
Topological Data Analysis in Time Series: Temporal Filtration and Application to Single-Cell Genomics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Quest from Topological Data Analysis
1.2. Quest from Single-Cell Resolution Data
1.3. Framework: Single-Cell Topological Simplicial Analysis (scTSA)
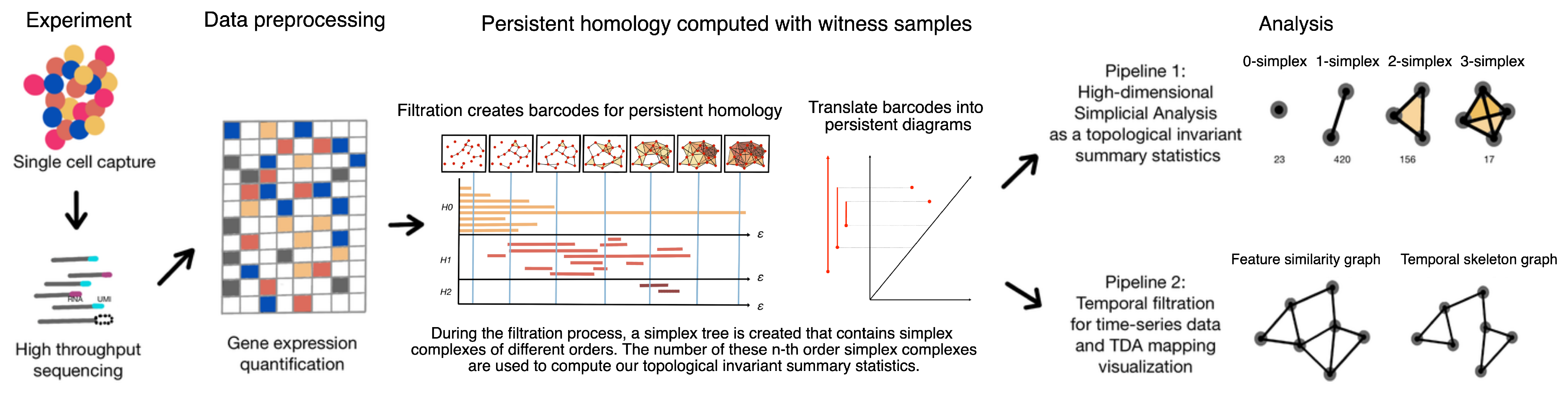
2. Materials and Methods
2.1. Single-Cell Data in the Point Cloud Space
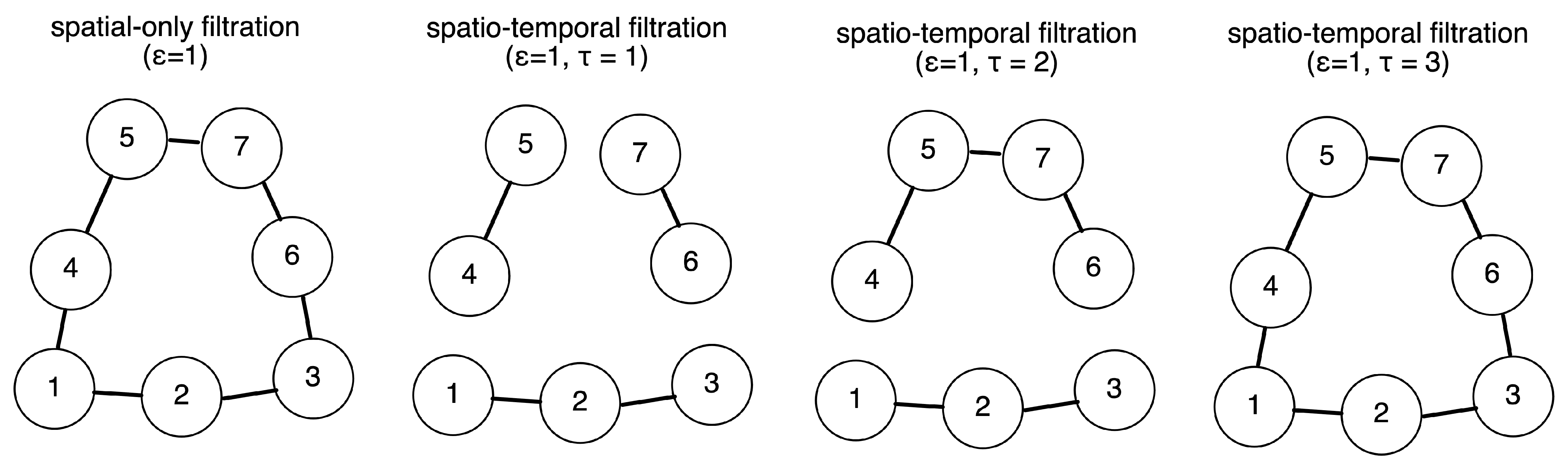
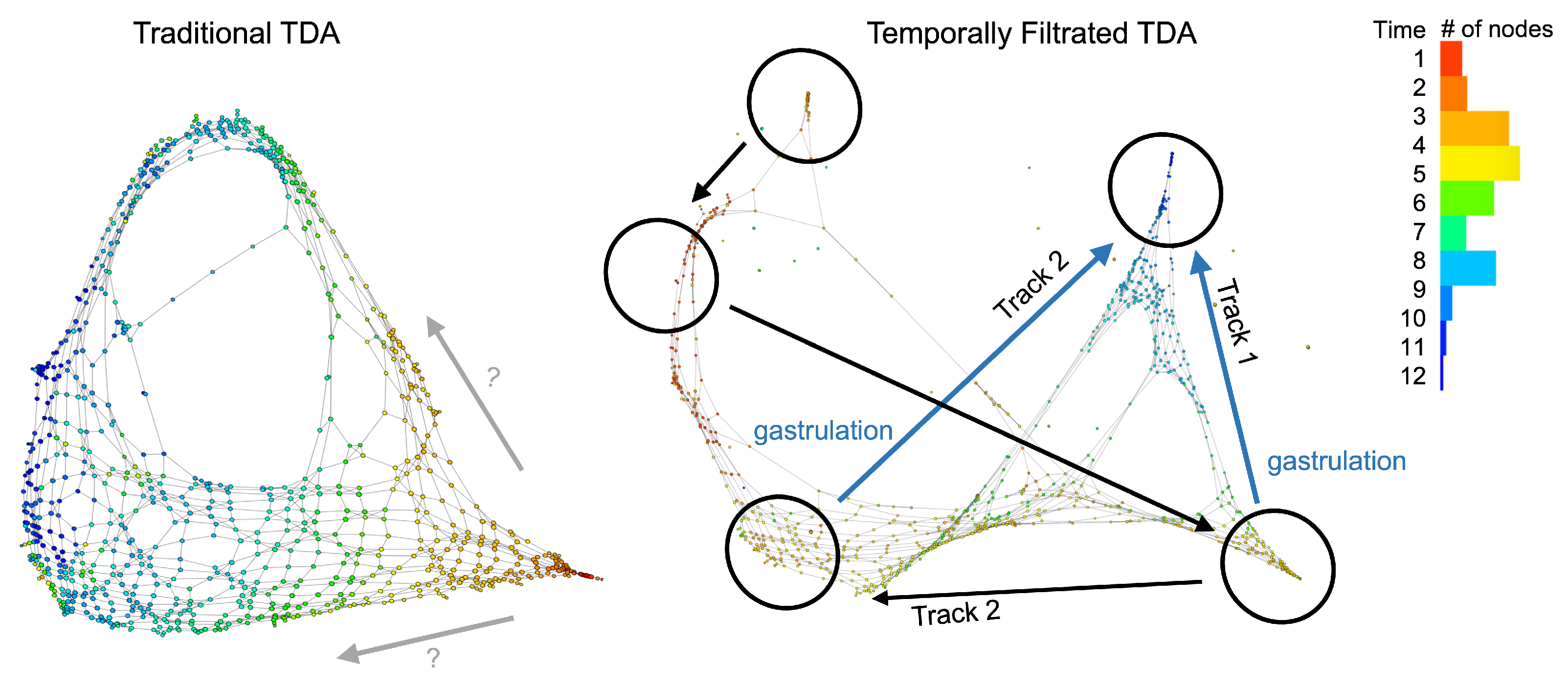
2.2. Definition of Simplicial and Temporal Filtration
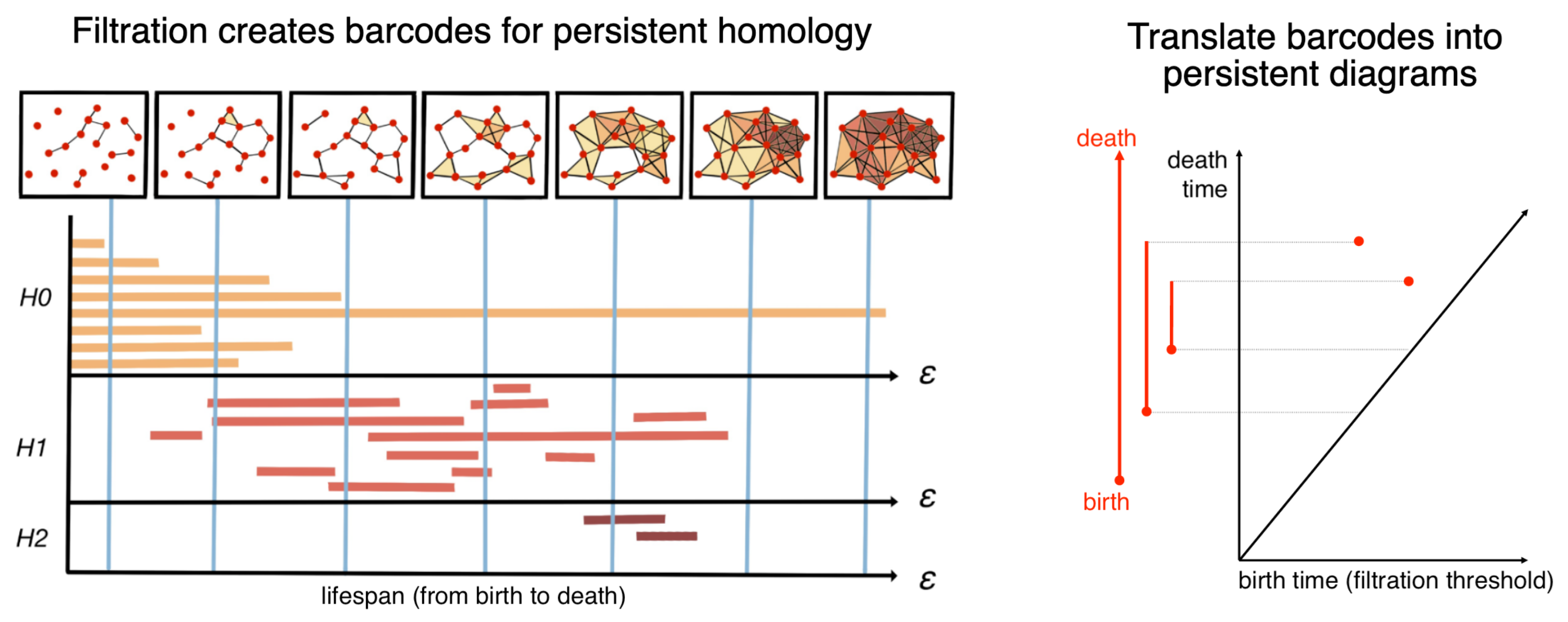
2.3. Topological Data Analysis with Persistent Homology
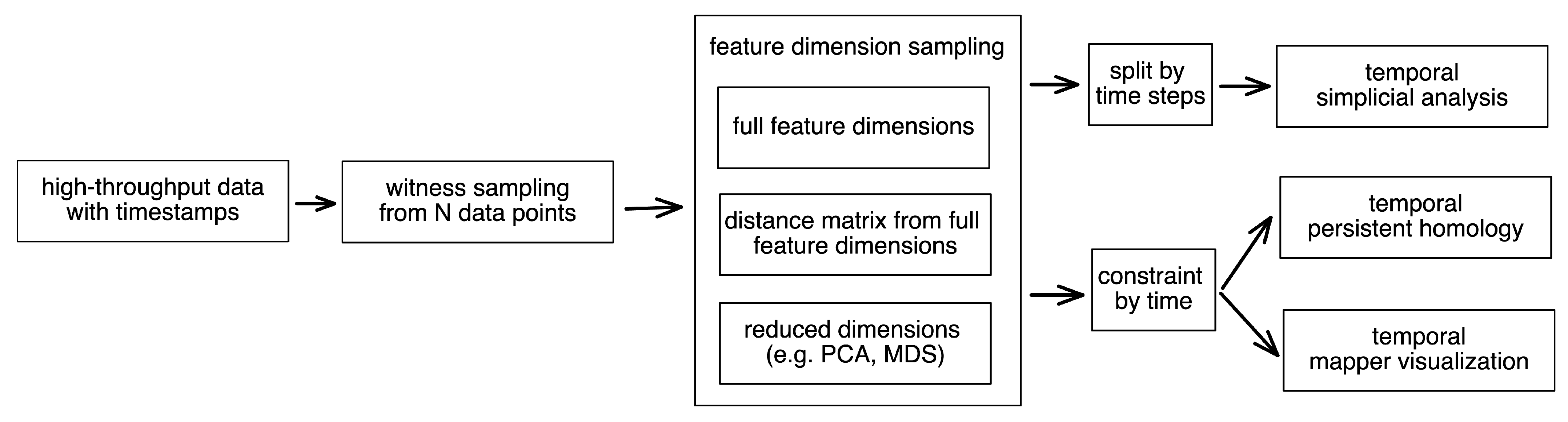
2.4. Empirical Simplicial Computation with Witness Sampling and Dimension Reduction
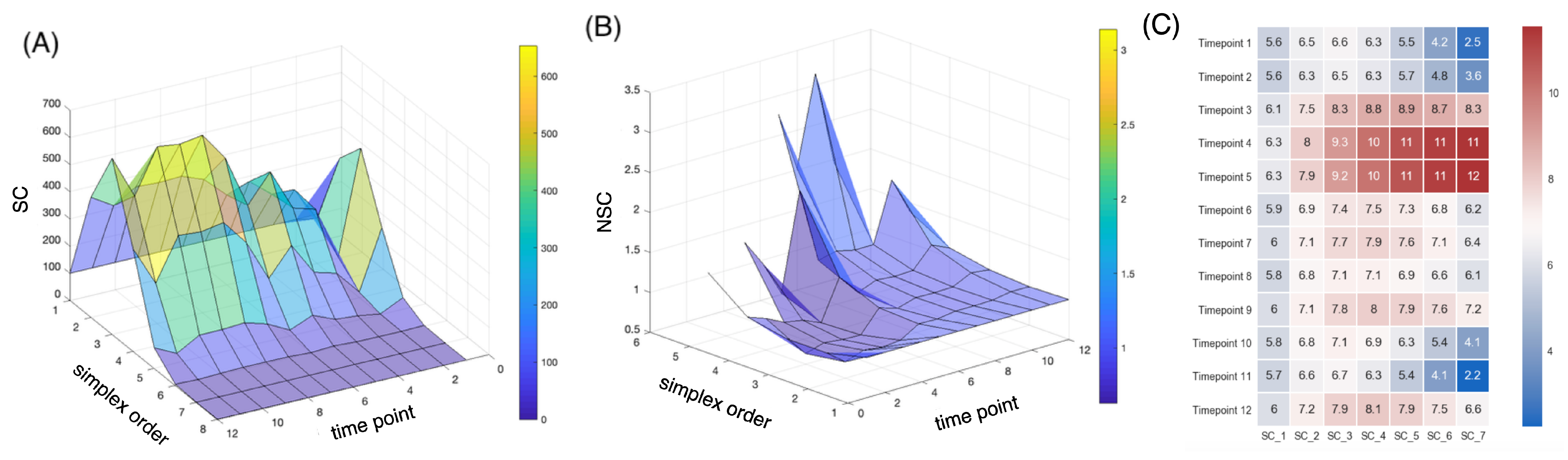
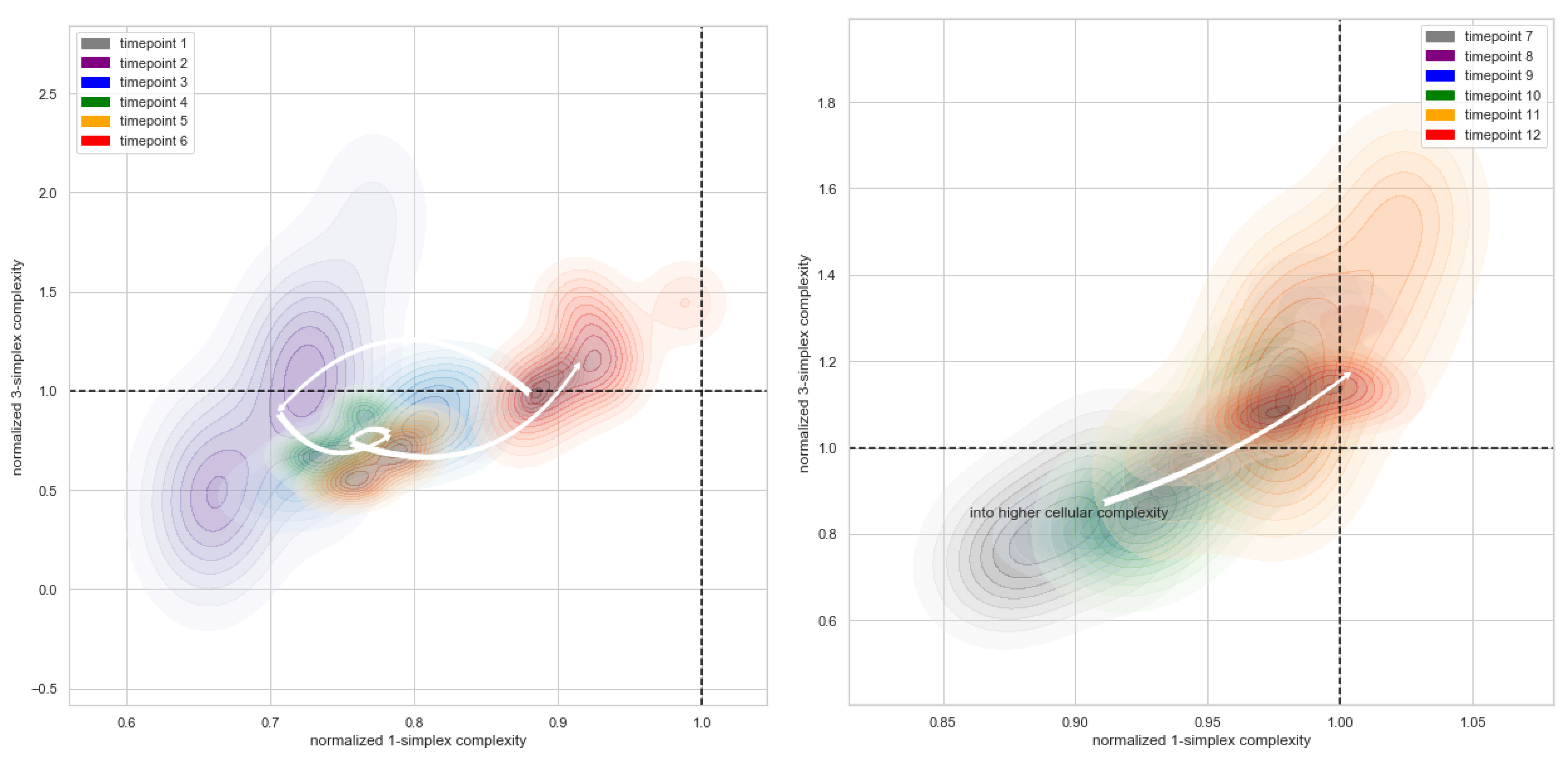
2.5. Topological Simplicial Analysis
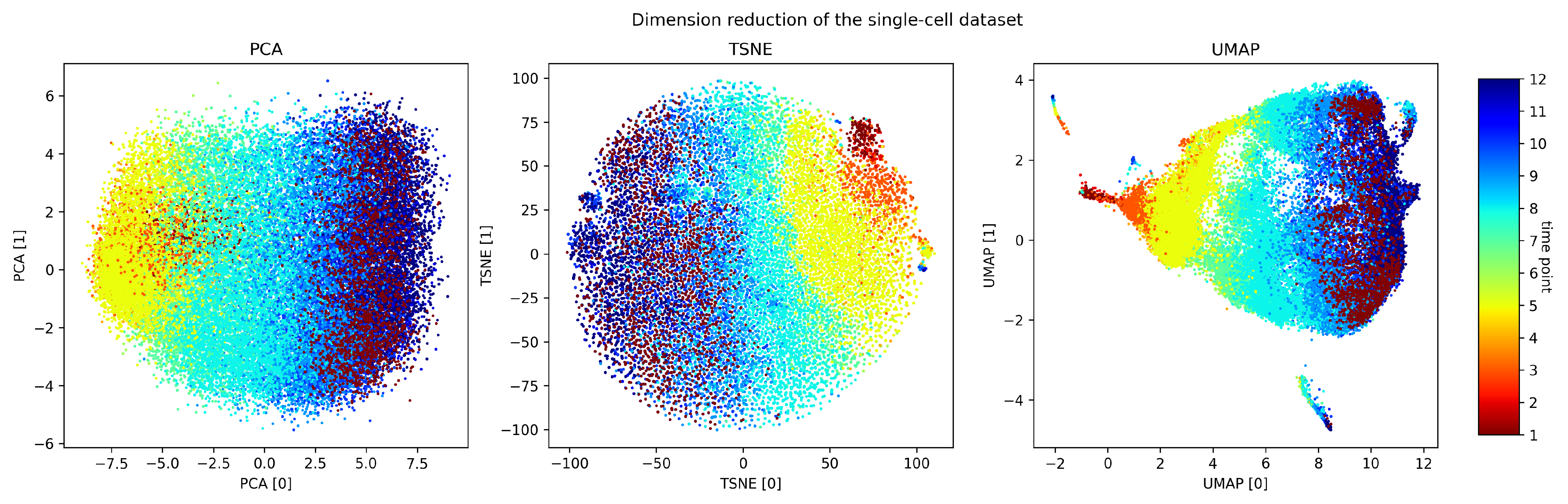
2.6. Topological Data Visualization with Low-Dimensional Mapping
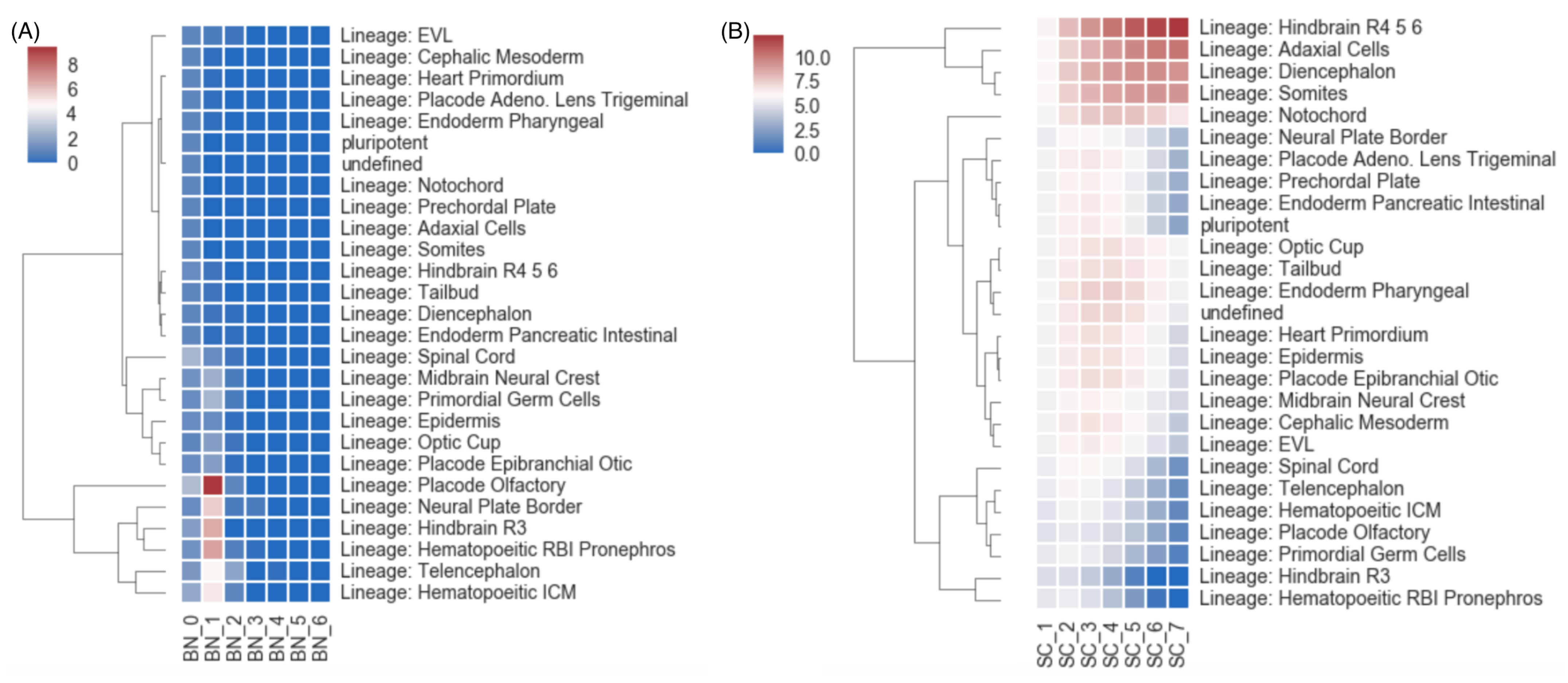
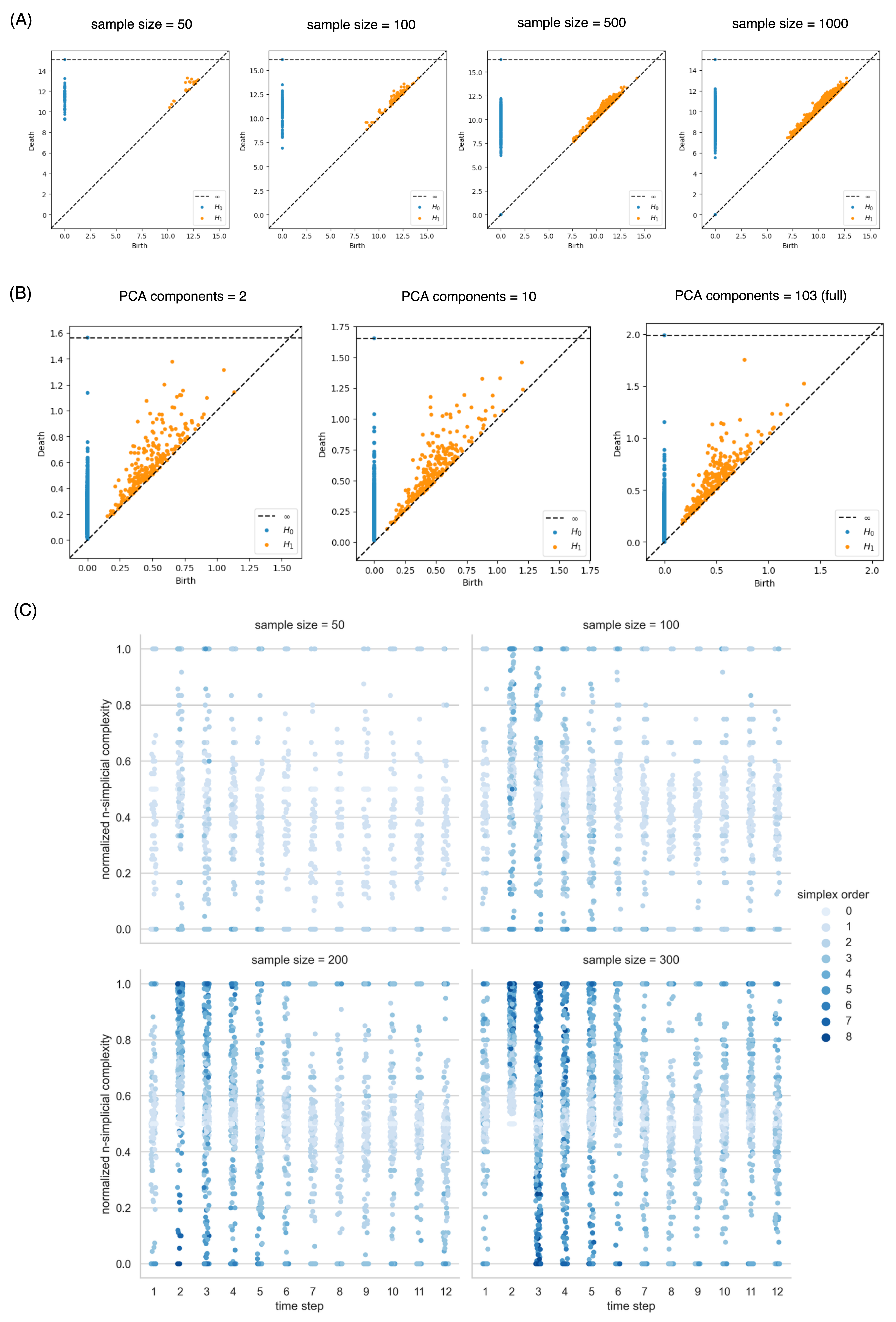
3. Results
4. Discussion
5. Conclusions
5.1. A Lack of Time Series Analytical Methods in Quantifying the Underlying Temporal Skeleton within the Manifold of the Similarities among Data Points
5.2. A Lack of Scalable Computational Methods for Characterizing Single-Cell Sequence Signals in the Scale of 10,000+ Data Points While Single-Cell Sequencing Data Have Dominated Bioinformatics in Recent Years
5.3. A Lack of Insight and Interpretation That Connects the Mathematical Language of Algebraic Topology for the Physical References to Biological Phenomena
5.4. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carlsson, G. Topology and data. Bull. Am. Math. Soc. 2009, 46, 255–308. [Google Scholar] [CrossRef]
- Crawford, L.; Monod, A.; Chen, A.X.; Mukherjee, S.; Rabadán, R. Predicting clinical outcomes in glioblastoma: An application of topological and functional data analysis. J. Am. Stat. Assoc. 2020, 115, 1139–1150. [Google Scholar] [CrossRef]
- Saggar, M.; Sporns, O.; Gonzalez-Castillo, J.; Bandettini, P.A.; Carlsson, G.; Glover, G.; Reiss, A.L. Towards a new approach to reveal dynamical organization of the brain using topological data analysis. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Phinyomark, A.; Ibanez-Marcelo, E.; Petri, G. Resting-state fMRI functional connectivity: Big data preprocessing pipelines and topological data analysis. IEEE Trans. Big Data 2017, 3, 415–428. [Google Scholar] [CrossRef]
- Amézquita, E.J.; Quigley, M.Y.; Ophelders, T.; Munch, E.; Chitwood, D.H. The shape of things to come: Topological data analysis and biology, from molecules to organisms. Dev. Dyn. 2020, 249, 816–833. [Google Scholar] [CrossRef]
- Topaz, C.M.; Ziegelmeier, L.; Halverson, T. Topological data analysis of biological aggregation models. PLoS ONE 2015, 10, e0126383. [Google Scholar] [CrossRef]
- Offroy, M.; Duponchel, L. Topological data analysis: A promising big data exploration tool in biology, analytical chemistry and physical chemistry. Anal. Chim. Acta 2016, 910, 1–11. [Google Scholar] [CrossRef]
- Chazal, F.; Michel, B. An introduction to topological data analysis: Fundamental and practical aspects for data scientists. arXiv 2017, arXiv:1710.04019. [Google Scholar] [CrossRef]
- Otter, N.; Porter, M.A.; Tillmann, U.; Grindrod, P.; Harrington, H.A. A roadmap for the computation of persistent homology. EPJ Data Sci. 2017, 6, 1–38. [Google Scholar] [CrossRef]
- Rizvi, A.H.; Camara, P.G.; Kandror, E.K.; Roberts, T.J.; Schieren, I.; Maniatis, T.; Rabadan, R. Single-cell topological RNA-seq analysis reveals insights into cellular differentiation and development. Nat. Biotechnol. 2017, 35, 551. [Google Scholar] [CrossRef]
- Carlsson, G. Topological pattern recognition for point cloud data. Acta Numer. 2014, 23, 289–368. [Google Scholar] [CrossRef]
- Gulati, G.S.; Sikandar, S.S.; Wesche, D.J.; Manjunath, A.; Bharadwaj, A.; Berger, M.J.; Ilagan, F.; Kuo, A.H.; Hsieh, R.W.; Cai, S.; et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 2020, 367, 405–411. [Google Scholar] [CrossRef]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 2021, 22, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Arneson, D.; Zhang, G.; Ying, Z.; Zhuang, Y.; Byun, H.R.; Ahn, I.S.; Gomez-Pinilla, F.; Yang, X. Single cell molecular alterations reveal target cells and pathways of concussive brain injury. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.Y.; Christensen, S.M.; Ghanta, S.; Jeong, J.C.; Bucur, O.; Glass, B.; Montaser-Kouhsari, L.; Knoblauch, N.W.; Bertos, N.; Saleh, S.M.; et al. Extensive rewiring of epithelial-stromal co-expression networks in breast cancer. Genome Biol. 2015, 16, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the mouse cell atlas by microwell-seq. Cell 2018, 172, 1091–1107. [Google Scholar] [CrossRef]
- Lin, B.; Kriegeskorte, N. Adaptive Geo-Topological Independence Criterion. arXiv 2018, arXiv:1810.02923. [Google Scholar]
- Lin, B. Geometric and Topological Inference for Deep Representations of Complex Networks. In Proceedings of the Web Conference 2022, Lyon, France, 25–29 April 2022. [Google Scholar]
- Reimann, M.W.; Nolte, M.; Scolamiero, M.; Turner, K.; Perin, R.; Chindemi, G.; Dłotko, P.; Levi, R.; Hess, K.; Markram, H. Cliques of neurons bound into cavities provide a missing link between structure and function. Front. Comput. Neurosci. 2017, 11, 48. [Google Scholar] [CrossRef]
- Lin, B. Cliques of single-cell RNA-seq profiles reveal insights into cell ecology during development and differentiation. In Proceedings of the ISMB, Basel, Switzerland, 22 July 2019. [Google Scholar]
- Gallaher, J.A.; Massey, S.C.; Hawkins-Daarud, A.; Noticewala, S.S.; Rockne, R.C.; Johnston, S.K.; Gonzalez-Cuyar, L.; Juliano, J.; Gil, O.; Swanson, K.R.; et al. From cells to tissue: How cell scale heterogeneity impacts glioblastoma growth and treatment response. PLoS Comput. Biol. 2020, 16, e1007672. [Google Scholar] [CrossRef]
- Amend, S.R.; Roy, S.; Brown, J.S.; Pienta, K.J. Ecological paradigms to understand the dynamics of metastasis. Cancer Lett. 2016, 380, 237–242. [Google Scholar] [CrossRef]
- Farrell, J.A.; Wang, Y.; Riesenfeld, S.J.; Shekhar, K.; Regev, A.; Schier, A.F. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 2018, 360, eaar3131. [Google Scholar] [CrossRef] [PubMed]
- Kalisky, T.; Quake, S.R. Single-cell genomics. Nat. Methods 2011, 8, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- De Silva, V.; Carlsson, G.E. Topological estimation using witness complexes. SPBG 2004, 4, 157–166. [Google Scholar]
- Ghrist, R. Barcodes: The persistent topology of data. Bull. Am. Math. Soc. 2008, 45, 61–75. [Google Scholar] [CrossRef]
- Cohen-Steiner, D.; Edelsbrunner, H.; Harer, J. Stability of persistence diagrams. In Proceedings of the Twenty-First Annual Symposium on Computational Geometry, Pisa, Italy, 6–8 June 2005; pp. 263–271. [Google Scholar]
- Botnan, M.B.; Lesnick, M. An introduction to multiparameter persistence. arXiv 2022, arXiv:2203.14289. [Google Scholar]
- Faure, A.J.; Schmiedel, J.M.; Lehner, B. Systematic analysis of the determinants of gene expression noise in embryonic stem cells. Cell Syst. 2017, 5, 471–484. [Google Scholar] [CrossRef]
- Adams, H.; Carlsson, G. On the nonlinear statistics of range image patches. SIAM J. Imaging Sci. 2009, 2, 110–117. [Google Scholar] [CrossRef][Green Version]
- Maria, C.; Boissonnat, J.D.; Glisse, M.; Yvinec, M. The gudhi library: Simplicial complexes and persistent homology. In Proceedings of the International Congress on Mathematical Software, Seoul, Korea, 5–9 August 2014; pp. 167–174. [Google Scholar]
- Bauer, U. Ripser: Efficient computation of Vietoris–Rips persistence barcodes. J. Appl. Comput. Topol. 2021, 5, 391–423. [Google Scholar]
- Sexton, H.; Johansson, M. Jplex. 2008. Available online: http://comptop.stanford.edu/programs/j (accessed on 15 August 2022).
- Erdős, P.; Rényi, A. On the evolution of random graphs. Publ. Math. Inst. Hung. Acad. Sci. 1960, 5, 17–60. [Google Scholar]
- Singh, G.; Mémoli, F.; Carlsson, G.E. Topological methods for the analysis of high dimensional data sets and 3d object recognition. In Proceedings of the Eurographics Symposium on Point-Based Graphics, Prague, Czech Republic, 2–3 September 2007; Volume 2. [Google Scholar]
- Mead, A. Review of the development of multidimensional scaling methods. J. R. Stat. Soc. Ser. Stat. 1992, 41, 27–39. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Gilbert, S.F.; Barresi, M.J.F. Developmental Biology; Oxford University Press: Oxford, UK, 2000. [Google Scholar]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, B. Topological Data Analysis in Time Series: Temporal Filtration and Application to Single-Cell Genomics. Algorithms 2022, 15, 371. https://doi.org/10.3390/a15100371
Lin B. Topological Data Analysis in Time Series: Temporal Filtration and Application to Single-Cell Genomics. Algorithms. 2022; 15(10):371. https://doi.org/10.3390/a15100371
Chicago/Turabian StyleLin, Baihan. 2022. "Topological Data Analysis in Time Series: Temporal Filtration and Application to Single-Cell Genomics" Algorithms 15, no. 10: 371. https://doi.org/10.3390/a15100371
APA StyleLin, B. (2022). Topological Data Analysis in Time Series: Temporal Filtration and Application to Single-Cell Genomics. Algorithms, 15(10), 371. https://doi.org/10.3390/a15100371