
Molecular Subtyping and Outlier Detection in Human Disease Using the Paraclique Algorithm



Abstract
1. Introduction
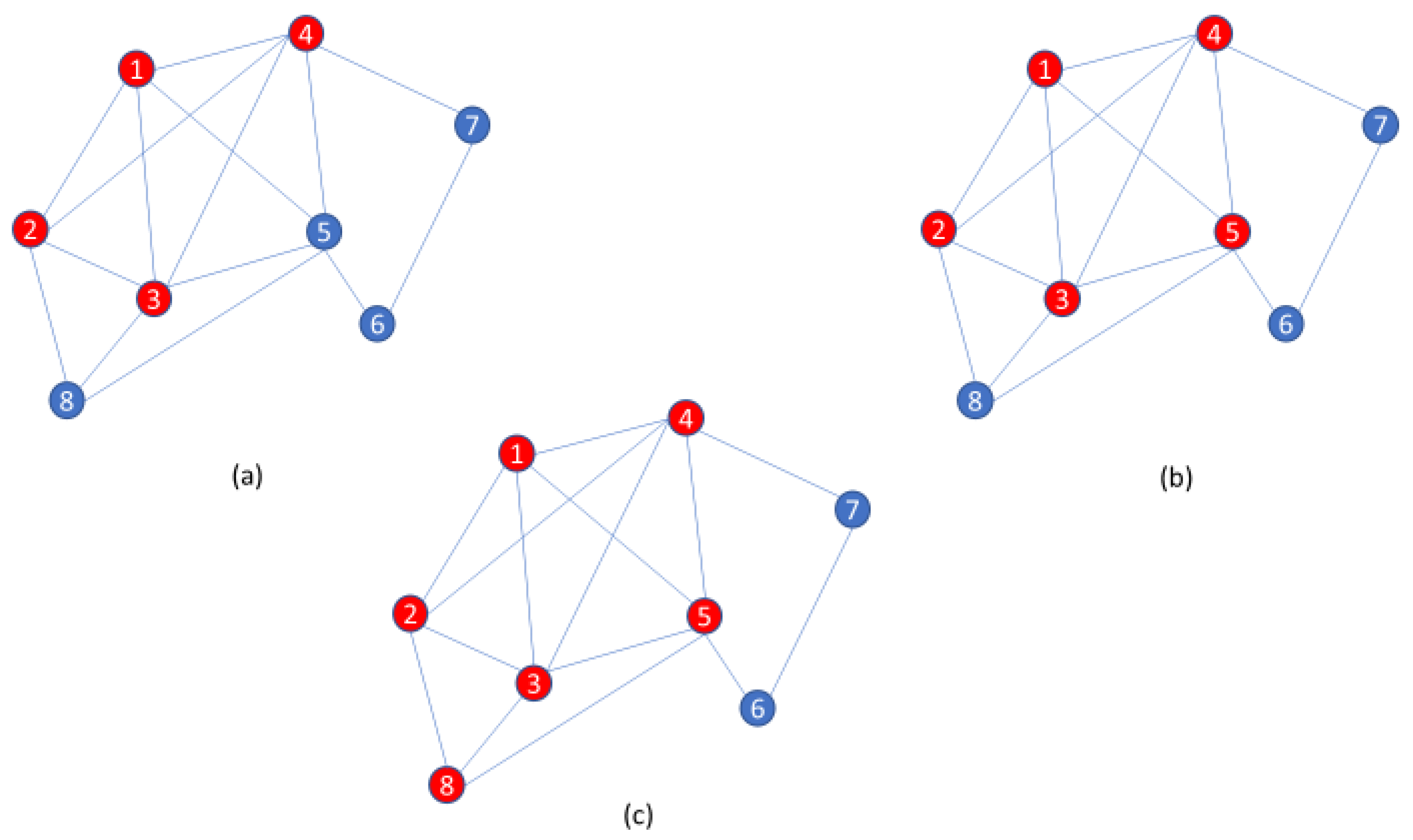
2. Methodology
3. Experimental Results
3.1. Discussion
3.2. A Search for Unrecognized Subtypes
3.2.1. Asthma
3.2.2. Breast Cancer
3.2.3. Chronic Lymphocytic Leukemia
3.2.4. Colorectal Cancer
3.3. Alignment with Previously Known Subtypes
3.3.1. Gastric Cancer
3.3.2. Non-Small Cell Lung Cancer
3.3.3. Comparison with Other Methods
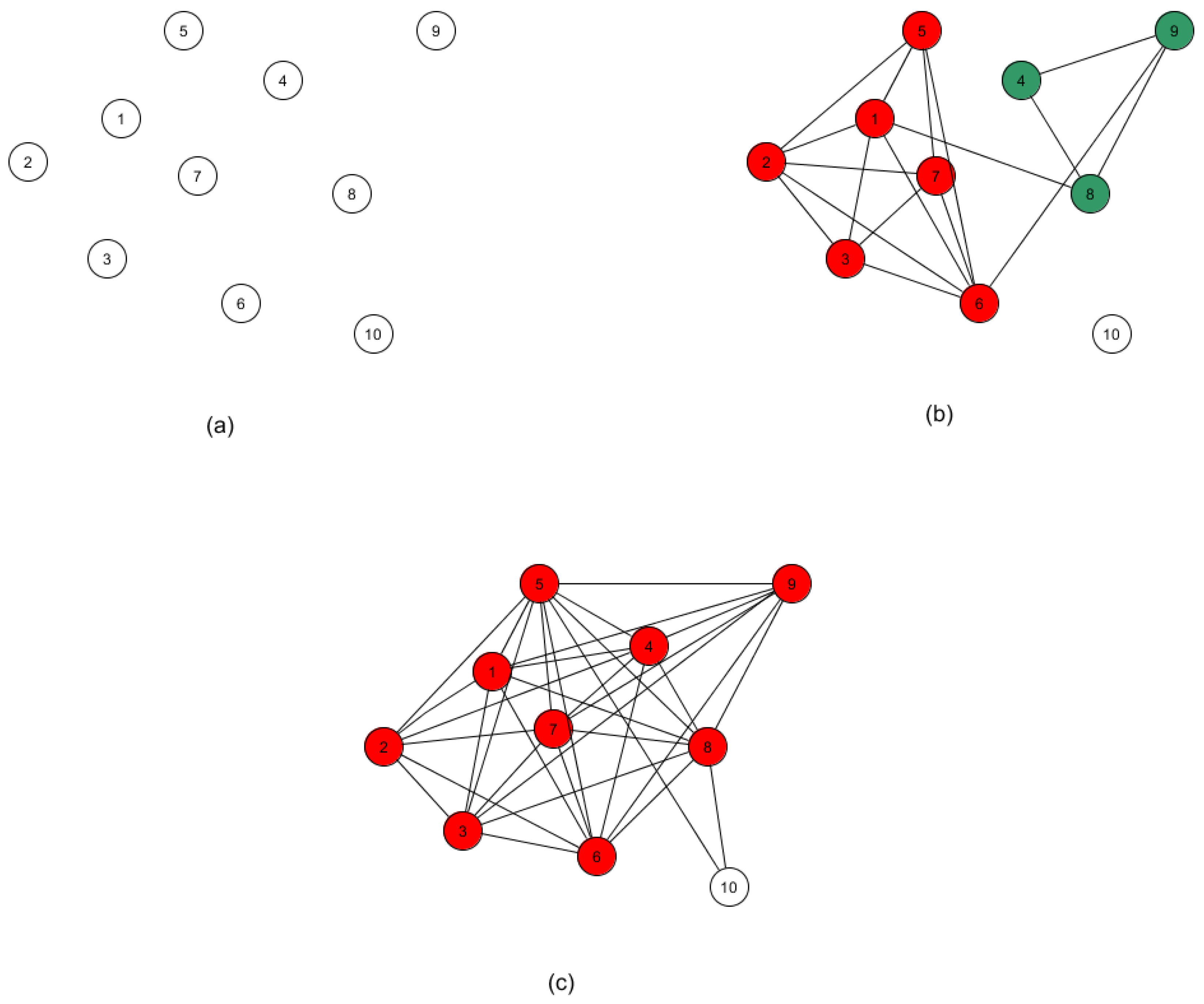
4. Outlier Detection
5. Summary, Discussion and Directions for Future Research
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savas, P.; Hughes, B.; Solomon, B. Targeted Therapy in Lung Cancer: IPASS and Beyond, Keeping Abreast of the Explosion of Targeted Therapies for Lung Cancer. J. Thorac. Dis. 2013, 5 (Suppl. 5), S579. [Google Scholar]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; de Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Leith, C.P.; Kopecky, K.J.; Godwin, J.; McConnell, T.; Slovak, M.L.; Chen, I.-M.; Head, D.R.; Appelbaum, F.R.; Willman, C.L. Acute myeloid leukemia in the elderly: Assessment of multidrug resistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably distinct responses to standard chemotherapy. A Southwest Oncology Group study. Blood J. Am. Soc. Hematol. 1997, 89, 3323–3329. [Google Scholar] [CrossRef]
- Balko, J.M.; Cook, R.S.; Vaught, D.B.; Kuba, M.G.; Miller, T.W.; Bhola, N.E.; Sanders, M.E.; Granja-Ingram, N.M.; Smith, J.J.; Meszoely, I.M.J.N.M. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat. Med. 2012, 18, 1052–1059. [Google Scholar] [CrossRef]
- Kuruvilla, M.E.; Lee, F.E.-H.; Lee, G.B. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233. [Google Scholar] [CrossRef]
- Di Fede, G.; Catania, M.; Maderna, E.; Ghidoni, R.; Benussi, L.; Tonoli, E.; Giaccone, G.; Moda, F.; Paterlini, A.; Campagnani, I. Molecular subtypes of Alzheimer’s disease. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Weiser, M.; Simon, J.M.; Kochar, B.; Tovar, A.; Israel, J.W.; Robinson, A.; Gipson, G.R.; Schaner, M.S.; Herfarth, H.H.; Sartor, R.B. Molecular classification of Crohn’s disease reveals two clinically relevant subtypes. Gut 2018, 67, 36–42. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Kohonen, T. The self-organizing map. Proc. IEEE 1990, 78, 1464–1480. [Google Scholar] [CrossRef]
- Luo, F.; Khan, L.; Bastani, F.; Yen, I.-L.; Zhou, J. A dynamically growing self-organizing tree (DGSOT) for hierarchical clustering gene expression profiles. Bioinformatics 2004, 20, 2605–2617. [Google Scholar] [CrossRef]
- Tamayo, P.; Slonim, D.; Mesirov, J.; Zhu, Q.; Kitareewan, S.; Dmitrovsky, E.; Lander, E.S.; Golub, T.R. Interpreting patterns of gene expression with self-organizing maps: Methods and application to hematopoietic differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 2907–2912. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Ambrogi, F.; Biganzoli, E.; Querzoli, P.; Ferretti, S.; Boracchi, P.; Alberti, S.; Marubini, E.; Nenci, I. Molecular subtyping of breast cancer from traditional tumor marker profiles using parallel clustering methods. Clin. Cancer Res. 2006, 12, 781–790. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wessman, J.; Paunio, T.; Tuulio-Henriksson, A.; Koivisto, M.; Partonen, T.; Suvisaari, J.; Turunen, J.A.; Wedenoja, J.; Hennah, W.; Pietiläinen, O.P. Mixture model clustering of phenotype features reveals evidence for association of DTNBP1 to a specific subtype of schizophrenia. Biol. Psychiatry 2009, 66, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Jay, J.J.; Eblen, J.D.; Zhang, Y.; Benson, M.; Perkins, A.D.; Saxton, A.M.; Voy, B.H.; Chesler, E.J.; Langston, M.A. A systematic comparison of genome-scale clustering algorithms. BMC Bioinform. 2012, 13, S7. [Google Scholar] [CrossRef]
- Chesler, E.J.; Langston, M.A. Combinatorial Genetic Regulatory Network Analysis Tools for High Throughput Transcriptomic Data. In Systems Biology and Regulatory Genomics; Eskin, E., Ed.; Springer: Berlin, Germany, 2006; pp. 150–165. [Google Scholar]
- Bron, C.; Kerbosch, J. Algorithm 457: Finding all cliques of an undirected graph. Commun. ACM 1973, 16, 575–577. [Google Scholar] [CrossRef]
- Palla, G.; Derényi, I.; Farkas, I.; Vicsek, T. Uncovering the overlapping community structure of complex networks in nature and society. Nature 2005, 435, 814–818. [Google Scholar] [CrossRef]
- Langston, M.A.; Perkins, A.D.; Saxton, A.M.; Scharff, J.A.; Voy, B.H. Innovative Computational Methods for Transcriptomic Data Analysis: A Case Study in the Use of FPT for Practical Algorithm Design and Implementation. Comput. J. 2008, 51, 26–38. [Google Scholar] [CrossRef]
- Schoenrock, A.; Samanfar, B.; Pitre, S.; Hooshyar, M.; Jin, K.; Phillips, C.A.; Wang, H.; Phanse, S.; Omidi, K.; Gui, Y.; et al. Efficient prediction of human protein-protein interactions at a global scale. BMC Bioinform. 2014, 15, 383. [Google Scholar] [CrossRef]
- Macartney-Coxson, D.; Benton, M.C.; Blick, R.; Stubbs, R.S.; Hagan, R.D.; Langston, M.A. Genome-wide DNA methylation analysis reveals loci that distinguish different types of adipose tissue in obese individuals. Clin. Epigenet. 2017, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Langston, M.A.; Levine, R.S.; Kilbourne, B.J.; Rogers, G.L.; Kershenbaum, A.D.; Baktash, S.H.; Coughlin, S.S.; Saxton, A.M.; Agboto, V.K.; Hood, D.B.; et al. Scalable combinatorial tools for health disparities research. Int. J. Environ. Res. Public Health 2014, 11, 10419–10443. [Google Scholar] [CrossRef]
- Grubb, M.C.; Kilbourne, B.J.; Kilbourne, C. Socioeconomic, Environmental and Geographic Factors and United States Lung Cancer Mortality, 1999–2009. Fam. Med. Community Health 2017, 5, 3–12. [Google Scholar] [CrossRef]
- Eblen, J.D.; Gerling, I.C.; Saxton, A.M.; Wu, J.; Snoddy, J.R.; Langston, M.A. Graph Algorithms for Integrated Biological Analysis, with Applications to Type 1 Diabetes Data; World Scientific: Singapore, 2009; pp. 207–222. [Google Scholar]
- Bruhn, S.; Barrenas, F.; Mobini, R.; Andersson, B.A.; Chavali, S.; Egan, B.S.; Hovig, E.; Sandve, G.K.; Langston, M.A.; Rogers, G.; et al. Increased expression of IRF4 and ETS1 in CD4+ cells from patients with intermittent allergic rhinitis. Allergy 2012, 67, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Palmer, O.M.P.; Rogers, G.; Yende, S.; Angus, D.C.; Clermont, G.; Langston, M.A. Graph Theoretical Analysis of Genome-Scale Data: Examination of Gene Activation Occurring in the Setting of Community-Acquired Pneumonia. Shock 2018, 50, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Voy, B.H.; Scharff, J.A.; Perkins, A.D.; Saxton, A.M.; Borate, B.; Chesler, E.J.; Branstetter, L.K.; Langston, M.A. Extracting Gene Networks for Low-Dose Radiation using Graph Theoretical Algorithms. PLoS Comput. Biol. 2006, 2, e89. [Google Scholar] [CrossRef]
- Bomze, I.; Budinich, M.; Pardalos, P.; Pelillo, M. The Maximum Clique Problem. In Handbook of Combinatorial Optimization; Du, D.-Z., Pardalos, P.M., Eds.; Kluwer Academic Publishers: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Hagan, R.D.; Langston, M.A.; Wang, K. Lower Bounds on Paraclique Density. Discret. Appl. Math. 2016, 204, 208–212. [Google Scholar] [CrossRef]
- Lu, Y.; Phillips, C.A.; Chesler, E.J.; Langston, M.A. Clique Selection and its Effect on Paraclique Enrichment: An Experimental Study. In Proceedings of the International Conference on Bioinformatics and Computational Biology, San Francisco, CA, USA, 23–25 March 2020. [Google Scholar]
- Graph Algorithms Pipeline for Pathway Analysis. Available online: https://grappa.eecs.utk.edu (accessed on 20 October 2020).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wang, K.; Phillips, C.A.; Saxton, A.M.; Langston, M.A. EntropyExplorer: An R package for computing and comparing differential Shannon entropy, differential coefficient of variation and differential expression. BMC Res. Notes 2015, 8, 832. [Google Scholar] [CrossRef]
- Perkins, A.D.; Langston, M.A. Threshold Selection in Gene Co-Expression Networks Using Spectral Graph Theory Techniques. BMC Bioinform. 2009, 10, S4. [Google Scholar] [CrossRef]
- Pablo-Fernández, E.D.; Lees, A.J.; Holton, J.L.; Warner, T.T. Prognosis and Neuropathologic Correlation of Clinical Subtypes of Parkinson Disease. JAMA Neurol. 2019, 76, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R. Type 2 Diabetes: A Multifaceted Disease. Diabetologia 2019, 62, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Bope, E.T.; Kellerman, R.D. Conn’s Current Therapy 2016; Elsevier Health Sciences: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Woodruff, P.G.; Boushey, H.A.; Dolganov, G.M.; Barker, C.S.; Yang, Y.H.; Donnelly, S.; Ellwanger, A.; Sidhu, S.S.; Dao-Pick, T.P.; Pantoja, C. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc. Natl. Acad. Sci. USA 2007, 104, 15858–15863. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, P.G. Subtypes of asthma defined by epithelial cell expression of messenger RNA and microRNA. Ann. Am. Thorac. Soc. 2013, 10, S186–S189. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.; Easton, D.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.; Weber, B.; Lenoir, G.; Chang-Claude, J. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef]
- Easton, D.; Bishop, D.; Ford, D.; Crockford, G. Genetic linkage analysis in familial breast and ovarian cancer: Results from 214 families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1993, 52, 678. [Google Scholar]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Pedraza, V.; Gomez-Capilla, J.A.; Escaramis, G.; Gomez, C.; Torné, P.; Rivera, J.M.; Gil, A.; Araque, P.; Olea, N.; Estivill, X. Gene expression signatures in breast cancer distinguish phenotype characteristics, histologic subtypes, and tumor invasiveness. Cancer 2010, 116, 486–496. [Google Scholar] [CrossRef]
- Srour, N.; Reymond, M.A.; Steinert, R. Lost in translation? A systematic database of gene expression in breast cancer. Pathobiology 2008, 75, 112–118. [Google Scholar] [CrossRef]
- Rudland, S.D.; Martin, L.; Roshanlall, C.; Winstanley, J.; Leinster, S.; Platt-Higgins, A.; Carroll, J.; West, C.; Barraclough, R.; Rudland, P. Association of S100A4 and osteopontin with specific prognostic factors and survival of patients with minimally invasive breast cancer. Clin. Cancer Res. 2006, 12, 1192–1200. [Google Scholar] [CrossRef]
- King, E.R.; Tung, C.S.; Tsang, Y.T.; Zu, Z.; Lok, G.T.; Deavers, M.T.; Malpica, A.; Wolf, J.K.; Lu, K.H.; Birrer, M.J. The anterior gradient homolog 3 (AGR3) gene is associated with differentiation and survival in ovarian cancer. Am. J. Surg. Pathol. 2011, 35, 904. [Google Scholar] [CrossRef]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitão, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011. [Google Scholar] [CrossRef]
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.-X. EpCAM and α-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008, 68, 1451–1461. [Google Scholar] [CrossRef]
- Rozman, C.; Montserrat, E. Chronic lymphocytic leukemia. N. Engl. J. Med. 1995, 333, 1052–1057. [Google Scholar] [CrossRef]
- Görgün, G.; Holderried, T.A.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J. Clin. Investig. 2005, 115, 1797–1805. [Google Scholar] [CrossRef]
- Wiestner, A.; Rosenwald, A.; Barry, T.S.; Wright, G.; Davis, R.E.; Henrickson, S.E.; Zhao, H.; Ibbotson, R.E.; Orchard, J.A.; Davis, Z. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood 2003, 101, 4944–4951. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.K.; Ward, E.; Kohler, B.A.; Eheman, C.; Zauber, A.G.; Anderson, R.N.; Jemal, A.; Schymura, M.J.; Lansdorp-Vogelaar, I.; Seeff, L.C. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010, 116, 544–573. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Hong, Y.; Downey, T.; Eu, K.W.; Koh, P.K.; Cheah, P.Y. A ‘metastasis-prone’signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin. Exp. Metastasis 2010, 27, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Belov, L.; Zhou, J.; Christopherson, R.I. Cell surface markers in colorectal cancer prognosis. Int. J. Mol. Sci. 2010, 12, 78–113. [Google Scholar] [CrossRef] [PubMed]
- Besson, D.; Pavageau, A.-H.; Valo, I.; Bourreau, A.; Bélanger, A.; Eymerit-Morin, C.; Moulière, A.; Chassevent, A.; Boisdron-Celle, M.; Morel, A. A quantitative proteomic approach of the different stages of colorectal cancer establishes OLFM4 as a new nonmetastatic tumor marker. Mol. Cell. Proteom. 2011, 10, M111-009712. [Google Scholar] [CrossRef]
- Huang, M.-Y.; Wang, H.-M.; Chang, H.-J.; Hsiao, C.-P.; Wang, J.-Y.; Lin, S.-R. Overexpression of S100B, TM4SF4, and OLFM4 genes is correlated with liver metastasis in Taiwanese colorectal cancer patients. DNA Cell Biol. 2012, 31, 43–49. [Google Scholar] [CrossRef]
- Chia, N.-Y.; Deng, N.; Das, K.; Huang, D.; Hu, L.; Zhu, Y.; Lim, K.H.; Lee, M.-H.; Wu, J.; Sam, X.X. Regulatory crosstalk between lineage-survival oncogenes KLF5, GATA4 and GATA6 cooperatively promotes gastric cancer development. Gut 2015, 64, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Tan, I.B.; Das, K.; Deng, N.; Zouridis, H.; Pattison, S.; Chua, C.; Feng, Z.; Guan, Y.K.; Ooi, C.H. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology 2013, 145, 554–565. [Google Scholar] [CrossRef]
- Wu, Y.; Grabsch, H.; Ivanova, T.; Tan, I.B.; Murray, J.; Ooi, C.H.; Wright, A.I.; West, N.P.; Hutchins, G.G.; Wu, J. Comprehensive genomic meta-analysis identifies intra-tumoural stroma as a predictor of survival in patients with gastric cancer. Gut 2013, 62, 1100–1111. [Google Scholar] [CrossRef]
- Kuner, R.; Muley, T.; Meister, M.; Ruschhaupt, M.; Buness, A.; Xu, E.C.; Schnabel, P.; Warth, A.; Poustka, A.; Sültmann, H. Global gene expression analysis reveals specific patterns of cell junctions in non-small cell lung cancer subtypes. Lung Cancer 2009, 63, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lazar, C.; Taminau, J.; Meganck, S.; Steenhoff, D.; Coletta, A.; Molter, C.; Schaetzen, V.D.; Duque, R.; Bersini, H.; Nowé, A. A Survey on Filter Techniques for Feature Selection in Gene Expression Microarray Analysis. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 30, 1106–1119. [Google Scholar] [CrossRef]
- Nguyen, T.; Tagett, R.; Diaz, D.; Draghici, S. A novel approach for data integration and disease subtyping. Genome Res. 2017, 27, 2025–2039. [Google Scholar] [CrossRef]
- Krishnagopal, S.; Coelln, R.V.; Shulman, L.M.; Girvan, M. Identifying and predicting Parkinson’s disease subtypes through trajectory clustering via bipartite networks. PLoS ONE 2020, 15, e0233296. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | GEO Accession | Patients | Probes | ||
|---|---|---|---|---|---|
| Case | Control | Initial | Filtered | ||
| Asthma | GSE4302 | 42 | 28 | 54,675 | 2322 |
| Breast Cancer | GSE10810 | 31 | 27 | 18,382 | 11,531 |
| Chronic Lymphocytic Leukemia | GSE8835 | 24 | 12 | 22,283 | 1338 |
| Colorectal Cancer | GSE9348 | 70 | 12 | 54,675 | 22,968 |
| Lung Cancer | GSE7670 | 27 | 27 | 22,283 | 7458 |
| Multiple Sclerosis | GDS3920 | 14 | 15 | 54,674 | 9844 |
| Pancreatic Cancer | GDS4102 | 36 | 16 | 54,613 | 23,711 |
| Parkinson’s Disease | GSE20141 | 10 | 8 | 54,674 | 6625 |
| Prostate Cancer | GSE6919 | 61 | 63 | 12,625 | 1531 |
| Psoriasis | GSE13355 | 58 | 58 | 54,675 | 29,407 |
| Schizophrenia | GSE17612 | 28 | 23 | 54,675 | 4250 |
| Type 2 Diabetes | GSE20966 | 10 | 10 | 61,294 | 93 |
| Disease | Subgroups Identified | Subgroup Sizes |
|---|---|---|
| Asthma | 3 | 31, 8, 3 |
| Breast Cancer | 2 | 22, 5 |
| Chronic Lymphocytic Leukemia | 2 | 4, 18 |
| Colorectal Cancer | 2 | 63, 5 |
| Lung Cancer | 2 | 21, 5 |
| Multiple Sclerosis | 2 | 11, 3 |
| Pancreatic Cancer | 2 | 31, 5 |
| Parkinson’s Disease | 1 | 8 |
| Prostate Cancer | 2 | 56, 3 |
| Psoriasis | 2 | 49, 5 |
| Schizophrenia | 2 | 19, 6 |
| Type 2 Diabetes | 1 | 9 |
| Dataset | GO Category | p-Value |
|---|---|---|
| Asthma GSE4302 | Oxireductase | 1.1 × 10−4 |
| Breast Cancer GSE10810 | Secreted | 1.0 × 10−13 |
| Chronic Lymphocytic Leukemia GSE8835 | Mhc ii | 2.4 × 10−15 |
| Colorectal Cancer GSE9348 | Translational elongation | 2.8 × 10−28 |
| Lung Cancer GSE7670 | Secreted | 7.7 × 10−10 |
| Multiple Sclerosis GDS3920 | Translational elongation | 1.9 × 10−34 |
| Pancreatic Cancer GDS4102 | Signal | 4.59 × 10−15 |
| Prostate Cancer GSE6919 | Translational elongation | 4.92 × 10−46 |
| Psoriasis GSE13355 | Immune response | 3.5 × 10−15 |
| Schizophrenia GSE17612 | Organelle membrane | 5.24 × 10−4 |
| Paraclique Results | ||||||
|---|---|---|---|---|---|---|
| Gastric Cancer | NSCLC | |||||
| Paraclique Sizes | Paraclique Sizes | |||||
| 29 | 16 | 26 | 12 | 8 | ||
| Subtype | Subtype | |||||
| proliferative | 1 | 12 | AC | 23 | 0 | 8 |
| invasive | 19 | 1 | SCC | 3 | 12 | 0 |
| metabolic | 9 | 3 | ||||
| k-Means Results | ||||||
| Gastric Cancer | NSCLC | |||||
| Cluster Sizes | Cluster Sizes | |||||
| 26 | 44 | 28 | 30 | |||
| Subtype | Subtype | |||||
| proliferative | 0 | 29 | AC | 18 | 22 | |
| invasive | 25 | 1 | SCC | 10 | 8 | |
| metabolic | 1 | 14 | ||||
| Hierarchical Clustering Results | ||||||
| Gastric Cancer | NSCLC | |||||
| Cluster Sizes | Cluster Sizes | |||||
| 33 | 37 | 9 | 49 | |||
| Subtype | Subtype | |||||
| proliferative | 0 | 29 | AC | 0 | 40 | |
| invasive | 22 | 4 | SCC | 9 | 9 | |
| metabolic | 11 | 4 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hagan, R.D.; Langston, M.A. Molecular Subtyping and Outlier Detection in Human Disease Using the Paraclique Algorithm. Algorithms 2021, 14, 63. https://doi.org/10.3390/a14020063
Hagan RD, Langston MA. Molecular Subtyping and Outlier Detection in Human Disease Using the Paraclique Algorithm. Algorithms. 2021; 14(2):63. https://doi.org/10.3390/a14020063
Chicago/Turabian StyleHagan, Ronald D., and Michael A. Langston. 2021. "Molecular Subtyping and Outlier Detection in Human Disease Using the Paraclique Algorithm" Algorithms 14, no. 2: 63. https://doi.org/10.3390/a14020063
APA StyleHagan, R. D., & Langston, M. A. (2021). Molecular Subtyping and Outlier Detection in Human Disease Using the Paraclique Algorithm. Algorithms, 14(2), 63. https://doi.org/10.3390/a14020063