Revisiting Chameleon Sequences in the Protein Data Bank


Abstract
:
1. Introduction
2. Materials and Methods
- For each PDB entry, extract all helix (H) and sheet (S) sequences as L-character strings (using the 1-character amino acid symbols) and save the PDBid, chain id, and starting residue number for each.
- For each PDB entry that includes more than one chain, sort the list of sequences and eliminate sequences that occur repeatedly in different chains (as it is assumed that such duplicates are the result of chains repeated in polymeric proteins).
- Add the character ‘H’ or ‘S’ (indicating helix or sheet) to the L+1th place to each sequence remaining.
- Once all PDB entries are read, sort the L-residue peptide sequences using all L+1 characters.
- Partition the sorted list into segments of identical sequences. If the first and last member of the partition has a different character at the L+1th position a chameleon is found.
- Perform the chameleon search starting with the longest possible, Lmax. It is set to 24 in the current implementation to be larger than the longest chameleon found, but it can be raised with minimal change in the code if needed (at the expense of using more memory).
- For all chameleon lengths L, Lmin ≤ L ≤ Lmax, create an L-character list consisting of: (a) all chameleons just found of length L; and (b) all L-character substrings of the already found chameleons. Lmin can be any positive number ≤ Lmax.
- Sort this combined list.
- Scan the sorted list. Whenever more than one identical string is found, and one of these was from the just found L-residue chameleon list, that chameleon should be dropped as it is part of a longer chameleon.
- 6.
- For all chameleon lengths L < Lmax create an L + 1-character list consisting of: (a) two copies of all occurrences of chameleons just found of length L, where the residue string is extended in both directions with the residue at that position of the protein the chameleon copy was found; and (b) all L + 1-character substrings of the already found chameleons.
- 7.
- Sort this combined list.
- 8.
- Scan the sorted list. Whenever only one identical string is found and it came from an L-residue chameleon that occurrence was a non-extendable one.
- (1)
- The number of occurrences of residue i in chameleons of length L NL,i divided by L × NCL, where NCL is the number of chameleons of exactly L residues, P1(L,i) = NL,i/( L × NCL) (or multiplied by 100 to obtain percentages).
- (2)
- P1(L,i) normalized by a measure of the overall propensity of residue i. There are different options:
- (2.1)
- P2(L,i) = P1(L,i)/0.05 (or 5 if percentages were used). This measure ignores the different probabilities of occurrences of the different amino acids. It was the measure used in [15].
- (2.2)
- P3(L,i) = P1(L,i)/P(i), where P(i) is the overall probability of occurrence of residue i.
- (2.3)
- P4(L,i) = P1(L,i)/PHS(i), where PHS(i) is the overall probability of occurrence of residue i in either a helix or in a sheet.
3. Results
4. Discussion
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Gnanakaran, S.; Nymeyer, H.; Portman, J.; Sanbonmatsu, K.Y.; Garcıa, A.E.; Tovchigrechko, A. Peptide folding simulations. Curr. Opin. Struct. Biol. 2003, 13, 68–174. [Google Scholar] [CrossRef]
- Rose, G.D.; Creamer, T.O. Protein folding: Predicting predicting. Proteins. Struct. Funct. Genet. 1994, 19, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.L.; Loogera, L.L. Extant fold-switching proteins are widespread. Proc. Natl. Acad. Sci. USA 2018, 115, 5968–5973. [Google Scholar] [CrossRef] [PubMed]
- Cohen, F.E. Protein misfolding and prion diseases. J. Mol. Biol. 1999, 283, 313–320. [Google Scholar] [CrossRef] [PubMed]
- DeToma, A.S.; Salamekh, S.; Ramamoorthy, A.; Lim, M.H. Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem. Soc. Rev. 2012, 41, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Eliezer, D. Biophysics of Parkinson’s disease: Structure and aggregation of alpha-synuclein. Curr. Protein Pept. Sci. 2009, 10, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, A.A. Complexity of protein folding. Bull. Math. Biol. 1993, 55, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.Y.; Fasman, G.D. Prediction of protein conformation. Biochemistry 1974, 13, 222–245. [Google Scholar] [CrossRef] [PubMed]
- Baldwinn, R.L.; Rose, G.D. Is protein folding hierarchic? II. Folding intermediates and transition states. Trends Biochem. Sci. 1999, 24, 185–191. [Google Scholar] [CrossRef]
- Sen, T.Z.; Cheng, H.; Kloczkowski, A.; Jernigan, R.L. A Consensus Data Mining secondary structure prediction by combining GOR V and Fragment Database Mining. Protein Sci. 2006, 15, 2499–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. On the use of sequence homologies to predict protein structure: Identical pentapeptides can have completely different conformations. Proc. Natl. Acad. Sci. USA 1984, 81, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Minor, D.L.; Kim, P.S. Context-dependent secondary structure formation of a designed protein sequence. Nature 1996, 380, 730–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezei, M. Chameleon sequences in the PDB. Prot. Eng. 1998, 11, 411–414. [Google Scholar] [CrossRef] [Green Version]
- Sudarsanam, S. Structural diversity of sequentially identical subsequences of proteins: Identical octapeptides can have different conformations. Proteins Struct. Funct. Genet. 1998, 30, 228–231. [Google Scholar] [CrossRef]
- Gendoo, D.M.A.; Harrison, P.M. Discordant and chameleon sequences: Their distribution and implications for amyloidogenicity. Protein Sci. 2011, 20, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Jaromczyk, J.W.; Xu, Y. Analysis of chameleon sequences and their implications in biological processes. Proteins Struct. Funct. Genet. 2007, 67, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kinch, L.N.; Karplus, P.A.; Grishin, N.V. ChSeq: A database of chameleon sequences. Protein Sci. 2015, 24, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Conte, L.; Ailey, B.; Hubbard, T.J.; Brenner, S.E.; Murzin, A.G.; Chothia, C. SCOP: A structural classification of proteins database. Nucleic Acids Res. 2000, 28, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Mezei, M. Simulaid: A simulation facilitator and analysis program. J. Comput. Chem. 2010, 31, 2658–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Gr. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schrodinger, L. The PyMOL Molecular Graphics System, Version 1.8.6.2; Schrödinger, LLC: New York, NY, USA, 2010.
- The Uniprot Consortium Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169.
- De Lucrezia, D.; Slanzi, D.; Poli, I.; Polticelli, F.; Minervini, G. Do natural proteins differ from random sequences polypeptides? Natural vs. random proteins classification using an evolutionary neural network. PLoS ONE 2012, 7, e36634. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
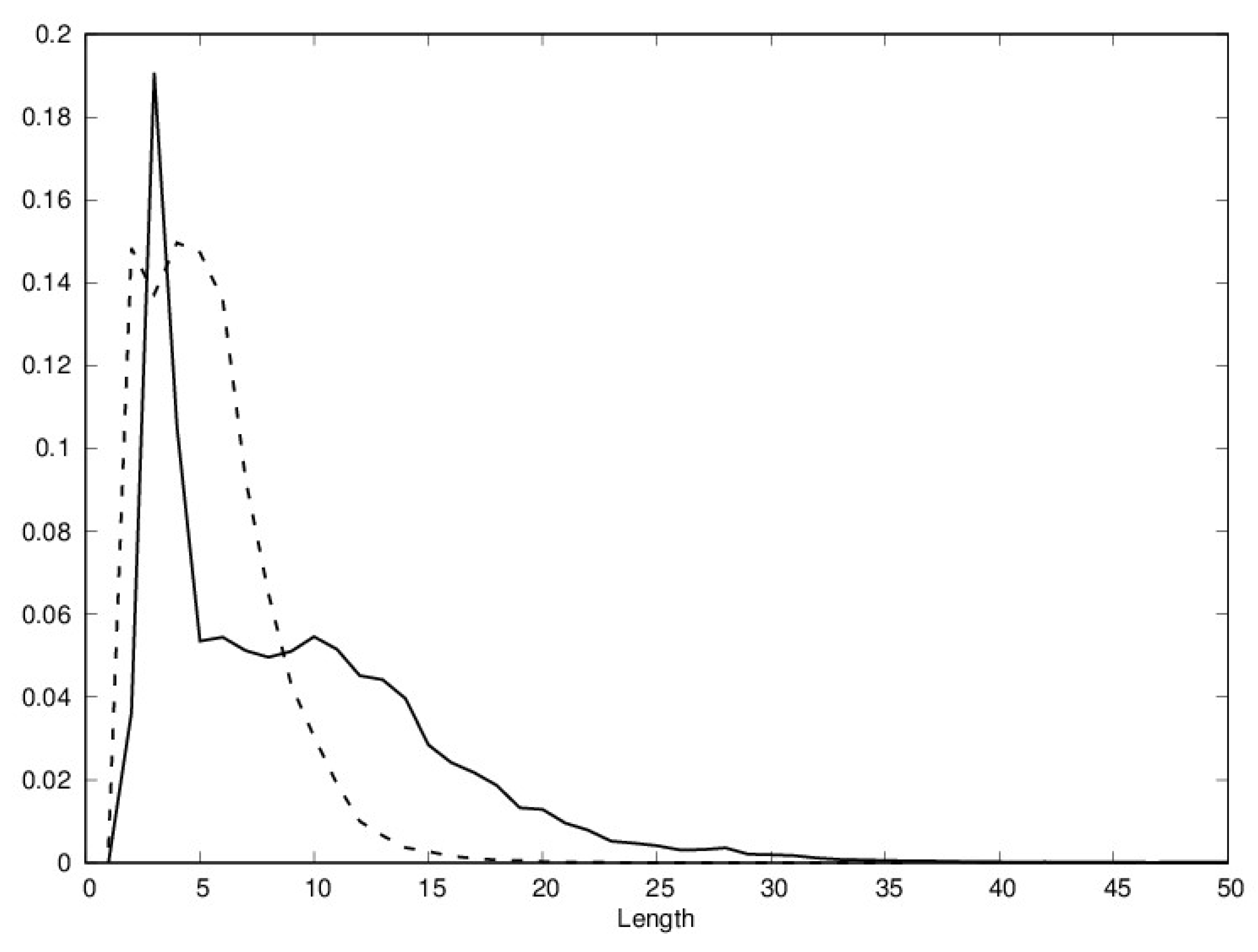{kind=link}
| 20 Residue Based | Low-Resolution (HP+−) | Low-Resolution (HPC) | |||||
|---|---|---|---|---|---|---|---|
| Length | Number of Sequences | NL(cham) | NL,X(cham) | NL(cham) | NL,X(cham) | NL(cham) | NL,X(cham) |
| 20 | 852,133 | 1 | 1 | 1 | 1 | 1 | 1 |
| 19 | 1,012,722 | 2 | 0 | 6 | 4 | 7 | 5 |
| 18 | 1,211,135 | 3 | 0 | 13 | 2 | 21 | 8 |
| 17 | 1,454,006 | 3 | 0 | 29 | 9 | 58 | 23 |
| 16 | 1,755,667 | 3 | 0 | 97 | 52 | 207 | 113 |
| 15 | 2,096,973 | 7 | 1 | 333 | 171 | 781 | 432 |
| 14 | 2,529,092 | 10 | 1 | 1245 | 693 | 3143 | 1824 |
| 13 | 3,055,102 | 14 | 1 | 440 | 2394 | 10,597 | 5506 |
| 12 | 3,685,170 | 18 | 0 | 13,359 | 6698 | 25,969 | 10,448 |
| 11 | 4,449,698 | 23 | 1 | 30,300 | 12,469 | 38,778 | 8658 |
| 10 | 5,380,037 | 32 | 4 | 45,328 | 13,386 | 32,363 | 2623 |
| 9 | 6,490,617 | 52 | 11 | 42,728 | 7408 | 16,527 | 265 |
| 8 | 7,827,484 | 149 | 79 | 26,482 | 2228 | 6390 | 18 |
| 7 | 9,444,433 | 2063 | 1822 | 11,499 | 314 | 2183 | 0 |
| 6 | 11,433,790 | 40,595 | 36,669 | 3769 | 14 | 729 | 0 |
| 5 | 13,801,708 | 249,407 | 187,803 | 1061 | 0 | 242 | 0 |
| PDBid1 | Ch1 | Residue1 | PDBid2 | Ch2 | Residue1 | Sequence | Length | Number |
|---|---|---|---|---|---|---|---|---|
| 1AMB | A | 3 | 5OQV | F | 3 | EFRHDSGYEVHHQKLVFFAE | 20 | 2 |
| 1VCL | B | 343 | 3W9T | F | 343 | VKVTASLSKAWTNSQ | 15 | 2 |
| 2KKW | A | 42 | 2N0A | G | 42 | SKTKEGVVHGVATV | 14 | 2 |
| 1VCL | B | 320 | 3W9T | F | 320 | AGVAVEVSSTIEK | 13 | 4 |
| 2LLM | A | 19 | 2BEG | D | 31 | IIGLMVGGVVI | 11 | 4 |
| 2R1B | B | 145 | 3MW2 | B | 138 | LGRVVDEWLL | 10 | 2 |
| 2MDK | A | 115 | 3IKK | A | 118 | MDSKLRCVFE | 10 | 7 |
| 1BA4 | A | 24 | 2MXU | G | 24 | VGSNKGAIIG | 10 | 3 |
| 1G2C | C | 28 | 4JHW | F | 133 | VSVLTSKVLD | 10 | 8 |
| 2QZV | B | 340 | 2ZV5 | A | 328 | EQQGLLLKA | 9 | 4 |
| 1AMB | A | 16 | 2LMN | E | 16 | KLVFFAEDV | 9 | 9 |
| 2GEJ | A | 143 | 4NC9 | C | 127 | KSLTLSVFQ | 9 | 2 |
| 5N8Y | G | 86 | 4KSO | D | 86 | LIGLDLLYG | 9 | 2 |
| 1AMB | A | 17 | 2OTK | C | 17 | LVFFAEDVG | 9 | 2 |
| 2KB8 | A | 16 | 5KO0 | A | 2 | LVHSSNNFG | 9 | 3 |
| 5XLO | D | 74 | 6B44 | E | 76 | PNLQTVDVA | 9 | 3 |
| 5EY7 | A | 152 | 2OWY | B | 123 | RSSTFAAIA | 9 | 5 |
| 1AMB | A | 18 | 2BEG | D | 18 | VFFAEDVGS | 9 | 5 |
| 1XQ8 | A | 70 | 2N0A | G | 70 | VVTGVTAVA | 9 | 3 |
| 2N2C | D | 28 | 6CFH | B | 2 | WGMMGMLAS | 9 | 3 |
| %(helix) | %(sheet) | %(helix)/%(aa) | %(sheet)/%(aa) | %(aa) | %(aa,Expasy) | |
|---|---|---|---|---|---|---|
| ALA | 11.10 | 6.25 | 1.386 | 0.780 | 8.01 | 8.25 |
| CYS | 1.16 | 1.95 | 0.855 | 1.442 | 1.35 | 1.37 |
| ASP | 4.97 | 3.07 | 0.882 | 0.545 | 5.64 | 5.45 |
| GLU | 8.87 | 4.51 | 1.342 | 0.682 | 6.61 | 6.75 |
| PHE | 4.05 | 5.58 | 1.036 | 1.427 | 3.91 | 3.86 |
| GLY | 3.59 | 4.94 | 0.484 | 0.665 | 7.42 | 7.07 |
| HIS | 2.15 | 2.26 | 0.798 | 0.842 | 2.69 | 2.27 |
| ILE | 6.10 | 9.62 | 1.087 | 1.715 | 5.61 | 5.96 |
| LYS | 6.50 | 4.74 | 1.100 | 0.802 | 5.91 | 5.84 |
| LEU | 11.89 | 10.23 | 1.319 | 1.135 | 9.02 | 9.66 |
| MET | 2.79 | 2.22 | 1.189 | 0.948 | 2.34 | 2.42 |
| ASN | 3.26 | 2.60 | 0.779 | 0.620 | 4.19 | 4.06 |
| PRO | 2.42 | 1.90 | 0.520 | 0.409 | 4.65 | 4.70 |
| GLN | 4.65 | 3.00 | 1.231 | 0.794 | 3.78 | 3.93 |
| ARG | 6.04 | 4.47 | 1.165 | 0.862 | 5.18 | 5.53 |
| SER | 4.88 | 5.40 | 0.773 | 0.856 | 6.31 | 6.56 |
| THR | 4.29 | 6.87 | 0.769 | 1.229 | 5.59 | 5.34 |
| VAL | 6.36 | 13.52 | 0.905 | 1.925 | 7.02 | 6.87 |
| TRP | 1.52 | 1.81 | 1.137 | 1.354 | 1.34 | 1.08 |
| TYR | 3.43 | 5.06 | 0.998 | 1.472 | 3.44 | 2.92 |
| res | N(cham) | %(cham) | %(HS) | %(cham)/%(HS) | %(cham)/%(aa) | %(cham)/5 |
|---|---|---|---|---|---|---|
| ALA | 102,472 | 8.74 | 9.32 | 0.94 | 1.09 | 1.75 |
| CYS | 12,346 | 1.05 | 1.45 | 0.73 | 0.78 | 0.21 |
| ASP | 37,854 | 3.23 | 4.27 | 0.76 | 0.57 | 0.65 |
| GLU | 68,719 | 5.86 | 7.27 | 0.81 | 0.89 | 1.17 |
| PHE | 62,609 | 5.34 | 4.61 | 1.16 | 1.37 | 1.07 |
| GLY | 54,815 | 4.67 | 4.09 | 1.14 | 0.63 | 0.93 |
| HIS | 20,950 | 1.79 | 2.19 | 0.82 | 0.66 | 0.36 |
| ILE | 108,171 | 9.22 | 7.39 | 1.25 | 1.64 | 1.84 |
| LYS | 60,089 | 5.12 | 5.85 | 0.88 | 0.87 | 1.02 |
| LEU | 143,655 | 12.25 | 11.28 | 1.09 | 1.36 | 2.45 |
| MET | 26,285 | 2.24 | 2.58 | 0.87 | 0.96 | 0.45 |
| ASN | 31,384 | 2.68 | 3.02 | 0.89 | 0.64 | 0.54 |
| PRO | 11,719 | 1.00 | 2.23 | 0.45 | 0.21 | 0.20 |
| GLN | 40,116 | 3.42 | 4.04 | 0.85 | 0.91 | 0.68 |
| ARG | 61,077 | 5.21 | 5.46 | 0.95 | 1.01 | 1.04 |
| SER | 63,667 | 5.43 | 5.07 | 1.07 | 0.86 | 1.09 |
| THR | 70,060 | 5.97 | 5.24 | 1.14 | 1.07 | 1.19 |
| VAL | 131,596 | 11.22 | 8.99 | 1.25 | 1.60 | 2.24 |
| TRP | 12,907 | 1.10 | 1.63 | 0.68 | 0.82 | 0.22 |
| TYR | 52,136 | 4.45 | 4.03 | 1.10 | 1.29 | 0.89 |
| N(cham) | %(cham) | %(HS) | %(cham)/%(HS) | %(cham)/%(aa) | %(cham)/5 | |
|---|---|---|---|---|---|---|
| H | 150,809 | 42.43 | 45.67 | 0.93 | 1.04 | 1.27 |
| P | 124,200 | 34.94 | 31.48 | 1.11 | 0.98 | 1.05 |
| C | 80,416 | 22.63 | 22.85 | 0.99 | 0.97 | 0.68 |
| N(cham) | %(cham) | %(HS) | %(cham)/%(HS) | %(cham)/%(aa) | %(cham)/5 | |
|---|---|---|---|---|---|---|
| H | 205,518 | 42.58 | 45.67 | 0.93 | 1.04 | 1.7 |
| O | 160,794 | 33.31 | 31.48 | 1.06 | 0.93 | 1.33 |
| N | 55,382 | 11.47 | 11.54 | 0.99 | 0.94 | 0.46 |
| P | 60,985 | 12.63 | 11.31 | 1.12 | 1.14 | 0.51 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezei, M. Revisiting Chameleon Sequences in the Protein Data Bank. Algorithms 2018, 11, 114. https://doi.org/10.3390/a11080114
Mezei M. Revisiting Chameleon Sequences in the Protein Data Bank. Algorithms. 2018; 11(8):114. https://doi.org/10.3390/a11080114
Chicago/Turabian StyleMezei, Mihaly. 2018. "Revisiting Chameleon Sequences in the Protein Data Bank" Algorithms 11, no. 8: 114. https://doi.org/10.3390/a11080114
APA StyleMezei, M. (2018). Revisiting Chameleon Sequences in the Protein Data Bank. Algorithms, 11(8), 114. https://doi.org/10.3390/a11080114