4.1. Reads, Error Symbols and Error-Free Intervals
From the experimental point of view, a sequencing read is the result of an assay on some polymer of nucleic acid. The output of the assay is the decoded sequence of monomers that compose the molecule. Three types of sequencing errors can occur:
substitutions,
deletions and
insertions. A substitution is a nucleotide that is different in the molecule and in the read, a deletion is a nucleotide that is present in the molecule but not in the read, and an insertion is a nucleotide that is absent in the molecule but present in the read. For our purpose, the focus is not the nucleotide sequence per se, but whether the nucleotides are correct. Thus, we need only four symbols to describe a read: one for each type of error, plus one for correct nucleotides. In this view, a read is a finite sequence of letters from an alphabet of four symbols.
Figure 1 shows the typical structure of a read.
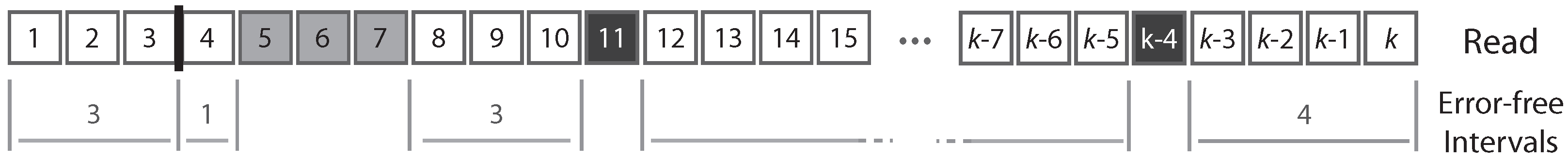
A read can be partitioned uniquely into maximal sequences of identical symbols referred to as “intervals”. Thus, reads can also be seen as sequences of either error-free intervals or error symbols (
Figure 2). As detailed below, this will allow us to control the size of the largest error-free interval.
These concepts established, we can compute seeding probabilities in the read mapping problem. We define an “exact -seed”, or simply a seed, as an exact match of minimum size between the read and the actual sequence of the molecule. In other words, an exact -seed is an error-free interval of size at least . Because of sequencing errors, it could be that the read contains no seed. In this case, the read cannot be mapped to the correct location if the mapping algorithm requires seeds of size or greater.
Our goal is to construct estimators of the probability that a read contains an exact -seed based on expected sequencing errors. For this, we will construct the weighted generating functions of reads that do not contain an exact -seed by decomposing them as sequences of either error symbols or error-free intervals of size less than . We will obtain their weighted generating functions from Theorem 1 and use Theorem 2 to approximate their probability of occurrence. With the weighted generating function of all the reads , and that of reads without an exact -seed , the probability that a read of size k has no exact -seed can be computed as , i.e., the total weight of reads of size k without seed divided by the total weight of reads of size k.
4.2. Substitutions Only
In the simplest model, we assume that errors can be only substitutions, and that they occur with the same probability
p for every nucleotide. Importantly, the model is not overly simple and it has some real applications. For instance, it describes reasonably well the error model of the Illumina platforms, where
p is around
[
26].
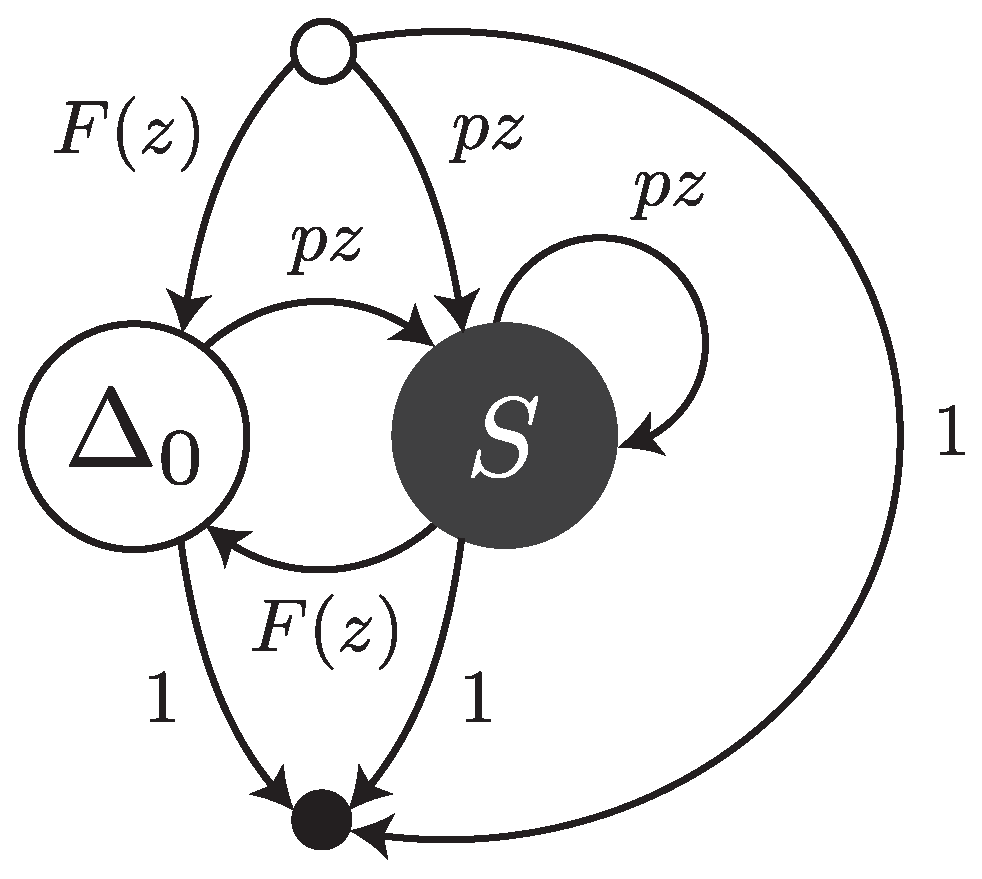
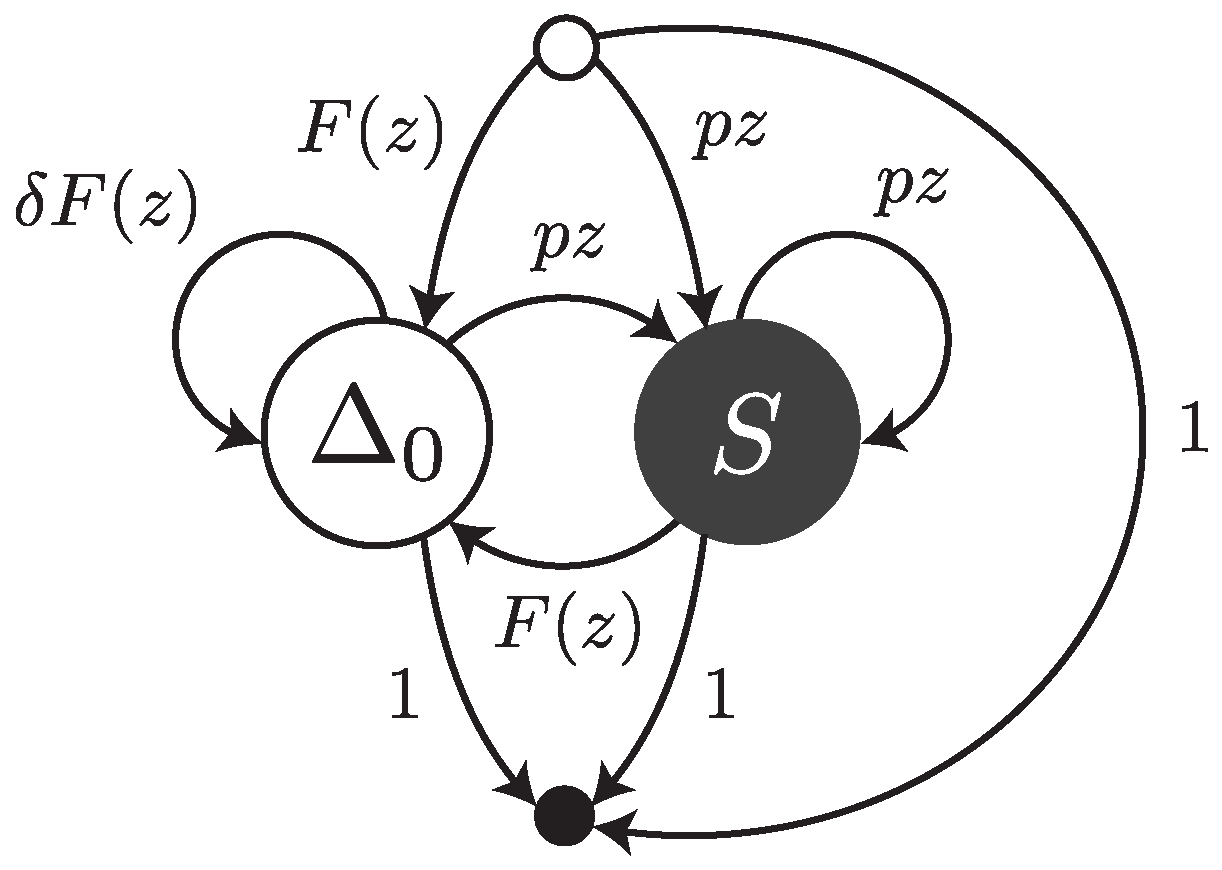
Under this error model, reads are sequences of single substitutions or error-free intervals. They can be thought of as walks on the transfer graph shown in
Figure 3. The symbol
stands for an error-free interval and the symbol
S stands for a single substitution.
and
are the weighted generating functions of error-free intervals and substitutions, respectively. The fact that an error-free interval cannot follow another error-free interval is a consequence of the definition: two consecutive intervals are automatically merged into a single one.
A substitution is a single nucleotide and thus has size 1. Because substitutions have probability
p, their weighted generating function is
. Conversely, the weighted generating function of correct nucleotides is
, where
. Error-free intervals are non-empty sequences of correct nucleotides, so by Proposition 1 their weighted generating function is
The transfer matrix of the graph shown in
Figure 3 is
With the notations of Theorem 1, we have
,
,
and
Applying Theorem 1,
, the weighted generating function of all reads is found from the formula
, which, in this case, translates to
where
is the determinant of
. Using equation (
8), this expression simplifies to
Since
, the total weight of reads of size
k is equal to 1 for any
. As a consequence,
and the probability that a read of size
k has no exact
-seed is equal to
. To find the weighted generating function of reads without an exact
-seed, we limit error-free intervals to a maximum size of
. To do this, we can replace
in expression (
9) by its truncation
. We obtain
Now applying Theorem 2 to the expression of above, we obtain the following proposition.
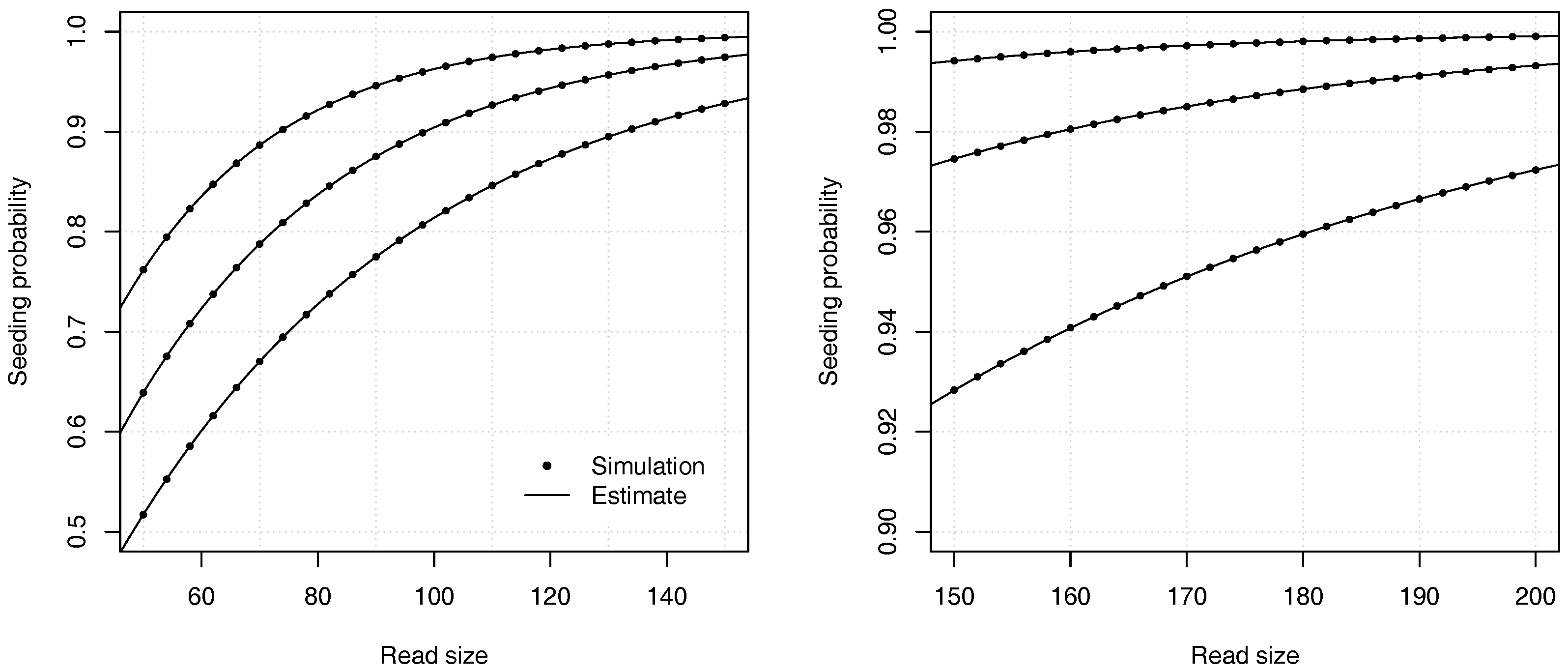
Proposition 2. The probability that a read of size k has no seed under the uniform substitutions model is asymptotically equivalent towhere is the root with smallest modulus of , and where If , expression (11) is undefined and the constant C should be computed as , where and are the respective numerator and denominator of in expression (10). Example 3. Approximate the probability that a read of size has no seed for and for a substitution rate . To find the dominant singularity of , we solve . We rewrite the equation as and use numerical bisection to obtain . Substituting this value in equation (11) yields , so the probability that a read contains no seed is approximately . For comparison, a 99% confidence interval obtained by performing 10 billion random simulations is . The computational cost of the analytic combinatorics approach is infinitesimal compared to the random simulations, and the precision is much higher for . Overall, the analytic combinatorics estimates are accurate.
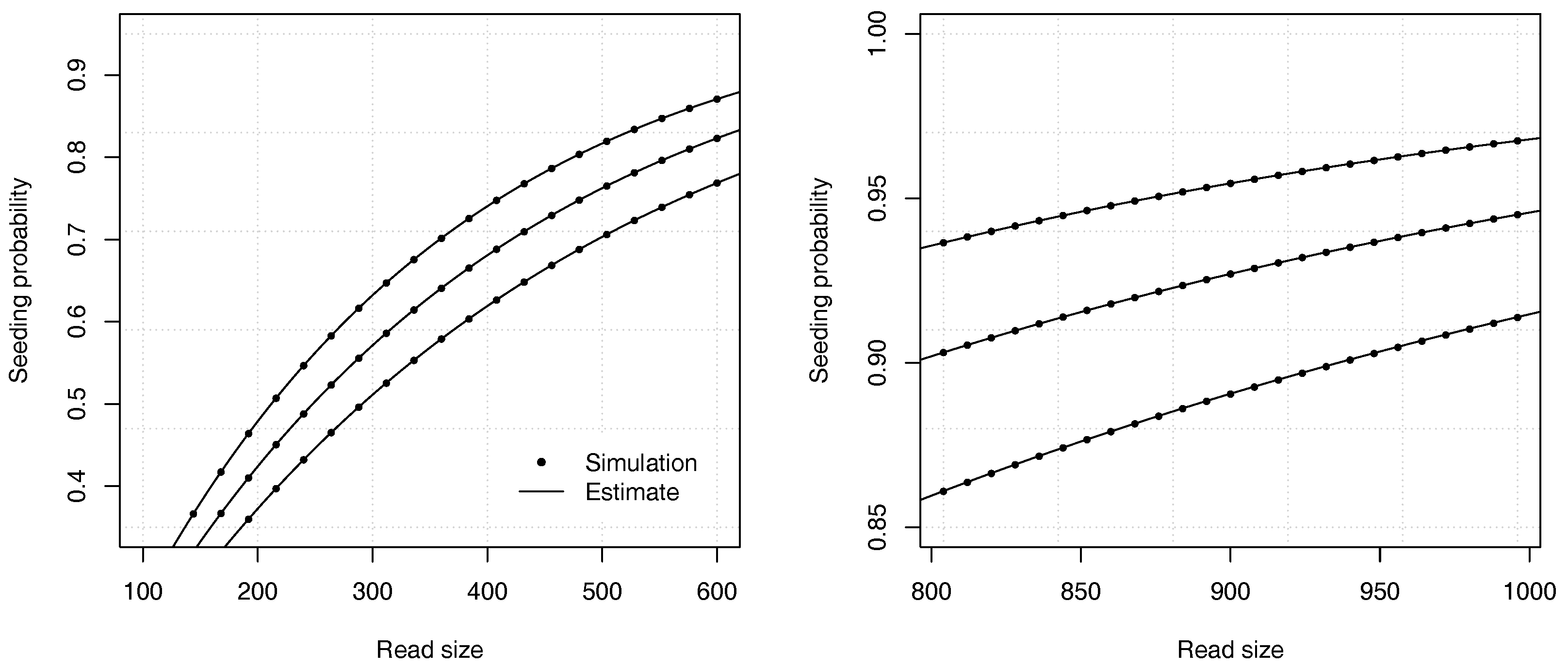
Figure 4 illustrates the precision of the estimates for different values of the error rate
p and of the read size
k.
One can also compute the probabilities by recurrence using Theorem 3, after replacing the term
by
in expression (
10). Denoting
as
, one obtains for every positive integer
4.3. Substitutions and Deletions
We now consider a model where errors can be deletions or substitutions, but not insertions. This case is not very realistic, but it will be useful to clarify how to construct reads with potential deletions. As in the case of uniform substitutions, we assume that every nucleotide call is false with a probability
p and true with a probability
. Here, we also assume that between every pair of decoded nucleotides in the read, an arbitrary number of nucleotides from the original molecule are deleted with probability
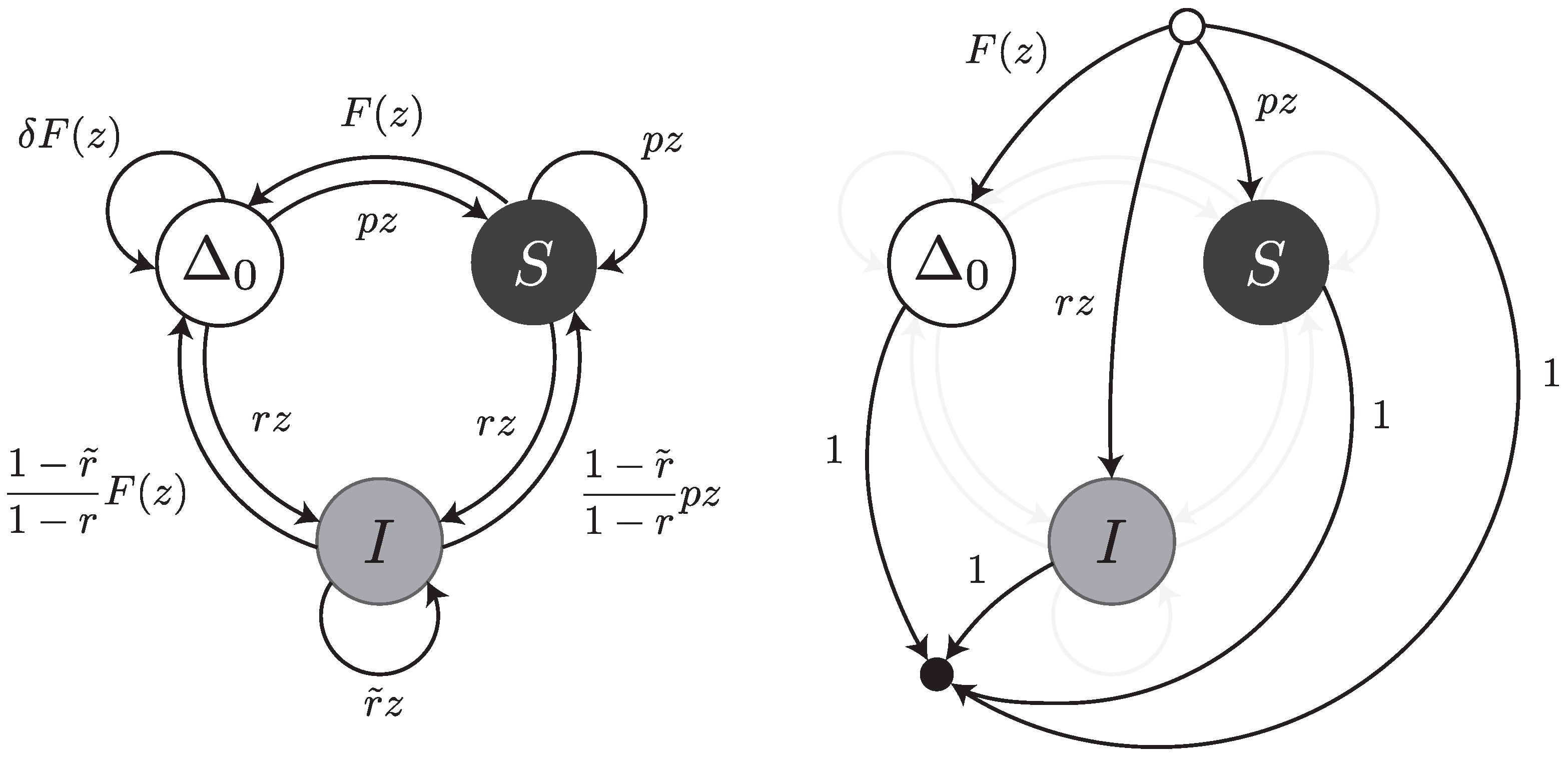
. Regardless of the number of deleted nucleotides, all the deletions are equivalent when the read is viewed as a sequence of error-free intervals or error symbols (see
Figure 2).
A deletion may be adjacent to a substitution, or lie between two correct nucleotides. In the first case, the deletion does not interrupt any error-free interval so it does not change the probability that the read contains a seed. For this reason, we ignore deletions next to substitutions. More precisely, we assume that they can occur, but whether they do has no importance for the problem.
Under this error model, a read can be thought of as a walk on the transfer graph shown in
Figure 5. The graph is almost the same as the one shown
Figure 3; the only difference is the edge labelled
from
to
. This edge represents the fact that an error-free interval can follow another one if a deletion with weighted generating function
is present in between (as illustrated for instance in
Figure 2).
The weighted generating function of error-free intervals
has a different expression from that of
Section 4.2. When the size of an error-free interval is 1, the weighted generating function is just
. For a size
, there are
“spaces” between the nucleotides, so the weighted generating function is
. Summing for all the possible sizes, we obtain the weighted generating function of error-free intervals as
The transfer matrix of the graph shown in
Figure 5 is
With the notations of Theorem 1,
,
,
and
From Theorem 1, the weighted generating function of all reads is
where
is the determinant of
. Using equation (
13), this expression simplifies to
As in
Section 4.2, the result is
, which means that the probability that a read of size
k contains no seed is equal to
. To find the weighted generating function of reads without an exact
-seed, we bound the size of error-free intervals to a maximum of
, i.e., we replace
by its truncation
. With this, the weighted generating function of reads without seed is
Applying Theorem 2 to this expression, we obtain the following proposition.
Proposition 3. The probability that a read of size k has no seed under the model of uniform substitutions and deletions is asymptotically equivalent to where is the root with smallest modulus of , and where If , expression (16) is undefined and the constant C should be computed as , where and are the respective numerator and denominator of in expression (15). Example 4. Approximate the probability that a read of size has no seed for , and . To find the dominant singularity of , we solve . We write it as and use numerical bisection to obtain . Now, substituting the obtained value in Equation (16) gives , so the probability is approximately . For comparison, a 99% confidence interval obtained by performing 10 billion random simulations is . Once again, the analytic combinatorics estimates are accurate.
Figure 6 illustrates the precision of the estimates for different values of the deletion rate
and of the read size
k.
The probabilities can also be computed by recurrence using Theorem 3, after replacing
by
in expression (
15). Denoting
as
, one obtains for every integer
4.4. Substitutions, Deletions and Insertions
Here, we consider a model where all types of errors are allowed (also referred to as the “full error model”). Introducing insertions brings two additional difficulties: the first is that a substitution is indistinguishable from an insertion followed by a deletion (or a deletion followed by an insertion). By convention, we will count all these cases as substitutions. As a consequence, a deletion can never be found next to an insertion. The second difficulty is that insertions usually come in bursts. This is also the case of deletions, but we could neglect it because this does not affect the size of the interval (all deletions have size 0).
To model insertion bursts, we need to assign a probability r to the first insertion, and a probability to all subsequent insertions of the burst. We will still denote the probability of a substitution p and that of a correct nucleotide q, but here . We will also assume that an insertion burst stops with probability at each position of the burst.
Under this error model, reads can be thought of as walks on the transfer graph shown in
Figure 7. To not overload the figure, the body of the transfer graph is represented on the left, and the head and tail vertices on the right. The symbols
,
S and
I stand for error-free intervals, single substitutions and single insertions, respectively. The terms
,
and
are the same as in
Section 4.3. The terms
and
are the weighted generating functions of the first inserted nucleotide and of all subsequent nucleotides of the insertion burst, respectively. The burst terminates with probability
and is followed by an error-free interval or by a substitution. The total weight of these two cases is
, so we need to further scale the weighted generating functions by a factor
.
The expression of the weighted generating function of error-free intervals
is the same as in
Section 4.3, namely
The transfer matrix of the graph shown in
Figure 7 is
With the notations of Theorem 1,
,
,
and
From Theorem 1, the weighted generating function of all reads is
, which is equal to
where
and
are second degree polynomials defined as
Substituting in (
18) the expressions of
,
and
, we find
Again, we obtain the simple expression
and the probability that a read of size
k contains no seed is
. To find the weighted generating function of reads without an exact
-seed, we replace
in expression (
18) by its truncated version
We obtain the following expression
where
and
are defined as in expression (
19).
Remark 2. Note that when , then and , expression (21) becomes This is expression (15), i.e., the model described in Section 4.3. When we also have , this expression further simplifies to This is expression (10), i.e., the model described in Section 4.2. In other words, the error models described previously are special cases of this error model. As in the previous sections, we can use Theorem 2 to obtain asymptotic approximations for the probability that the reads contain no seed.
Proposition 4. The probability that a read of size k has no seed under the error model with substitutions, deletions and insertions is asymptotically equivalent towhere is the root with smallest modulus of the polynomial and If , then and . Otherwise, If , then divides the numerator and the denominator, which should be simplified to remain coprime. In this case, Theorem 2 should be applied to the simplified rational function.
Example 5. Approximate the probability that a read of size has no seed for , , , and . With these values, and . We need to solve . We rewrite the equation as and use bisection to solve it numerically, yielding . From expression (22), we obtain , so the probability that a read contains no seed is approximately . For comparison, a 99% confidence interval obtained by performing 10 billion random simulations is . Once again, the analytic combinatorics estimates are accurate.
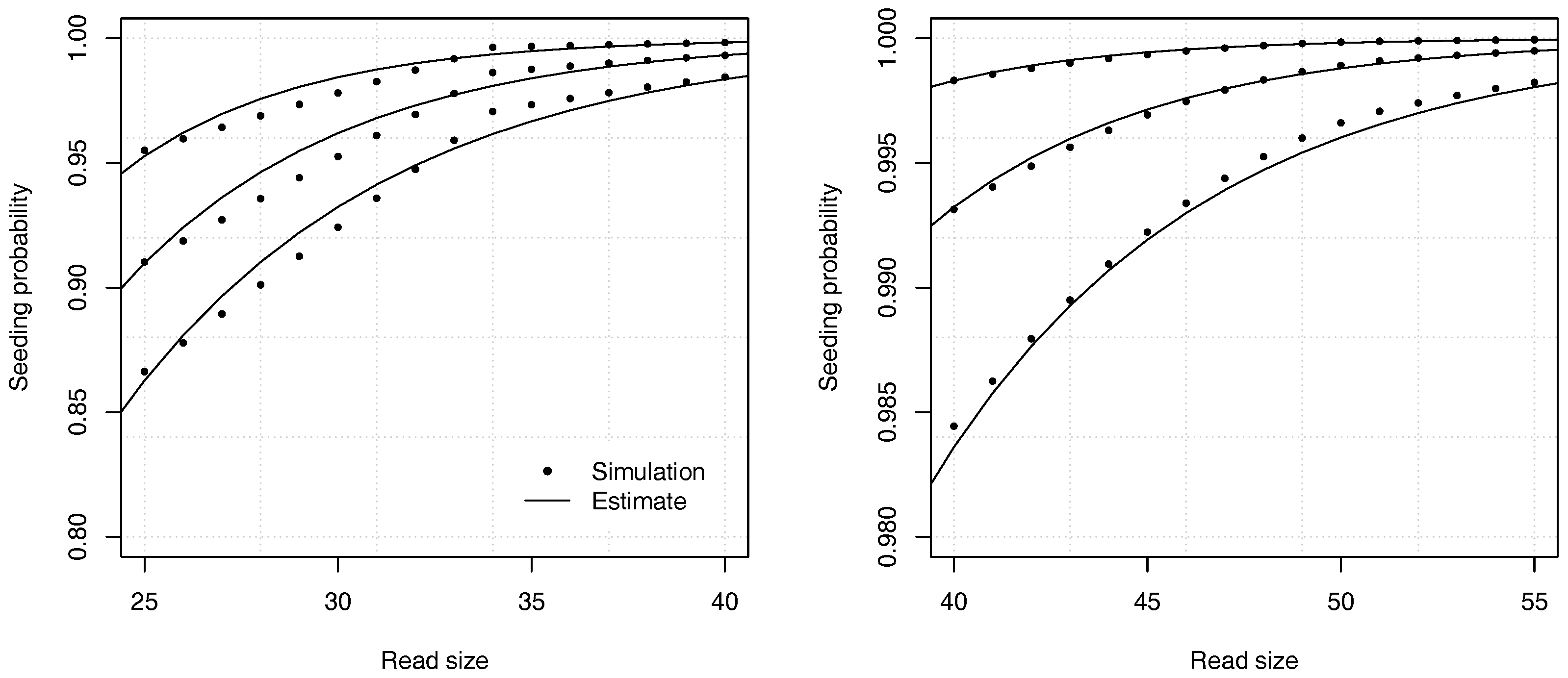
Figure 8 illustrates the precision of the estimates for different values of the insertion rate
r and of the read size
k.
The probabilities can also be computed by recurrence using Theorem 3, after replacing
by
in expression (
21). Denoting
as
, one obtains for every integer
4.5. Accuracy of the Approximations
So far, all the examples showed that the analytic combinatorics approximations are accurate. Indeed, the main motivation for our approach is to find estimates that converge exponentially fast to the target value. To find out whether we can use the approximations in place of the true values, we need to describe the behavior of the estimates in the worst conditions. The approximations become more accurate as the size of the sequence increases, i.e., as the reads become longer. This is somewhat inconvenient: the read size is usually fixed by the technology or by the problem at hand, so the user does not have easy ways to improve the accuracy. Overall, the approximations described above tend to be less accurate for short reads.
Another aspect is convergence speed. The proof of Theorem 2 shows that the rate of convergence is fastest when the dominant singularity has a significantly smaller
modulus than the other singularities. Conversely, convergence is slowest when at least one other singularity is almost as close to 0. The worst case for the approximation is thus when the reads are small and when the parameters are such that singularities have relatively close
moduli. It can be shown that, for the error model of uniform substitutions, this corresponds to small values of the error rate
p (see
Appendix A).
In practical terms, the situation above describes the specifications of the Illumina technology, where errors are almost always substitutions, occurring at a frequency around 1% on current instruments. Since the reads are often around 50 nucleotides, the analytic combinatorics estimates of the seeding probabilities are typically less accurate than suggested in the previous sections.
Figure 9 shows the accuracy of the estimates in one of the worst cases. The analytic combinatorics estimates are clearly distinct from the simulation estimates at the chosen scale, but the absolute difference is never higher than approximately
(and lower for read sizes above 40). Whether this error is acceptable depends on the problem. Often
p itself must be estimated, which is a more serious limitation on the precision than the convergence speed of the estimates. In most practical applications, the approximation error of Theorem 2 can be tolerated even in the worst case, but it is important to bear in mind that it may not be negligible for reads of size 50 or lower. If this level of precision is insufficient, the best option is to compute the coefficients by recurrence.
For long reads, the approximations rapidly gain in accuracy. Importantly, the calculations are also numerically stable, even for very long reads and for very high values of the error rate. To explore the behavior of the estimates, they were computed over a wide range of conditions, and compared to the values obtained by computing recurrence (
12).
The value of
is the
exact probability that a read of size
k does not contain a seed in the uniform substitution error model, when the probability of substitution is equal to
p. However, because the numbers are represented with finite precision, the computed value of
is also inexact in practice.
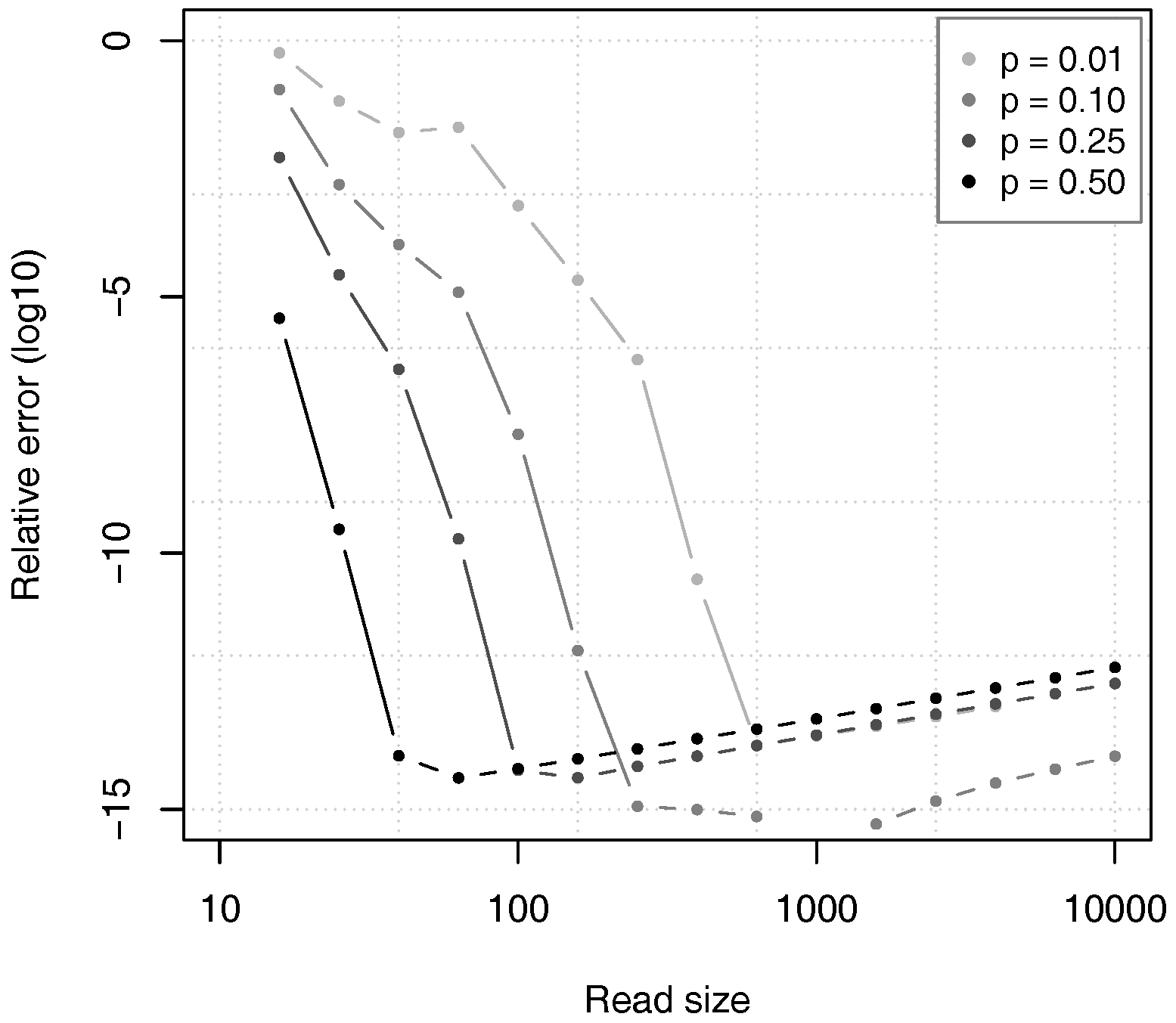
Figure 10 shows the relative error of the estimates given by Proposition 2, as compared to the value of
computed through equation (
12) in double precision arithmetic. For all the tested values of
p, the relative error first decreases to approximately
and then slowly rises. The reason is that the theoretical accuracy of the estimates increases with the read size
k, as justified by Theorem 2, but the errors in numerical approximations also increase and they finally dominate the error.
In spite of this artefact, it is clear that the relative error of the estimates remains low for very large read sizes. This means that the estimates are sufficiently close to the exact value, and that the calculations are numerically stable.
Figure 10 also confirms that the value of
p has a large influence on the convergence speed of the approximation. For high values of
p, the relative error drops faster than for low values of
p, as argued above.
The main reason for the numerical stability of the estimates is that the polynomial equations to solve can be properly represented with double precision numbers. For instance, in the extreme case of seeds of size 30 with a substitution rate , the leading term of in Proposition 2 is . This is several orders of magnitude above the machine epsilon (approximately equal to on most computers), sufficient to guarantee that this term will not underflow during the calculations, and thus that the dominant singularity will be computed with an adequate precision. The same applies for the other error models, as long as the rate of sequencing errors remains above , which is the case in the vast majority of practical applications.
In summary, the estimates presented above are accurate and numerically stable. In the case of short reads with low error rate, the precision may be limiting for some applications, but the approximations can be replaced by exact solutions. The approach presented here is thus a practical solution for computing seeding probabilities.


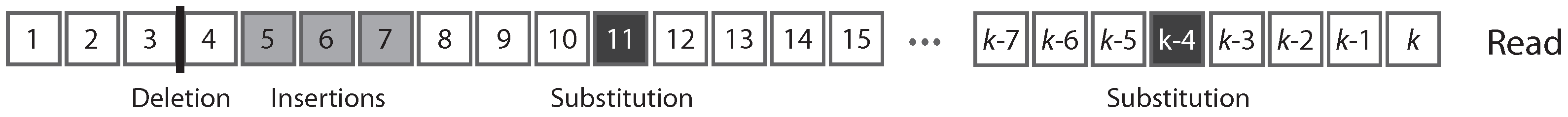{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









