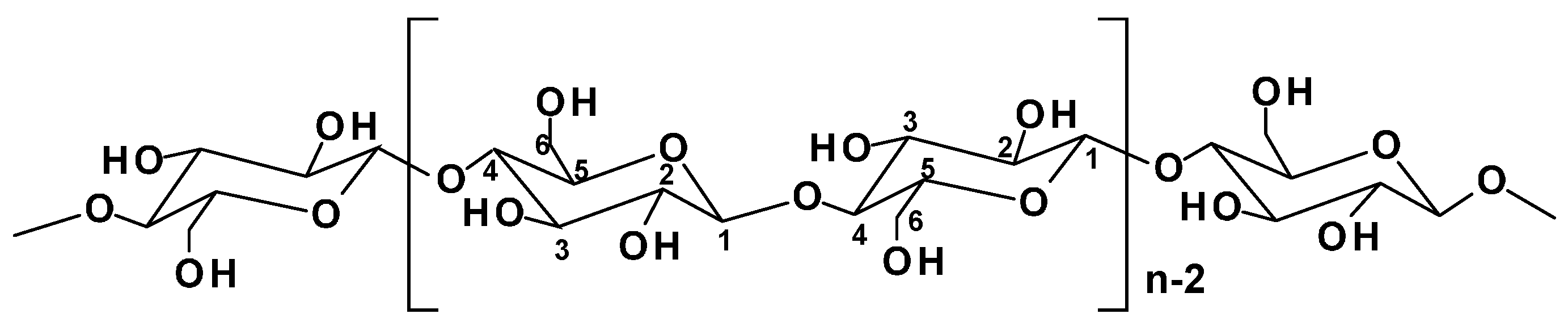
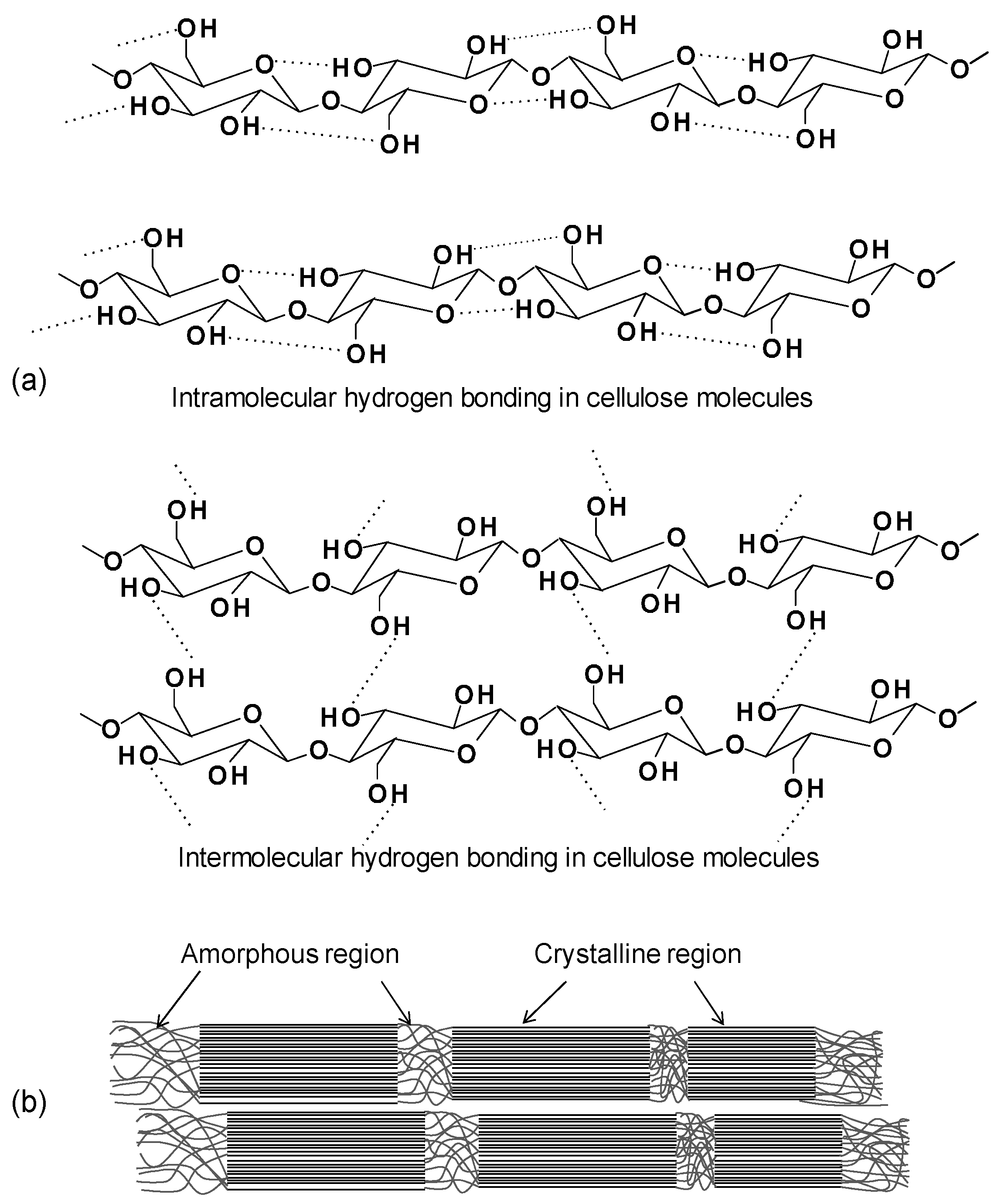
2.1.1. Cellulose
2.1.2. Nanocellulose
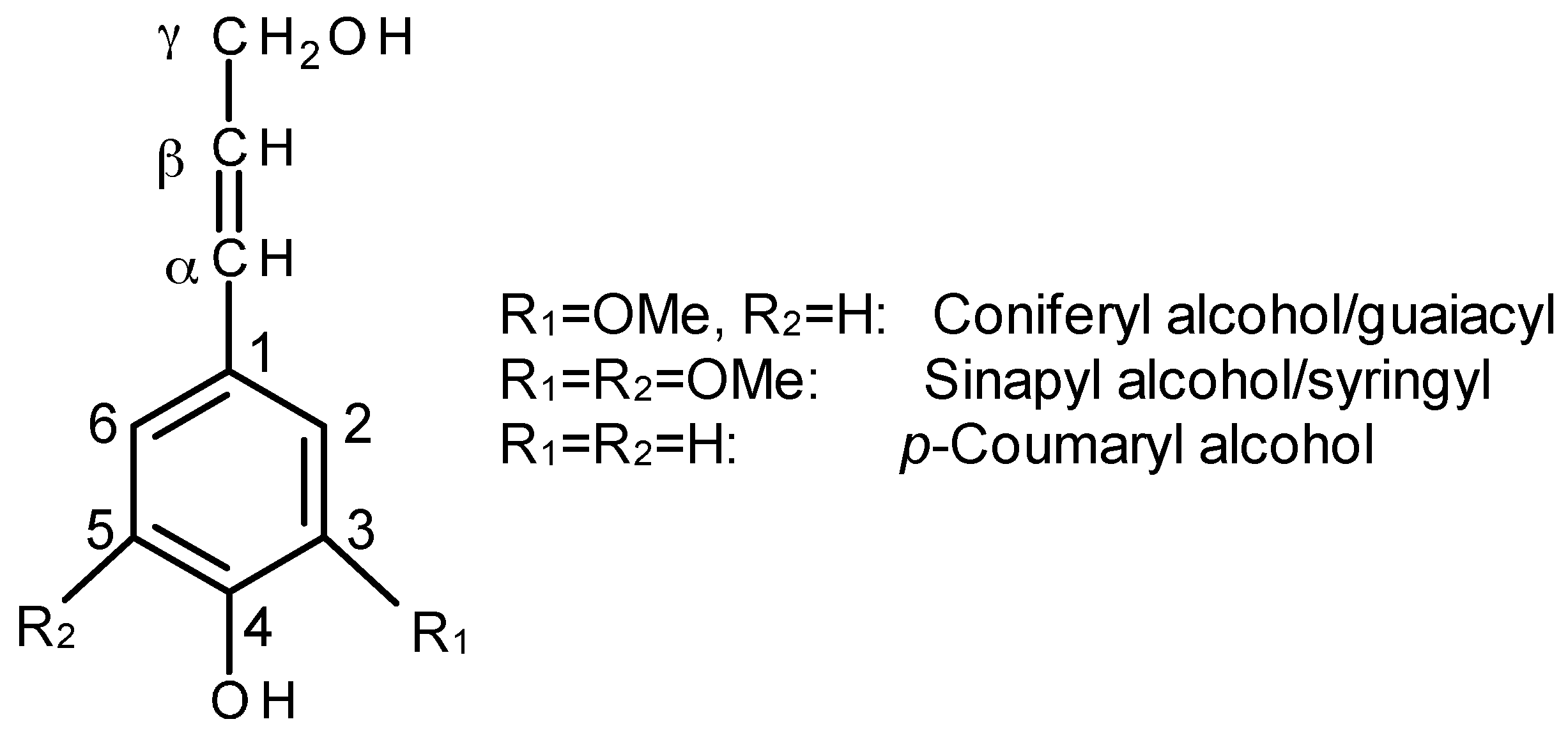
2.1.3. Lignin
2.2. Bacterial Cellulose
2.3. Biofibers from Other Sources
3. Biocomposites
3.1. Matrix Materials
3.1.1. Petroleum Based Polymer Matrix
3.1.2. Bio-Based Polymer Matrix
3.2. Green Composites
3.2.1. PHA Based Biocomposites
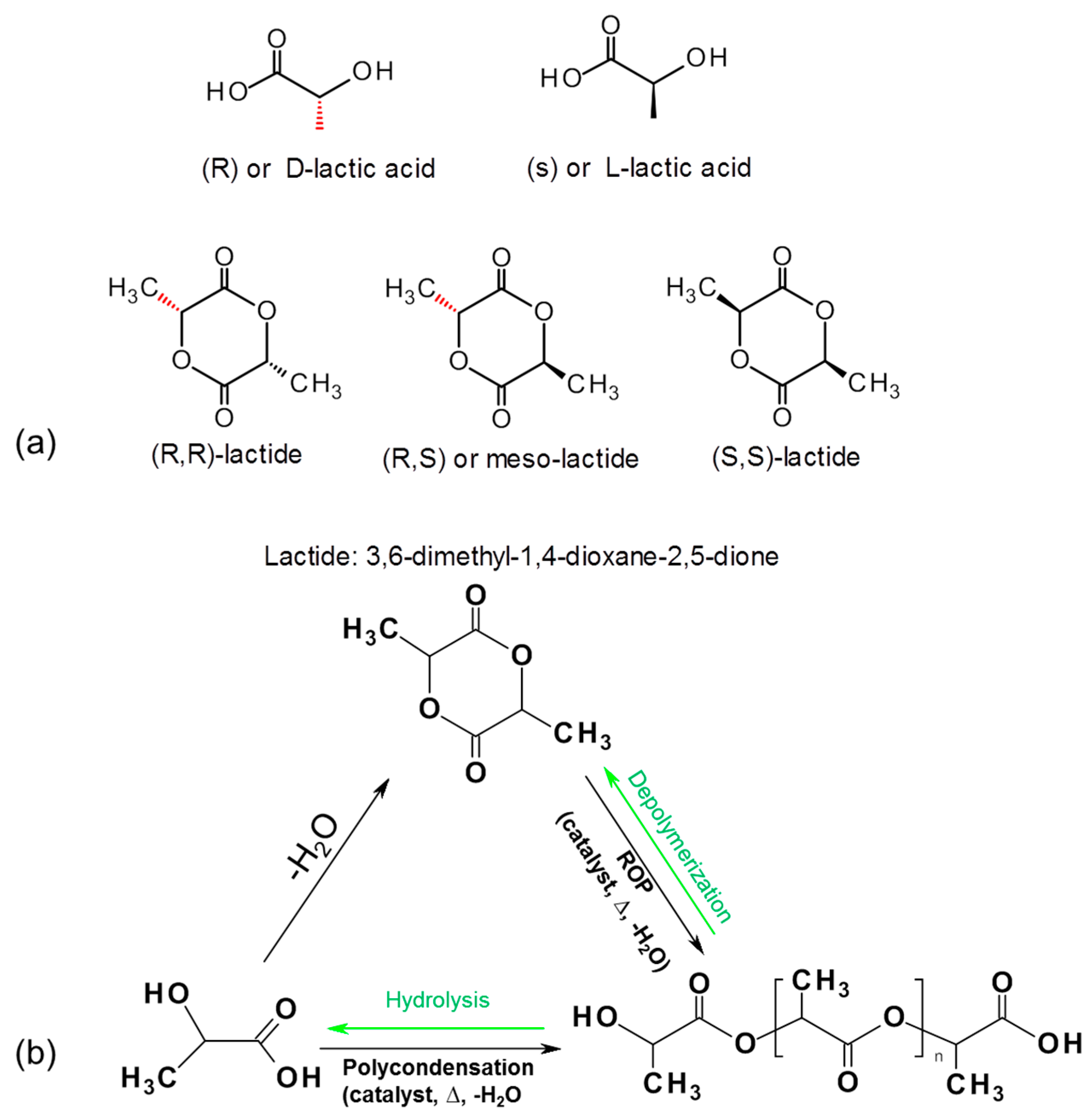
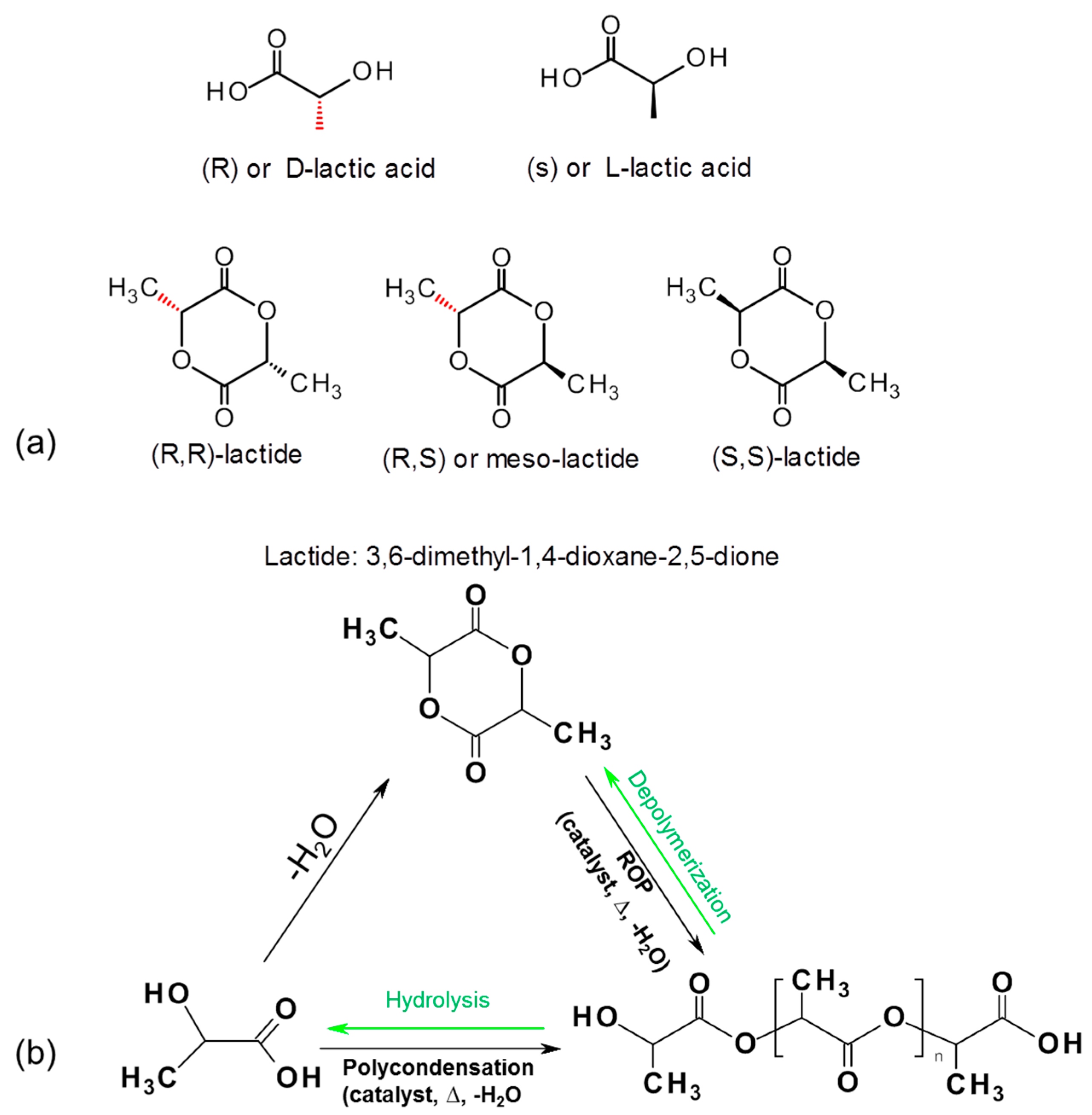
3.2.2. PLA Based Biocomposites
3.2.3. Other Commonly Studied Biocomposites
3.2.4. Approaches to Improve the Performance of Biocomposites by Modifications
4. Grafting Modifications of Biofibers for Biocomposites
4.1. Grafting Techniques
4.2. Grafting via Living Polymerizaiton Technique
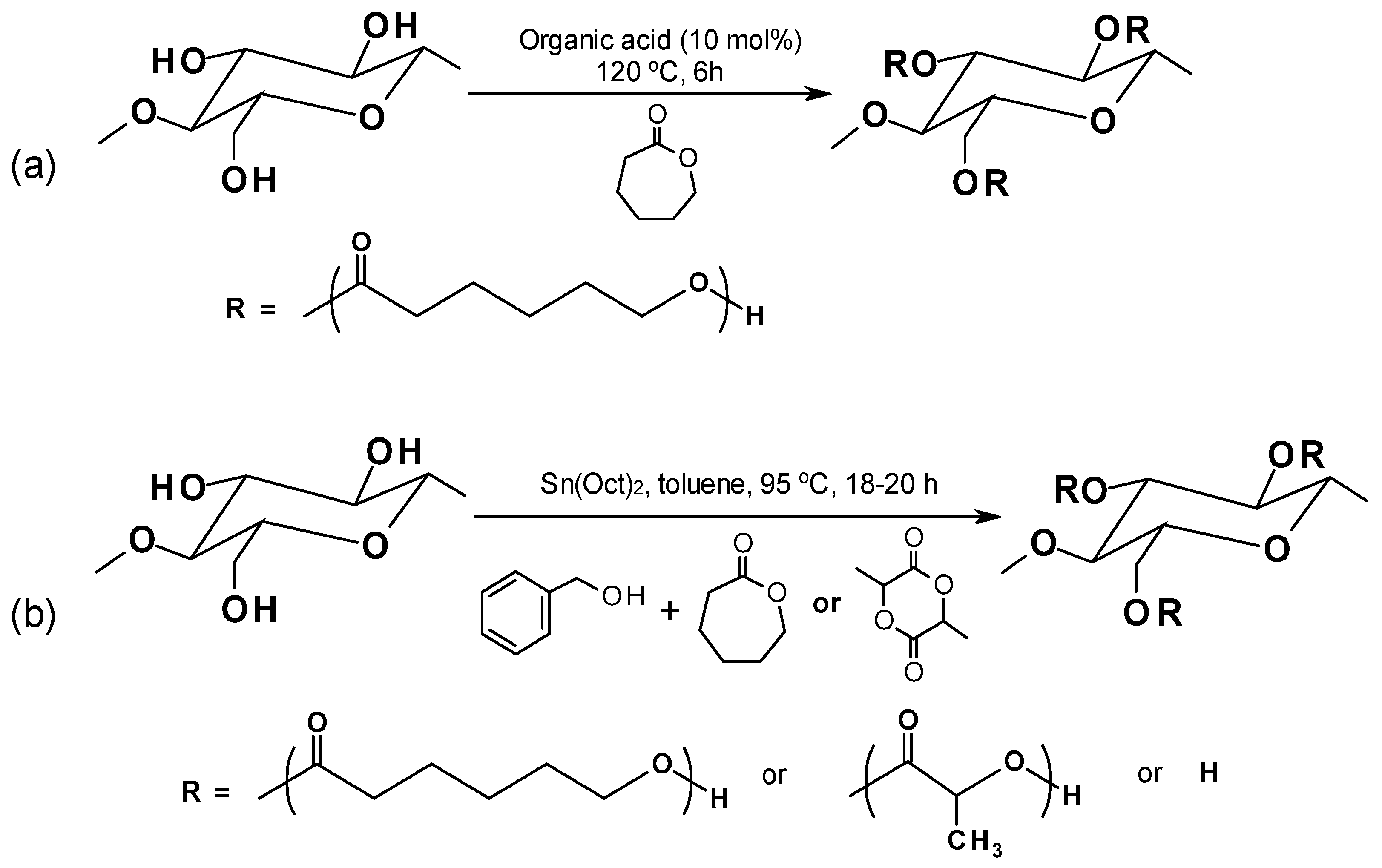
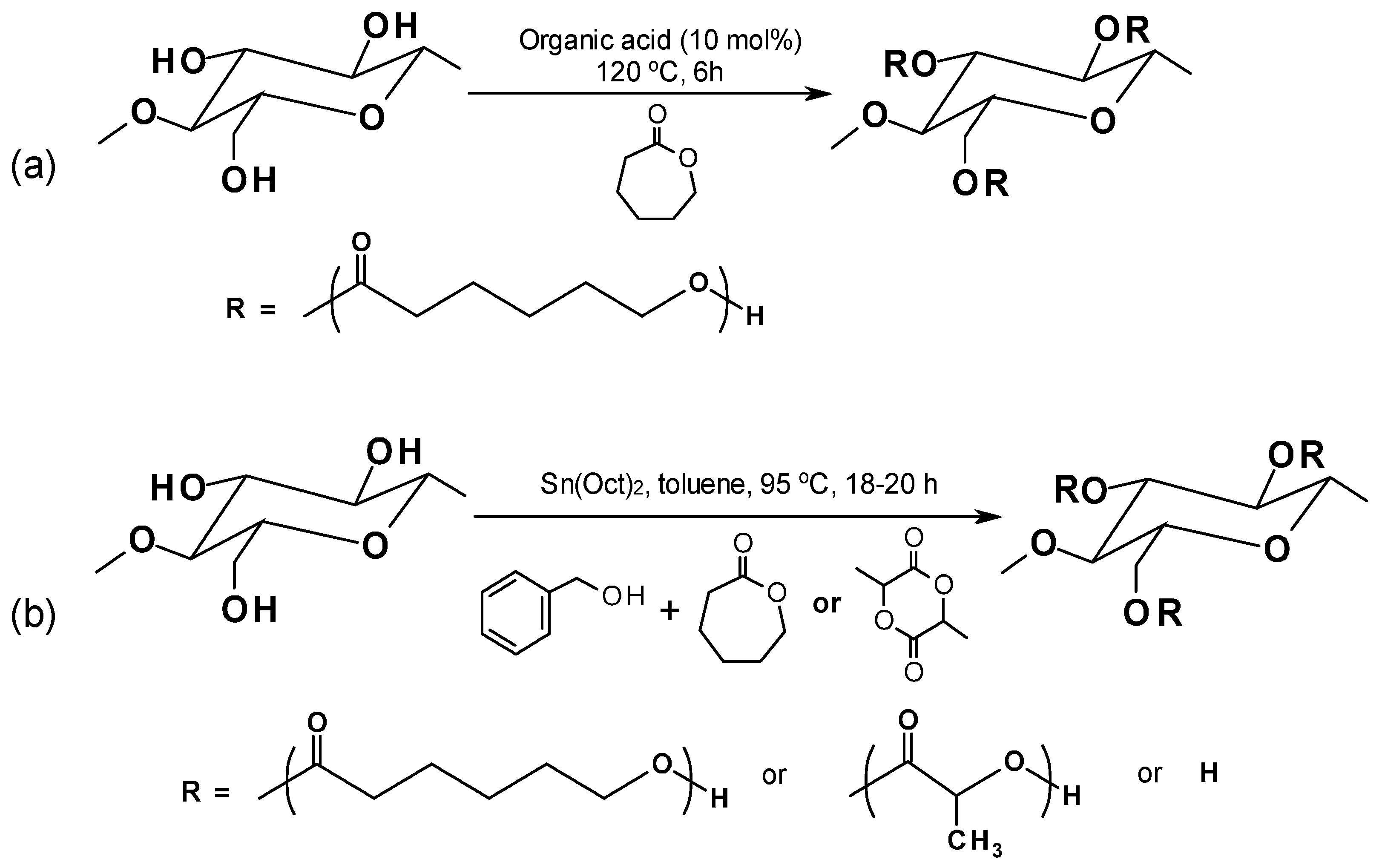
4.2.1. Ring Opening Polymerization for Biocomposites
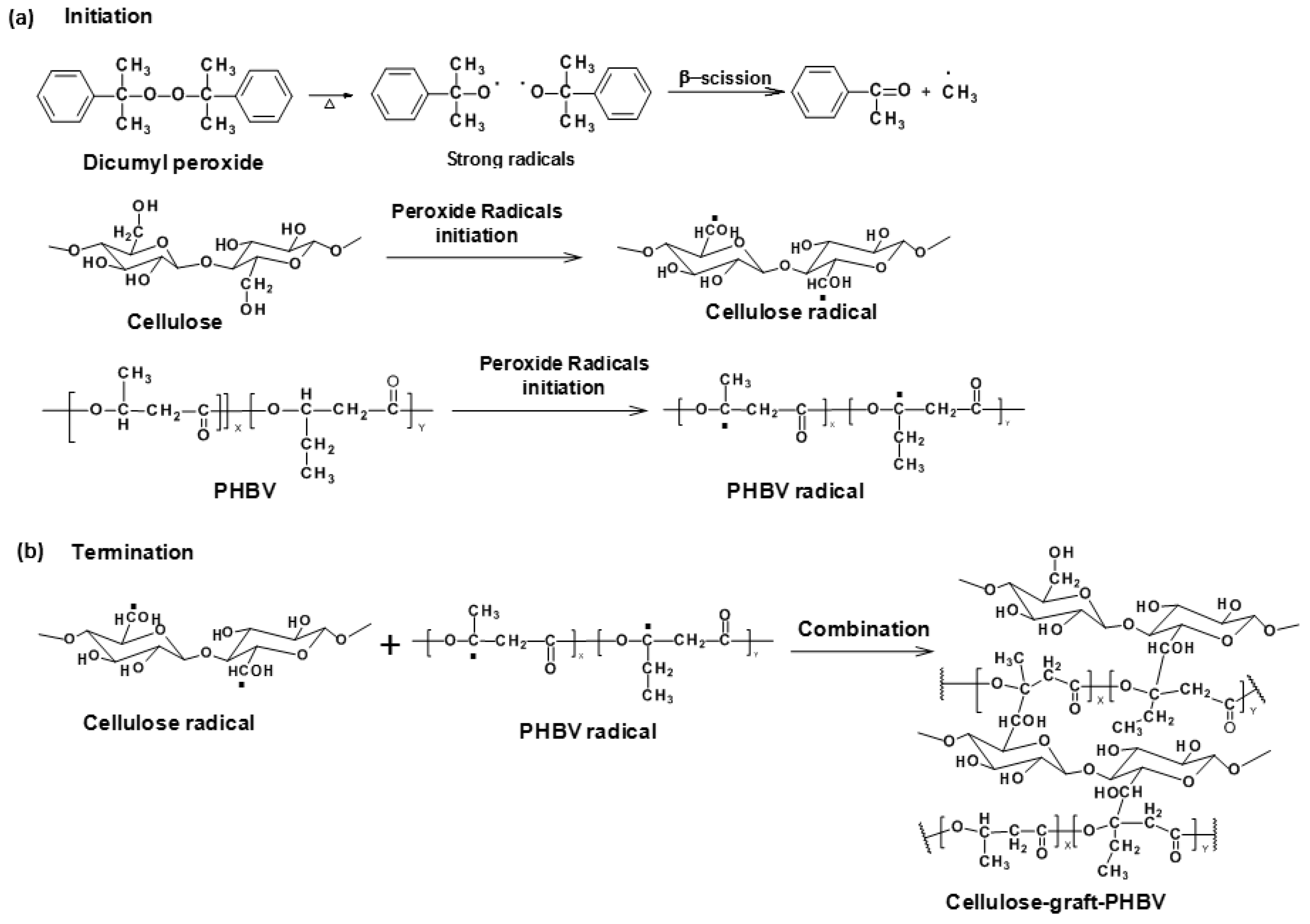
4.2.2. Free Radical Grafting
4.3. Grafting via Coupling Agent
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Li, J.; Hunt, J.F.; Gong, S.; Cai, Z. High strength wood-based sandwich panels reinforced with fiberglass and foam. BioResources 2014, 9, 1898–1913. [Google Scholar] [CrossRef]
- Faruk, O.; Bledzki, A.K.; Fink, H.-P.; Sain, M. Progress report on natural fiber reinforced composites. Macromol. Mater. Eng. 2014, 299, 9–26. [Google Scholar] [CrossRef]
- Dhand, V.; Mittal, G.; Rhee, K.Y.; Park, S.-J.; Hui, D. A short review on basalt fiber reinforced polymer composites. Compos. B Eng. 2015, 73, 166–180. [Google Scholar] [CrossRef]
- Li, J.; Hunt, J.F.; Gong, S.; Cai, Z. Improved fatigue performance for wood-based structural panels using slot and tab construction. Compos. A Appl. Sci. Manuf. 2016, 82, 235–242. [Google Scholar] [CrossRef]
- Li, J.; Hunt, J.F.; Gong, S.; Cai, Z. Testing and evaluation of a slot and tab construction technique for light-weight wood-fiber-based structural panels under bending. J. Test. Eval. 2016, 44, 1–10. [Google Scholar] [CrossRef]
- Sun, L.; Wu, Q.; Xie, Y.; Song, K.; Lee, S.; Wang, Q. Thermal decomposition of fire-retarded wood flour/polypropylene composites. J. Therm. Anal. Calorim. 2016, 123, 309–318. [Google Scholar] [CrossRef]
- Li, J.; Hunt, J.F.; Gong, S.; Cai, Z. Simplified analytical model and balanced design approach for light-weight wood-based structural panel in bending. Compos. Struct. 2016, 136, 16–24. [Google Scholar] [CrossRef]
- European and global markets: 2012 and future trends. Bioplastics Magazine, May/June 2014.
- Saha, S. Bio Composites Market: Asia Pacific Industry Analysis and Opportunity Assessment 2014–2020. Future Market Insights. Available online: http://www.Futuremarketinsights.Com/reports/asia-pacific-bio-composites-market (accessed on 12 February 2016).
- Pilla, S. Handbook of Bioplastics and Biocomposites Engineering Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; Volume 81. [Google Scholar]
- Roy, D.; Semsarilar, M.; Guthrie, J.T.; Perrier, S. Cellulose modification by polymer grafting: A review. Chem. Soc. Rev. 2009, 38, 2046–2064. [Google Scholar] [CrossRef] [PubMed]
- Carlmark, A.; Larsson, E.; Malmström, E. Grafting of cellulose by ring-opening polymerisation—A review. Eur. Polym. J. 2012, 48, 1646–1659. [Google Scholar] [CrossRef]
- Faruk, O.; Bledzki, A.K.; Fink, H.-P.; Sain, M. Biocomposites reinforced with natural fibers: 2000–2010. Prog. Polym. Sci. 2012, 37, 1552–1596. [Google Scholar] [CrossRef]
- John, M.; Thomas, S. Biofibres and biocomposites. Carbohydr. Polym. 2008, 71, 343–364. [Google Scholar] [CrossRef]
- Rowell, R. Handbook of Wood Chemistry and Wood Composites; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Barnett, J.; Bonham, V.A. Cellulose microfibril angle in the cell wall of wood fibres. Biol. Rev. 2004, 79, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Clemons, C.M.; Caulfield, D.F. Natural fibers. In Functional Fillers for Plastics; Xanthos, M., Ed.; Wiley-VCH: Weinheim, Germany, 2005; pp. 195–206. [Google Scholar]
- Hamelinck, C.N.; Hooijdonk, G.V.; Faaij, A.P.C. Ethanol from lignocellulosic biomass: Techno-economic performance in short-, middle- and long-term. Biomass Bioenergy 2005, 28, 384–410. [Google Scholar] [CrossRef]
- Siqueira, G.; Bras, J.; Dufresne, A. Cellulosic bionanocomposites: A review of preparation, properties and applications. Polymers 2010, 2, 728–765. [Google Scholar] [CrossRef]
- Komuraiah, A.; Kumar, N.S.; Prasad, B.D. Chemical composition of natural fibers and its influence on their mechanical properties. Mech. Compos. Mater. 2014, 50, 359–376. [Google Scholar] [CrossRef]
- Mwaikambo, L.Y.; Ansell, M.P. Mechanical properties of alkali treated plant fibres and their potential as reinforcement materials. I. Hemp fibres. J. Mater. Sci. 2006, 41, 2483–2496. [Google Scholar] [CrossRef]
- Dungani, R.; Khalil, H.P.S.A.; Sumardi, I.; Suhaya, Y.; Sulistyawati, E.; Islam, M.N.; Saraya, N.L.M.; Aprilia, N.A.S. Non-wood renewable materials: Properties improvement and its application. In Biomass and Bioenergy: Applications; Hakeem, K.R., Jawaid, M., Rashid, U., Eds.; Springer: Basel, Switzerland, 2014. [Google Scholar]
- Mwaikambo, L.Y. Review of the history, properties and application of plant fibres. Afr. J. Sci. Technol. 2006, 7, 120–133. [Google Scholar]
- Hon, D.N.-S.; Shiraishi, N. Wood and Cellulosic Chemistry, Second Edition, Revised, and Expanded; Marcel Dekker, Inc.: New York, NY, USA, 2001. [Google Scholar]
- Kadla, J.F.; Gilbert, R.D. Cellulose structure: A review. Cell. Chem. Technol. 2000, 34, 197–216. [Google Scholar]
- Klemm, D.; Heublein, B.; Fink, H.-P.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Sugiyama, J.; Chanzy, H.; Langan, P. Crystal structure and hydrogen bonding system in cellulose iα from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2003, 125, 14300–14306. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Langan, P.; Chanzy, H. Crystal structure and hydrogen-bonding system in cellulose iβ from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2002, 124, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, L.A. Lignification and lignin topochemistry—An ultrastructural view. Phytochemistry 2001, 57, 859–873. [Google Scholar] [CrossRef]
- Xu, X.; Liu, F.; Jiang, L.; Zhu, J.Y.; Haagenson, D.; Wiesenborn, D.P. Cellulose nanocrystals vs. Cellulose nanofibrils: A comparative study on their microstructures and effects as polymer reinforcing agents. ACS Appl. Mater. Interf. 2013, 5, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Habibi, Y.; Lucia, L.A.; Rojas, O.J. Cellulose nanocrystals: Chemistry, self-assembly, and applications. Chem. Rev. 2010, 110, 3479–3500. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Wu, Q.; Zhang, Z.; Ren, S.; Lei, T.; Dooley, K.M.; Liu, D.; Janes, M.E. Fabricating electrospun nanofibers with antimicrobial capability: A facile route to recycle biomass tar. Fuel 2015, 150, 123–130. [Google Scholar] [CrossRef]
- Šturcová, A.; Davies, G.R.; Eichhorn, S.J. Elastic modulus and stress-transfer properties of tunicate cellulose whiskers. Biomacromolecules 2005, 6, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, K.; Kobayashi, M. Theoretical evaluation of three-dimensional elastic constants of native and regenerated celluloses: Role of hydrogen bonds. Polymer 1991, 32, 1516–1526. [Google Scholar] [CrossRef]
- Sabo, R.C.; Elhajjar, R.F.; Clemons, C.M.; Pillai, K.M. Characterization and processing of nanocellulose thermosetting composites. In Handbook of Polymer Nanocomposites. Processing, Performance and Application; Pandey, J.K., Takagi, H., Nakagaito, A.N., Kim, H.-J., Eds.; Springer: Berlin, Germany; Heidelberg, Germany, 2015; pp. 265–295. [Google Scholar]
- Li, M.-C.; Wu, Q.; Song, K.; Lee, S.; Qing, Y.; Wu, Y. Cellulose nanoparticles: Structure–morphology–rheology relationships. ACS Sustain. Chem. Eng. 2015, 3, 821–832. [Google Scholar] [CrossRef]
- Lin, N.; Huang, J.; Dufresne, A. Preparation, properties and applications of polysaccharide nanocrystals in advanced functional nanomaterials: A review. Nanoscale 2012, 4, 3274–3294. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, U.P.; Sabo, R.; Reiner, R.S.; Clemons, C.M.; Rudie, A.W. Spatially resolved characterization of cellulose nanocrystal-polypropylene composite by confocal raman microscopy. Appl. Spectrosc. 2012, 66, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Takano, K.; Nakamae, K. Elastic modulus of the crystalline regions of cellulose polymorphs. J. Polym. Sci. B Polym. Phys. 1995, 33, 1647–1651. [Google Scholar] [CrossRef]
- Moon, R.J.; Beck, S.; Rudie, A.W. Cellulose Nanocrystals—A Material with Unique Properties and Many Potential Applications; Postek, M.T., Moon, R.J., Rudie, A.W., Bilodeau, M.A., Eds.; TAPPI Press: Peachtree Corners, GA, USA, 2013; pp. 9–12. [Google Scholar]
- Chen, L.; Wang, Q.; Hirth, K.; Baez, C.; Agarwal, U.P.; Zhu, J.Y. Tailoring the yield and characteristics of wood cellulose nanocrystals (cnc) using concentrated acid hydrolysis. Cellulose 2015, 22, 1753–1762. [Google Scholar] [CrossRef]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose nanomaterials review: Structure, properties and nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3944. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Iwamoto, S.; Yano, H. Obtaining cellulose nanofibers with a uniform width of 15 nm from wood. Biomacromolecules 2007, 8, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kimura, S.; Nishiyama, Y.; Isogai, A. Cellulose nanofibers prepared by tempo-mediated oxidation of native cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef] [PubMed]
- Brodin, I. Chemical Properties and Thermal Behaviour of Kraft Lignins. Master’s Thesis, Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, Stockholm, Sweden, 2009. [Google Scholar]
- Thakur, V.K.; Thakur, M.K.; Gupta, R.K. Graft copolymers of natural fibers for green composites. Carbohydr. Polym. 2014, 104, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Peng, Y.; Cao, J.; Chen, Y. Comparison on properties of lignocellulosic flour/polymer composites by using wood, cellulose, and lignin flours as fillers. Compos. Sci. Technol. 2014, 103, 1–7. [Google Scholar] [CrossRef]
- Bahi, A.; Goudarzi, A.; Cho, M.; Lin, L.; Karaaslan, M.; Ko, F. Fabrication and application of lignin-lignin composites. In Design, Manufacturing and Applications of Composites, Proceedings of the Tenth Joint Canada-Japan Workshop on Composites, Vancouver, BC, Canada, 19–21 August 2014; DEStech Publications, Inc.: Lancaster, PA, USA, 2015; p. 195. [Google Scholar]
- Mousavioun, P.; Halley, P.J.; Doherty, W.O.S. Thermophysical properties and rheology of phb/lignin blends. Ind. Crop. Prod. 2013, 50, 270–275. [Google Scholar] [CrossRef]
- Pohjanlehto, H.; Setala, H.M.; Kiely, D.E.; McDonald, A.G. Lignin xylaric acid polyurethane based polymer network systems: Preparation and characterization. J. Appl. Polym. Sci. 2013, 131. [Google Scholar] [CrossRef]
- Camargo, F.A.; Innocentini-Mei, L.H.; Lemes, A.P.; Moraes, S.G.; Duran, N. Processing and characterization of composites of poly(3-hydroxybutyrate-co-hydroxyvalerate) and lignin from sugar cane bagasse. J. Compos. Mater. 2011, 46, 417–425. [Google Scholar] [CrossRef]
- Chung, Y.-L.; Olsson, J.V.; Li, R.J.; Frank, C.W.; Waymouth, R.M.; Billington, S.L.; Sattely, E.S. A renewable lignin–lactide copolymer and application in biobased composites. ACS Sustain. Chem. Eng. 2013, 1, 1231–1238. [Google Scholar] [CrossRef]
- Singha, A.; Shama, A.; Thakur, V.K. Graft copolymerization of acrylonitrile onto saccharum cilliari fiber. e-Polymers 2009, 9, 1258–1269. [Google Scholar] [CrossRef]
- Thakur, V.; Thakur, M.; Singha, A. Free radical–induced graft copolymerization onto natural fibers. Int. J. Polym. Anal. Charact. 2013, 18, 430–438. [Google Scholar] [CrossRef]
- Thakur, V.; Singha, A.; Thakur, M. Pressure induced synthesis of ea grafted saccaharum cilliare fibers. Int. J. Polym. Mater. 2014, 63, 17–22. [Google Scholar] [CrossRef]
- Sen, S.; Patil, S.; Argyropoulos, D.S. Thermal properties of lignin in copolymers, blends, and composites: A review. Green Chem. 2015, 17, 4862–4887. [Google Scholar] [CrossRef]
- Thakur, V.K.; Thakur, M.K.; Raghavan, P.; Kessler, M.R. Progress in green polymer composites from lignin for multifunctional applications: A review. ACS Sustain. Chem. Eng. 2014, 2, 1072–1092. [Google Scholar] [CrossRef]
- Iguchi, M.; Yamanaka, S.; Budhiono, A. Review bacterial cellulose—A masterpiece of nature’s arts. J. Mater. Sci. 2000, 35, 261–270. [Google Scholar] [CrossRef]
- Ifuku, S.; Nogi, M.; Abe, K.; Handa, K.; Nakatsubo, F.; Yano, H. Surface modification of bacterial cellulose nanofibers for property enhancement of optically transparent composites: Dependence on acetyl-group ds. Biomacromolecules 2007, 8, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- Butchosa, N.; Brown, C.; Larsson, P.T.; Berglund, L.A.; Bulone, V.; Zhou, Q. Nanocomposites of bacterial cellulose nanofibers and chitin nanocrystals: Fabrication, characterization and bactericidal activity. Green Chem. 2013, 15, 3404–3413. [Google Scholar] [CrossRef]
- Arslan, H.; Hazer, B.; Yoon, S.C. Grafting of poly(3-hydroxyalkanoate) and linoleic acid onto chitosan. J. Appl. Polym. Sci. 2007, 103, 81–89. [Google Scholar] [CrossRef]
- Yu, G.-E.; Morin, F.G.; Nobes, G.A.R.; Marchessault, R.H. Degree of acetylation of chitin and extent of grafting phb on chitosan determined by solid state 15n nmr. Macromolecules 1999, 32, 518–520. [Google Scholar] [CrossRef]
- Kikkawa, Y.; Fukuda, M.; Kimura, T.; Kashiwada, A.; Matsuda, K.; Kanesato, M.; Wada, M.; Imanaka, T.; Tanaka, T. Atomic force microscopic study of chitinase binding onto chitin and cellulose surfaces. Biomacromolecules 2014, 15, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Vroman, I.; Tighzert, L. Biodegradable polymers. Materials 2009, 2, 307–344. [Google Scholar] [CrossRef]
- Fortunati, E.; Armentano, I.; Zhou, Q.; Iannoni, A.; Saino, E.; Visai, L.; Berglund, L.A.; Kenny, J.M. Multifunctional bionanocomposite films of poly(lactic acid), cellulose nanocrystals and silver nanoparticles. Carbohydr. Polym. 2012, 87, 1596–1605. [Google Scholar] [CrossRef]
- Mushi, N.E.; Butchosa, N.; Salajkova, M.; Zhou, Q.; Berglund, L.A. Nanostructured membranes based on native chitin nanofibers prepared by mild process. Carbohydr. Polym. 2014, 112, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ifuku, S.; Saimoto, H. Chitin nanofibers: Preparations, modifications, and applications. Nanoscale 2012, 4, 3308–3318. [Google Scholar] [CrossRef] [PubMed]
- Mushi, N.E.; Butchosa, N.; Zhou, Q.; Berglund, L.A. Nanopaper membranes from chitin–protein composite nanofibers—Structure and mechanical properties. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Mekonnen, T.; Mussone, P.; Khalil, H.; Bressler, D. Progress in bio-based plastics and plasticizing modifications. J. Mater. Chem. 2013, 1, 13379–13398. [Google Scholar] [CrossRef]
- Chen, G.-Q.; Patel, M.K. Plastics derived from biological sources: Present and future: A technical and environmental review. Chem. Rev. 2012, 112, 2082–2099. [Google Scholar] [CrossRef] [PubMed]
- Ojijo, V.; Sinha Ray, S.; Sadiku, R. Toughening of biodegradable polylactide/poly(butylene succinate-co-adipate) blends via in situ reactive compatibilization. ACS Appl. Mater. Interf. 2013, 5, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Bugnicourt, E.; Cinelli, P.; Lazzeri, A.; Alvarez, V. Polyhydroxyalkanoate (pha): Review of synthesis, characteristics, processing and potential applications in packaging. Express Polym. Lett. 2014, 8, 791–808. [Google Scholar] [CrossRef]
- Lasprilla, A.J.R.; Martinez, G.A.R.; Lunelli, B.H.; Jardini, A.L.; Filho, R.M. Poly-lactic acid synthesis for application in biomedical devices—A review. Biotechnol. Adv. 2012, 30, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Laycock, B.; Halley, P.; Pratt, S.; Werker, A.; Lant, P. The chemomechanical properties of microbial polyhydroxyalkanoates. Prog. Polym. Sci. 2013, 38, 536–583. [Google Scholar] [CrossRef]
- Wei, L.; McDonald, A.G. Accelerated weathering studies on the bioplastic, poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Polym. Degrad. Stab. 2016, 126, 93–100. [Google Scholar] [CrossRef]
- Wei, L.; Liang, S.; Coats, E.R.; McDonald, A.G. Valorization of residual bacterial biomass waste after polyhydroxyalkanoate isolation by hydrothermal treatment. Bioresour. Technol. 2015, 198, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Gumel, A.M.; Annuar, M.S.M.; Chisti, Y. Recent advances in the production, recovery and applications of polyhydroxyalkanoates. J. Polym. Environ. 2013, 21, 580–605. [Google Scholar] [CrossRef]
- Khanna, S.; Srivastava, A.K. Recent advances in microbial polyhydroxyalkanoates. Process Biochem. 2005, 40, 607–619. [Google Scholar] [CrossRef]
- Ienczak, J.L.; Schmidell, W.; de Aragão, G.M.F. High-cell-density culture strategies for polyhydroxyalkanoate production: A review. J. Ind. Microbiol. Biotechnol. 2013, 40, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.S.K.; Ghai, R.; Kalia, V.C. Polyhydroxyalkanoates: An overview. Bioresour. Technol. 2003, 87, 137–146. [Google Scholar] [CrossRef]
- Suriyamongkol, P.; Weselake, R.; Narine, S.; Moloney, M.; Shah, S. Biotechnological approaches for the production of polyhydroxyalkanoates in microorganisms and plants—A review. Biotechnol. Adv. 2007, 25, 148–175. [Google Scholar] [CrossRef] [PubMed]
- Urtuvia, V.; Villegas, P.; González, M.; Seeger, M. Bacterial production of the biodegradable plastics polyhydroxyalkanoates. Int. J. Biol. Macromol. 2014, 70, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Jendrossek, D.; Pfeiffer, D. New insights in the formation of polyhydroxyalkanoate granules (carbonosomes) and novel functions of poly(3-hydroxybutyrate). Environ. Microbiol. 2014, 16, 2357–2373. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.M.L.; Lemos, P.C.; Serafim, L.S.; Oliveira, C.; Eiroa, M.; Albuquerque, M.G.E.; Ramos, A.M.; Oliveira, R.; Reis, M.A.M. Recent advances in polyhydroxyalkanoate production by mixed aerobic cultures: From the substrate to the final product. Macromol. Biosci. 2006, 6, 885–906. [Google Scholar] [CrossRef] [PubMed]
- Sudesh, K.; Abe, H.; Doi, Y. Synthesis, structure and properties of polyhydroxyalkanoates: Biological polyesters. Prog. Polym. Sci. 2000, 25, 1503–1555. [Google Scholar] [CrossRef]
- Wei, L.; Guho, N.M.; Coats, E.R.; McDonald, A.G. Characterization of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) biosynthesized by mixed microbial consortia fed fermented dairy manure. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Wei, L.; McDonald, A.G. Peroxide induced cross-linking by reactive melt processing of two biopolyesters: Poly(3-hydroxybutyrate) and poly(l-lactic acid) improve their melting processability. J. Appl. Polym. Sci. 2014, 132. [Google Scholar] [CrossRef]
- Wei, L.; McDonald, A.G. Thermophysical properties of bacterial poly(3-hydroxybutyrate): Characterized by tma, dsc, and tmdsc. J. Appl. Polym. Sci. 2014, 132. [Google Scholar] [CrossRef]
- Chanprateep, S. Current trends in biodegradable polyhydroxyalkanoates. J. Biosci. Bioeng. 2010, 110, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Södergård, A.; Stolt, M. Properties of lactic acid based polymers and their correlation with composition. Prog. Polym. Sci. 2002, 27, 1123–1163. [Google Scholar] [CrossRef]
- Wool, R.; Sun, X.S. Bio-Based Polymers and Composites; Academic Press: New York, NY, USA, 2011. [Google Scholar]
- Witzke, D.R.; Narayan, R.; Kolstad, J.J. Reversible kinetics and thermodynamics of the homopolymerization of l-lactide with 2-ethylhexanoic acid tin (ii) salt. Macromolecules 1997, 30, 7075–7085. [Google Scholar] [CrossRef]
- Narayan, R.; Wu, W.M.; Criddle, C.S. Lactide Production from Thermal Depolymerization of PLA with Applications to Production of PLA or Other Bioproducts. U.S. Patent 20,130,023,674, 15 March 2012. [Google Scholar]
- Narayan, R.; Shi, X.; Graiver, D. PLA recycling via thermal depolymerization. Bioplastics Magazine, May/June 2013. [Google Scholar]
- Terzopoulou, Z.N.; Papageorgiou, G.Z.; Papadopouloub, E.; Athanassiadou, E.; Alexopoulou, E.; Bikiaris, D.N. Green composites prepared from aliphatic polyesters and bast fibers. Ind. Crop. Prod. 2015, 68, 60–79. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Misra, M.; Drzal, L.T. Sustainable bio-composites from renewable resources: Opportunities and challenges in the green materials world. J. Polym. Environ. 2002, 10, 19–26. [Google Scholar] [CrossRef]
- Guan, J.; Hanna, M.A. Selected morphological and functional properties of extruded acetylated starch-polylactic acid foams. Ind. Eng. Chem. Res. 2005, 44, 3106–3115. [Google Scholar] [CrossRef]
- Godbole, S.; Gote, S.; Latkar, M.; Chakrabarti, T. Preparation and characterization of biodegradable poly-3-hydroxybutyrate-starch blend films. Bioresour. Technol. 2003, 86, 33–37. [Google Scholar] [CrossRef]
- Yu, L.; Dean, K.; Li, L. Polymer blends and composites from renewable resources. Prog. Polym. Sci. 2006, 31, 576–602. [Google Scholar] [CrossRef]
- Barud, H.S.; Souza, J.L.; Santos, D.B.; Crespi, M.S.; Ribeiro, C.A.; Messaddeq, Y.; Ribeiro, S.J.L. Bacterial cellulose/poly(3-hydroxybutyrate) composite membranes. Carbohydr. Polym. 2011, 83, 1279–1284. [Google Scholar] [CrossRef]
- Avella, M.; Bogoeva-Gaceva, G.; Buzõarovska, A.; Emanuela Errico, M.; Gentile, G.; Grozdanov, A. Poly (3-hydroxybutyrate-co-3-hydroxyvalerate)-based biocomposites reinforced with kenaf fibers. J. Appl. Polym. Sci. 2007, 104, 3192–3200. [Google Scholar] [CrossRef]
- Avella, M.; Martuscelli, E.; Raimo, M. Properties of blends and composites based on poly(3-hydroxy)butyrate (phb) and poly(3-hydroxybutyrate-hydroxyvalerate) (phbv) copolymers. J. Mater. Sci. 2000, 35, 523–545. [Google Scholar] [CrossRef]
- Wu, C.-S. Preparation and characterization of polyhydroxyalkanoate bioplastic-based green renewable composites from rice husk. J. Polym. Environ. 2014, 22, 384–392. [Google Scholar] [CrossRef]
- Wang, T.; Drzal, L.T. Cellulose-nanofiber-reinforced poly(lactic acid) composites prepared by a water-based approach. ACS Appl. Mater. Interfaces 2012, 4, 5079–5085. [Google Scholar] [CrossRef] [PubMed]
- Zarrinbakhsh, N.; Misra, M.; Mohanty, A.K. Biodegradable green composites from distiller’s dried grains with solubles (DDGS) and a polyhydroxy(butyrate-co-valerate) (PHBV)-based bioplastic. Macromol. Mater. Eng. 2011, 296, 1035–1045. [Google Scholar] [CrossRef]
- Madbouly, S.A.; Schrader, J.A.; Srinivasan, G.; Liu, K.; McCabe, K.G.; Grewell, D.; Graves, W.R.; Kessler, M.R. Biodegradation behavior of bacterial-based polyhydroxyalkanoate (PHA) and DDGS composites. Green Chem. 2014, 16, 1911–1920. [Google Scholar] [CrossRef]
- Wei, L.; Liang, S.; McDonald, A.G. Thermophysical properties and biodegradation behavior of green composites made from polyhydroxybutyrate and potato peel waste fermentation residue. Ind. Crop. Prod. 2015, 69, 91–103. [Google Scholar] [CrossRef]
- Shanks, R.A.; Hodzic, A.; Wong, S. Thermoplastic biopolyester natural fiber composites. J. Appl. Polym. Sci. 2004, 91, 2114–2121. [Google Scholar] [CrossRef]
- Barkoula, N.M.; Garkhail, S.K.; Peijs, T. Biodegradable composites based on flax/polyhydroxybutyrate and its copolymer with hydroxyvalerate. Ind. Crop. Prod. 2010, 31, 34–42. [Google Scholar] [CrossRef]
- Srithep, Y.; Ellingham, T.; Peng, J.; Sabo, R.; Clemons, C.; Turng, L.-S.; Pilla, S. Melt compounding of poly (3-hydroxybutyrate-co-3-hydroxyvalerate)/nanofibrillated cellulose nanocomposites. Polym. Degrad. Stab. 2013, 98, 1439–1449. [Google Scholar] [CrossRef]
- Coats, E.R.; Loge, F.J.; Wolcott, M.P.; Englund, K.; McDonald, A.G. Production of natural fiber reinforced thermoplastic composites through the use of polyhydroxybutyrate-rich biomass. Bioresour. Technol. 2008, 99, 2680–2686. [Google Scholar] [CrossRef] [PubMed]
- Oksman, K.; Skrifvars, M.; Selin, J.F. Natural fibres as reinforcement in polylactic acid (PLA) composites. Compos. Sci. Technol. 2003, 63, 1317–1324. [Google Scholar] [CrossRef]
- Dong, Y.; Ghataura, A.; Takagi, H.; Haroosh, H.J.; Nakagaito, A.N.; Lau, K.-T. Polylactic acid (PLA) biocomposites reinforced with coir fibres: Evaluation of mechanical performance and multifunctional properties. Compos. A Appl. Sci. Manuf. 2014, 63, 76–84. [Google Scholar] [CrossRef]
- Masirek, R.; Kulinski, Z.; Chionna, D.; Piorkowska, E.; Pracella, M. Composites of poly (l-lactide) with hemp fibers: Morphology and thermal and mechanical properties. J. Appl. Polym. Sci. 2007, 105, 255–268. [Google Scholar] [CrossRef]
- Wong, S.; Shanks, R.; Hodzic, A. Interfacial improvements in poly(3-hydroxybutyrate)-flax fibre composites with hydrogen bonding additives. Compos. Sci. Technol. 2004, 64, 1321–1330. [Google Scholar] [CrossRef]
- Huda, M.S.; Drzal, L.T.; Misra, M.; Mohanty, A.K.; Williams, K.; Mielewski, D.F. A study on biocomposites from recycled newspaper fiber and poly(lactic acid). Ind. Eng. Chem. Res. 2005, 44, 5593–5601. [Google Scholar] [CrossRef]
- Sullivan, E.; Moon, R.; Kalaitzidou, K. Processing and characterization of cellulose nanocrystals/polylactic acid nanocomposite films. Materials 2015, 8, 8106–8116. [Google Scholar] [CrossRef]
- Li, Y.; Sun, X. Mechanical and thermal properties of biocomposites from poly(lactic acid) and ddgs. J. Appl. Polym. Sci. 2011, 121, 589–597. [Google Scholar] [CrossRef]
- Tomé, L.C.; Pinto, R.J.B.; Trovatti, E.; Freire, C.S.R.; Silvestre, A.J.D.; Neto, C.P.; Gandini, A. Transparent bionanocomposites with improved properties prepared from acetylated bacterial cellulose and poly(lactic acid) through a simple approach. Green Chem. 2011, 13, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Botta, L.; Fiore, V.; Scalici, T.; Valenza, A.; Roberto, R. New polylactic acid composites reinforced with artichoke fibers. Materials 2015, 8, 7770–7779. [Google Scholar] [CrossRef] [Green Version]
- Acioli-Moura, R.; Sun, X.S. Thermal degradation and physical aging of poly(lactic acid) and its blends with starch. Polym. Eng. Sci. 2008, 48, 829–836. [Google Scholar] [CrossRef]
- Satyanarayana, K.G.; Arizaga, G.G.C.; Wypych, F. Biodegradable composites based on lignocellulosic fibers—An overview. Prog. Polym. Sci. 2009, 34, 982–1021. [Google Scholar] [CrossRef]
- Arrieta, M.; Fortunati, E.; Dominici, F.; Rayón, E.; López, J.; Kenny, J.M. PLA-PHB/cellulose based films: Mechanical, barrier and disintegration properties. Polym. Degrad. Stab. 2014, 107, 139–149. [Google Scholar] [CrossRef]
- Hejna, A.; Formela, K.; Saeb, M.R. Processing, mechanical and thermal behavior assessments of polycaprolactone/agricultural wastes biocomposites. Ind. Crop. Prod. 2015, 76, 725–733. [Google Scholar] [CrossRef]
- Wang, D.-W.; Xu, Y.-J.; Li, X.; Huang, C.-M.; Huang, K.-S.; Wang, C.-K.; Yeh, J.-T. Mechanical retention and waterproof properties of bacterial cellulose-reinforced thermoplastic starch biocomposites modified with sodium hexametaphosphate. Materials 2015, 8, 3168–3194. [Google Scholar] [CrossRef]
- Dufresne, A.; Kellerhals, M.B.; Witholt, B. Transcrystallization in MCL-PHAS/cellulose whiskers composites. Macromolecules 1999, 32, 7396–7401. [Google Scholar] [CrossRef]
- Kalia, S.; Dufresne, A.; Cherian, B.M.; Kaith, B.S.; Avérous, L.; Njuguna, J.; Nassiopoulos, E. Cellulose-based bio- and nanocomposites: A review. Int. J. Polym. Sci. 2011, 2011, 1–35. [Google Scholar] [CrossRef]
- Wei, L.; McDonald, A.G.; Freitag, C.; Morrell, J.J. Effects of wood fiber esterification on properties, weatherability and biodurability of wood plastic composites. Polym. Degrad. Stab. 2013, 98, 1348–1361. [Google Scholar] [CrossRef]
- Lu, J.Z.; Wu, Q.; McNabb, H.S. Chemical coupling in wood fiber and polymer composites: A review of coupling agents and treatments. Wood Fiber Sci. 2000, 32, 88–104. [Google Scholar]
- McDonald, A.G.; Ma, L. Plastic moldable pine fiber by benzylation. In Wood: Types, Properties, and Uses (Environmental Science, Engineering and Technology); Botannini, L.F., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2010; pp. 181–192. [Google Scholar]
- Lu, J.Z.; Wu, Q.; Negulescu, I.I. Wood-fiber/high-density-polyethylene composites: Coupling agent performance. J. Appl. Polym. Sci. 2005, 96, 93–102. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Gassan, J. Composites reinforced with cellulose based fibres. Prog. Polym. Sci. 1999, 24, 221–274. [Google Scholar] [CrossRef]
- Li, X.; Tabil, L.G.; Panigrahi, S. Chemical treatments of natural fiber for use in natural fiber-reinforced composites: A review. J. Polym. Environ. 2007, 15, 25–33. [Google Scholar] [CrossRef]
- Jerome, C.; Lecomte, P. Recent advances in the synthesis of aliphatic polyesters by ring-opening polymerization. Adv. Drug Deliv. Rev. 2008, 60, 1056–1076. [Google Scholar] [CrossRef] [PubMed]
- Hafrén, J.; Córdova, A. Direct organocatalytic polymerization from cellulose fibers. Macromol. Rapid Commun. 2005, 26, 82–86. [Google Scholar] [CrossRef]
- Lönnberg, H.; Zhou, Q.; Brumer, H.; Teeri, T.T.; Malmström, E.; Hult, A. Grafting of cellulose fibers with poly(e-caprolactone) and poly(l-lactic acid) via ring-opening polymerization. Biomacromolecules 2006, 7, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, A. Nanocellulose: A new ageless bionanomaterial. Mater. Today 2013, 16, 220–227. [Google Scholar] [CrossRef]
- Habibi, Y.; Goffin, A.-L.; Schiltz, N.; Duquesne, E.; Dubois, P.; Dufresne, A. Bionanocomposites based on poly(ε-caprolactone)-grafted cellulose nanocrystals by ring-opening polymerization. J. Mater. Chem. 2008, 18, 5002–5010. [Google Scholar] [CrossRef]
- Yuan, W.; Yuan, J.; Zhang, F.; Xie, X. Syntheses, characterization, and in vitro degradation of ethyl cellulose-graft-poly(ε-caprolactone)-block-poly(l-lactide) copolymers by sequential ring-opening polymerization. Biomacromolecules 2007, 8, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Goffin, A.-L.; Raquez, J.-M.; Duquesne, E.; Siqueira, G.; Habibi, Y.; Dufresne, A.; Dubois, P. From interfacial ring-opening polymerization to melt processing of cellulose nanowhisker-filled polylactide-based nanocomposites. Biomacromolecules 2011, 12, 2456–2465. [Google Scholar] [CrossRef] [PubMed]
- Boujemaoui, A.; Carlsson, L.; Malmström, E.; Lahcini, M.; Berglund, L.; Sehaqui, H.; Carlmark, A. Facile preparation route for nanostructured composites: Surface-initiated ring-opening polymerization of ε-caprolactone from high-surface-area nanopaper. ACS Appl. Mater. Interfaces 2012, 4, 3191–3198. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.K.; Thakur, M.K.; Gupta, R.K. Rapid synthesis of graft copolymers from natural cellulose fibers. Carbohydr. Polym. 2013, 98, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.K.; Thakur, M.K.; Gupta, R.K. Graft copolymers from cellulose: Synthesis, characterization and evaluation. Carbohydr. Polym. 2013, 97, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yao, K.; Korich, A.L.; Li, S.; Ma, S.; Ploehn, H.J.; Iovine, P.M.; Wang, C.; Chu, F.; Tang, C. Combining renewable gum rosin and lignin: Towards hydrophobic polymer composites by controlled polymerization. J. Polym. Sci. A Polym. Chem. 2011, 49, 3728–3738. [Google Scholar] [CrossRef]
- Majoinen, J.; Walther, A.; McKee, J.R.; Kontturi, E.; Aseyev, V.; Malho, J.M.; Ruokolainen, J.; Ikkala, O. Polyelectrolyte brushes grafted from cellulose nanocrystals using cu-mediated surface-initiated controlled radical polymerization. Biomacromolecules 2011, 12, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Zoppe, J.O.; Habibi, Y.; Rojas, O.J.; Venditti, R.A.; Johansson, L.-S.; Efimenko, K.; Osterberg, M.; Laine, J. Poly(n-isopropylacrylamide) brushes grafted from cellulose nanocrystals via surface-initiated single-electron transfer living radical polymerization. Biomacromolecules 2010, 11, 2683–2691. [Google Scholar] [CrossRef] [PubMed]
- Parambath Kanoth, B.; Claudino, M.; Johansson, M.; Berglund, L.A.; Zhou, Q. Biocomposites from natural rubber: Synergistic effects of functionalized cellulose nanocrystals as both reinforcing and cross-linking agents via free-radical thiol–ene chemistry. ACS Appl. Mater. Interfaces 2015, 7, 16303–16310. [Google Scholar] [CrossRef] [PubMed]
- Rowell, R.M.; Young, R.A.; Rowell, J. Paper and Composites from Agro-Based Resources; CRC Press: Boca Raton, FL, USA, 1996. [Google Scholar]
- El-Naggar, A.M.; El-Hosamy, M.B.; Zahran, A.H.; Zondy, M.H. Surface morphology/mechanical/dyeability properties of radiation-grafted sisal fibres. Am. Dyestuff Rep. 1992, 81, 40–44. [Google Scholar]
- Gupta, V.K.; Pathania, D.; Agarwal, S.; Sharma, S. Amputation of congo red dye from waste water using microwave induced grafted luffa cylindrica cellulosic fiber. Carbohydr. Polym. 2014, 111, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; Pathania, D.; Priya, B.; Singha, A.S.; Sharma, G. Microwave induced synthesis of graft copolymer of binary vinyl monomer mixtures onto delignified grewia optiva fiber: Application in dye removal. Front. Chem. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Stark, N.M.; McDonald, A.G. Interfacial improvements in biocomposites based on poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) bioplastics reinforced and grafted with α-cellulose fibers. Green Chem. 2015, 17, 4800–4814. [Google Scholar] [CrossRef]
- Wei, L.; McDonald, A.G.; Stark, N.M. Grafting of bacterial polyhydroxybutyrate (PHB) onto cellulose via in situ reactive extrusion with dicumyl peroxide. Biomacromolecules 2015, 16, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Dhar, P.; Tarafder, D.; Kumar, A.; Katiyar, V. Thermally recyclable polylactic acid/cellulose nanocrystal films through reactive extrusion process. Polymer 2016, 87, 268–282. [Google Scholar] [CrossRef]
- Hu, L.; Stevanovic, T.; Rodrigue, D. Unmodified and esterified kraft lignin-filled polyethylene composites: Compatibilization by free-radical grafting. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Csikós, Á.; Faludi, G.; Domján, A.; Renner, K.; Móczó, J.; Pukánszky, B. Modification of interfacial adhesion with a functionalized polymer in pla/wood composites. Eur. Polym. J. 2015, 68, 592–600. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, G.; Bras, J.; Dufresne, A. Cellulose whiskers versus microfibrils: Influence of the nature of the nanoparticle and its surface functionalization on the thermal and mechanical properties of nanocomposites. Biomacromolecules 2009, 10, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Magniez, K.; Voda, A.S.; Kafi, A.A.; Fichini, A.; Guo, Q.; Fox, B.L. Overcoming interfacial affinity issues in natural fiber reinforced polylactide biocomposites by surface adsorption of amphiphilic block copolymers. ACS Appl. Mater. Interfaces 2013, 5, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Samain, X.; Langlois, V.; Renard, E.; Lorang, G. Grafting biodegradable polyesters onto cellulose. J. Appl. Polym. Sci. 2011, 121, 1183–1192. [Google Scholar] [CrossRef]
- Yu, H.-Y.; Qin, Z.-Y.; Wang, L.-F.; Zhou, Z. Crystallization behavior and hydrophobic properties of biodegradable ethyl cellulose-g-poly(3-hydroxybutyrate-co-3-hydroxyvalerate): The influence of the side-chain length and grafting density. Carbohydr. Polym. 2012, 87, 2447–2454. [Google Scholar] [CrossRef]
- Essabir, H.; Bensalah, M.O.; Rodrigue, D.; Bouhfid, R.; Qaiss, A.E.K. Biocomposites based on argan nut shell and a polymer matrix: Effect of filler content and coupling agent. Carbohydr. Polym. 2016, 143, 70–83. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Fiber | Cellulose | Hemicellulose | Lignin | References |
|---|---|---|---|---|
| Black locust | 41.61 | 17.66 | 26.70 | [18] |
| Hybrid poplar | 44.70 | 18.55 | 26.44 | [18] |
| Eucalyptus | 49.50 | 13.07 | 27.71 | [18] |
| Pine | 44.55 | 21.90 | 27.67 | [18] |
| Switchgrass | 31.98 | 25.19 | 18.13 | [18] |
| Bagasse | 54.3–55.2 | 16.8–29.7 | 24.3–25.3 | [19] |
| Bamboo | 34.50 | 20.50 | 26.00 | [20,21] |
| Rattan | 35.6–52.9 | 22.8–34.7 | 21.0–22.0 | [22] |
| Flax | 70.50 | 16.50 | 2.50 | [20,21] |
| Hemp | 70–92 | 18-22 | 3–5 | [23] |
| Kenaf | 53.50 | 21.00 | 17.00 | [20,21] |
| Jute | 67.00 | 16.00 | 9.00 | [20,21] |
| Oil palm | 14.3–65.2 | 12.5–38.7 | 17.3–26.5 | [22] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, L.; McDonald, A.G. A Review on Grafting of Biofibers for Biocomposites. Materials 2016, 9, 303. https://doi.org/10.3390/ma9040303
Wei L, McDonald AG. A Review on Grafting of Biofibers for Biocomposites. Materials. 2016; 9(4):303. https://doi.org/10.3390/ma9040303
Chicago/Turabian StyleWei, Liqing, and Armando G. McDonald. 2016. "A Review on Grafting of Biofibers for Biocomposites" Materials 9, no. 4: 303. https://doi.org/10.3390/ma9040303
APA StyleWei, L., & McDonald, A. G. (2016). A Review on Grafting of Biofibers for Biocomposites. Materials, 9(4), 303. https://doi.org/10.3390/ma9040303