A Complete Transcriptional Landscape Analysis of Pinus elliottii Engelm. Using Third-Generation Sequencing and Comparative Analysis in the Pinus Phylogeny



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and RNA Preparation
2.2. Illumina RNA-Seq Library Construction
2.3. PacBio Long-Read Sequencing, Raw Data Processing and Differential Expression Analysis
2.4. lncRNA, SSR, CDS and Alternative Splice Identifications from the PacBio Sequencing
2.5. Functional Annotation of PacBio Isoforms
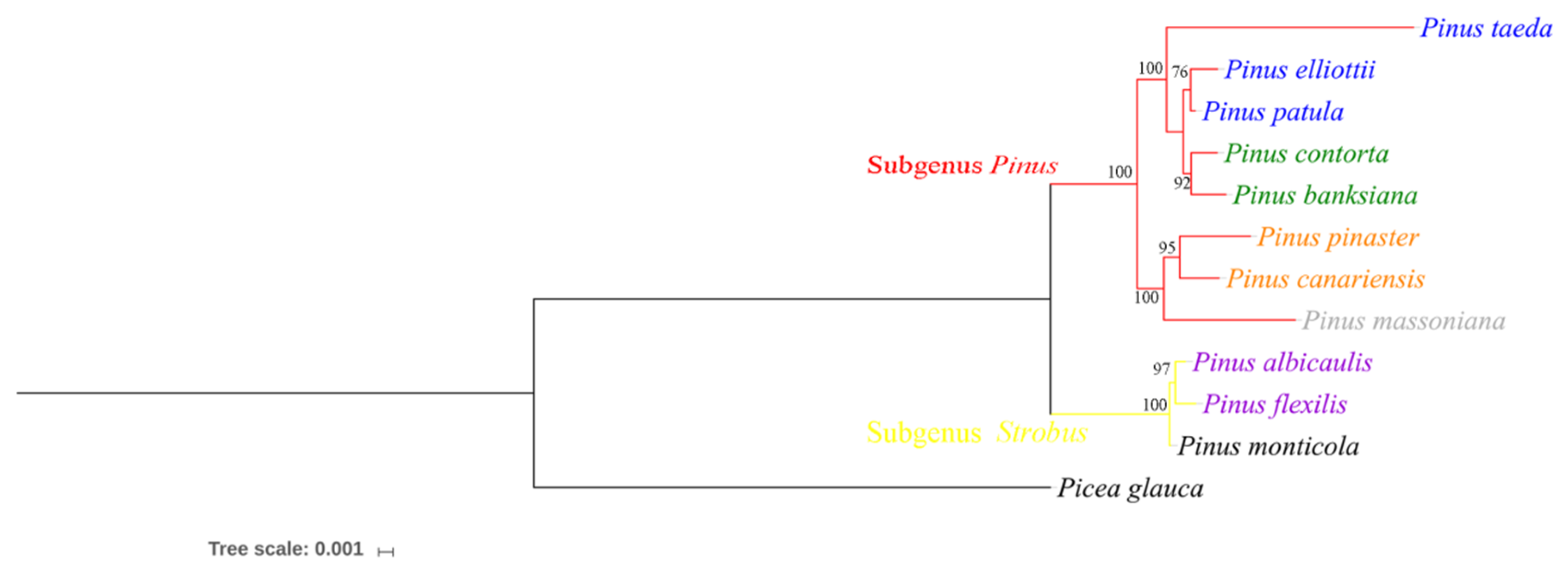
2.6. Phylogenetic Analysis of the Genus Pinus
2.7. Data Availability Statement
3. Results
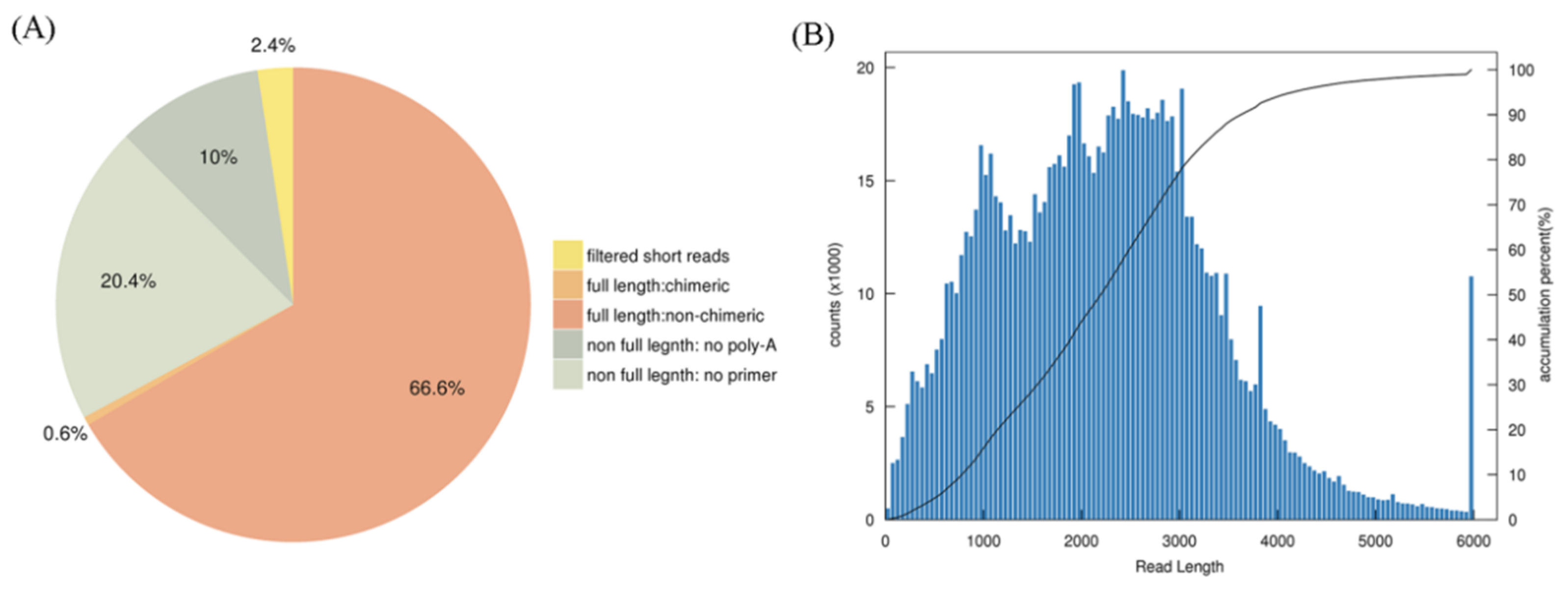
3.1. Third-Generation Sequencing of the Full-Length Transcriptome and the Second-Generation Sequencing
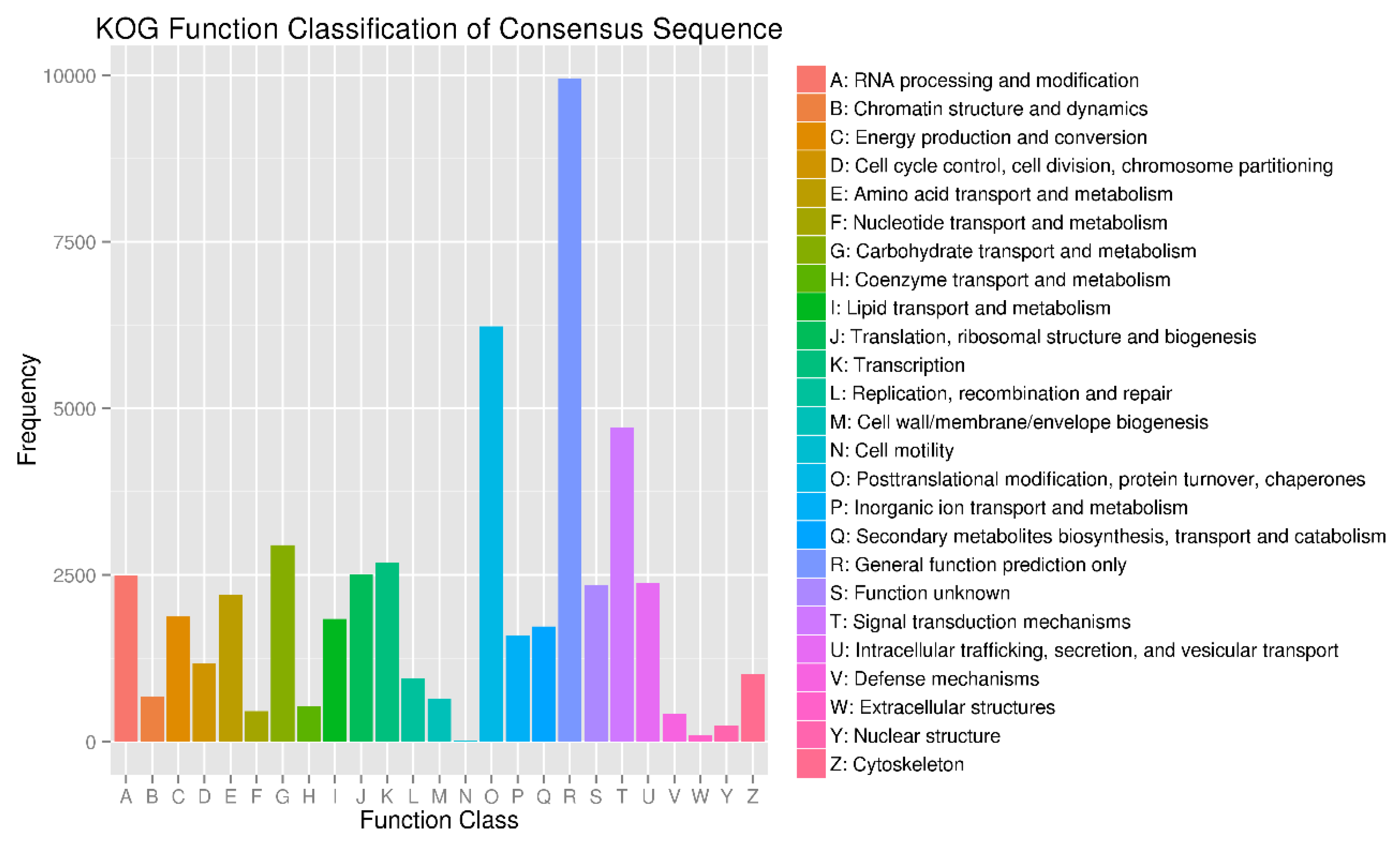
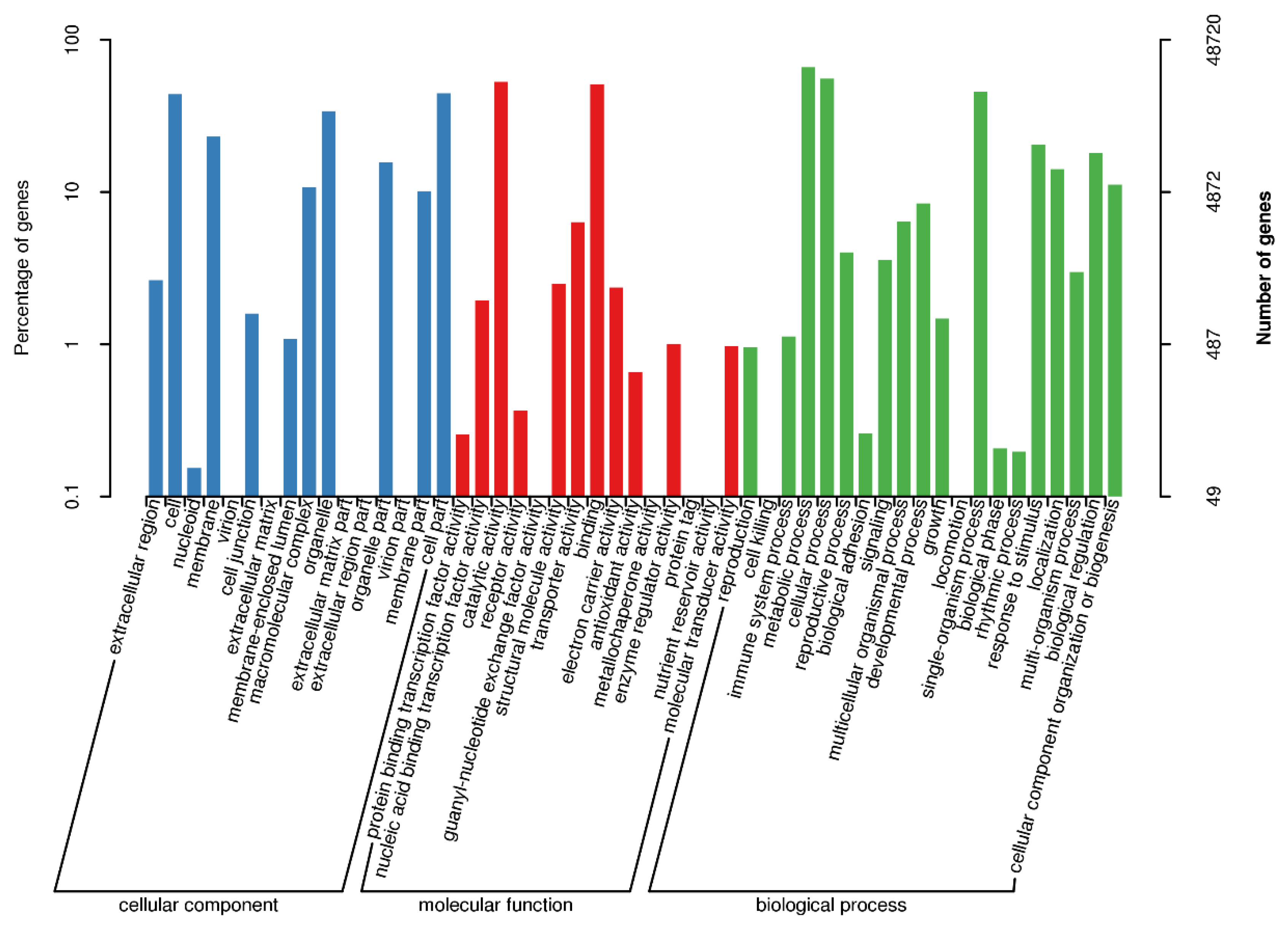
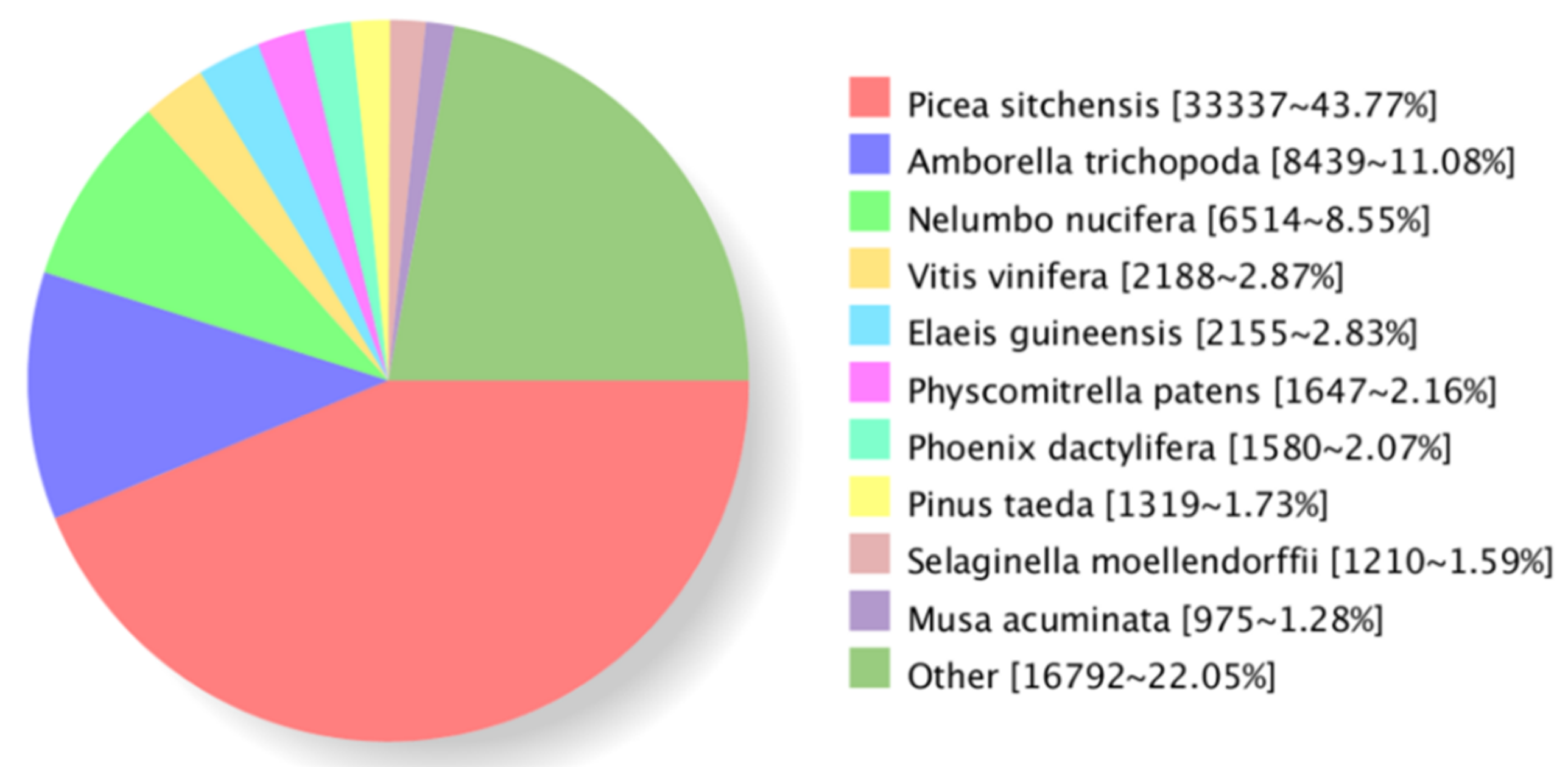
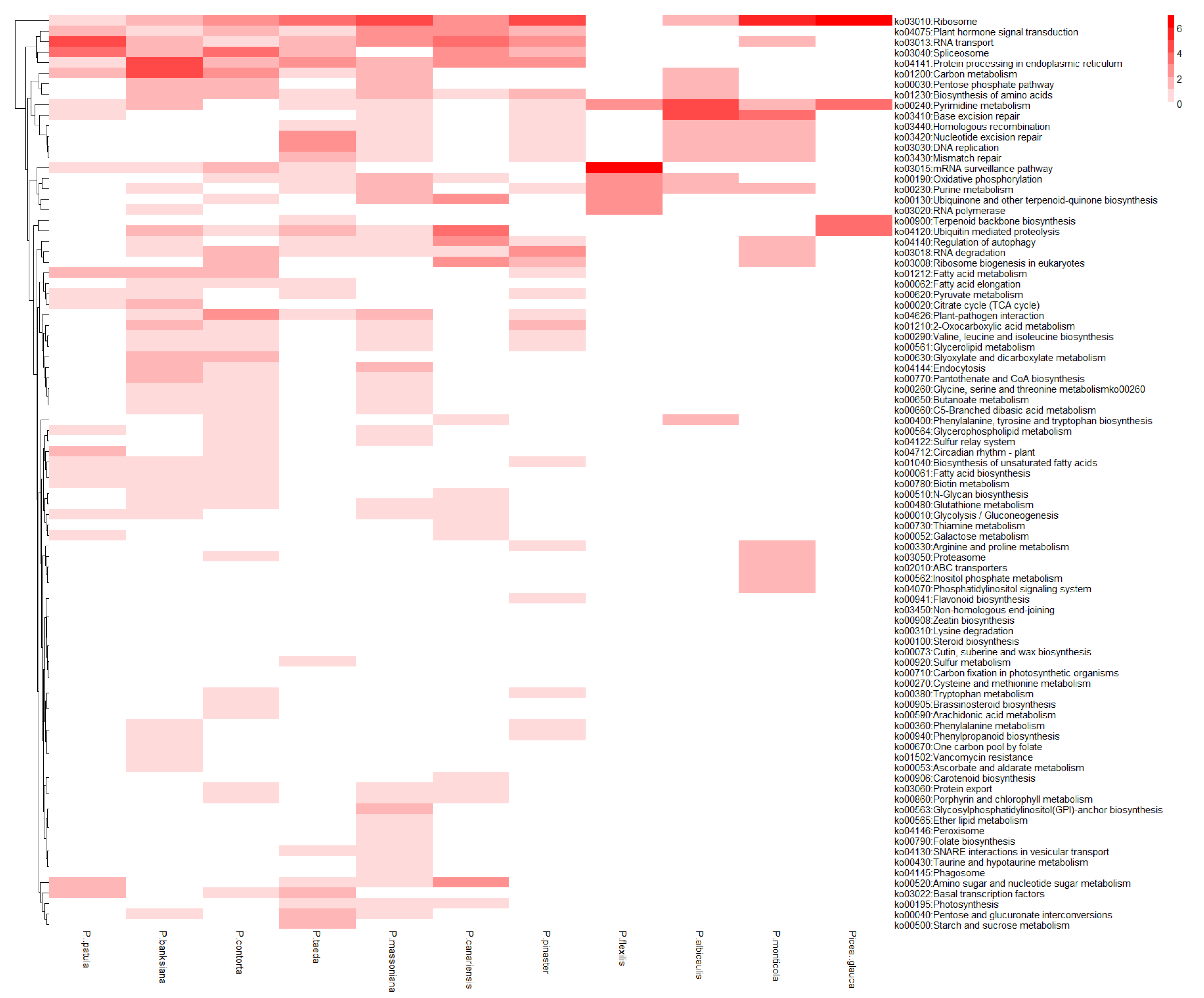
3.2. Functional Annotation of the Transcriptome
3.3. lncRNA, SSR, CDS, and Alternative Splice Identification
3.4. Phylogenetic Analysis of the Genus Pinus
3.5. Functional Genes under a Positive Selection of P. elliottii
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SMRT | Sequencing by single-molecule, real-time |
| TGS | Third-generation sequencing |
| SGS | Second-generation sequencing |
| CS | Coding sequences |
| SSR | Simple sequence repeat |
| AS | Alternative splicing |
| ROIs | Reads of insert |
| FLNC | Full-length non-chimeric |
| IEC | Iterative clustering for error correction |
| COG | Clusters of Orthologous Groups |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KOG | euKaryotic Ortholog Groups |
| Pfam | Protein family |
| NR | NCBI nonredundant protein sequences |
| EggNOG | Evolutionary genealogy of genes: Non-supervised Orthologous Groups |
Appendix A
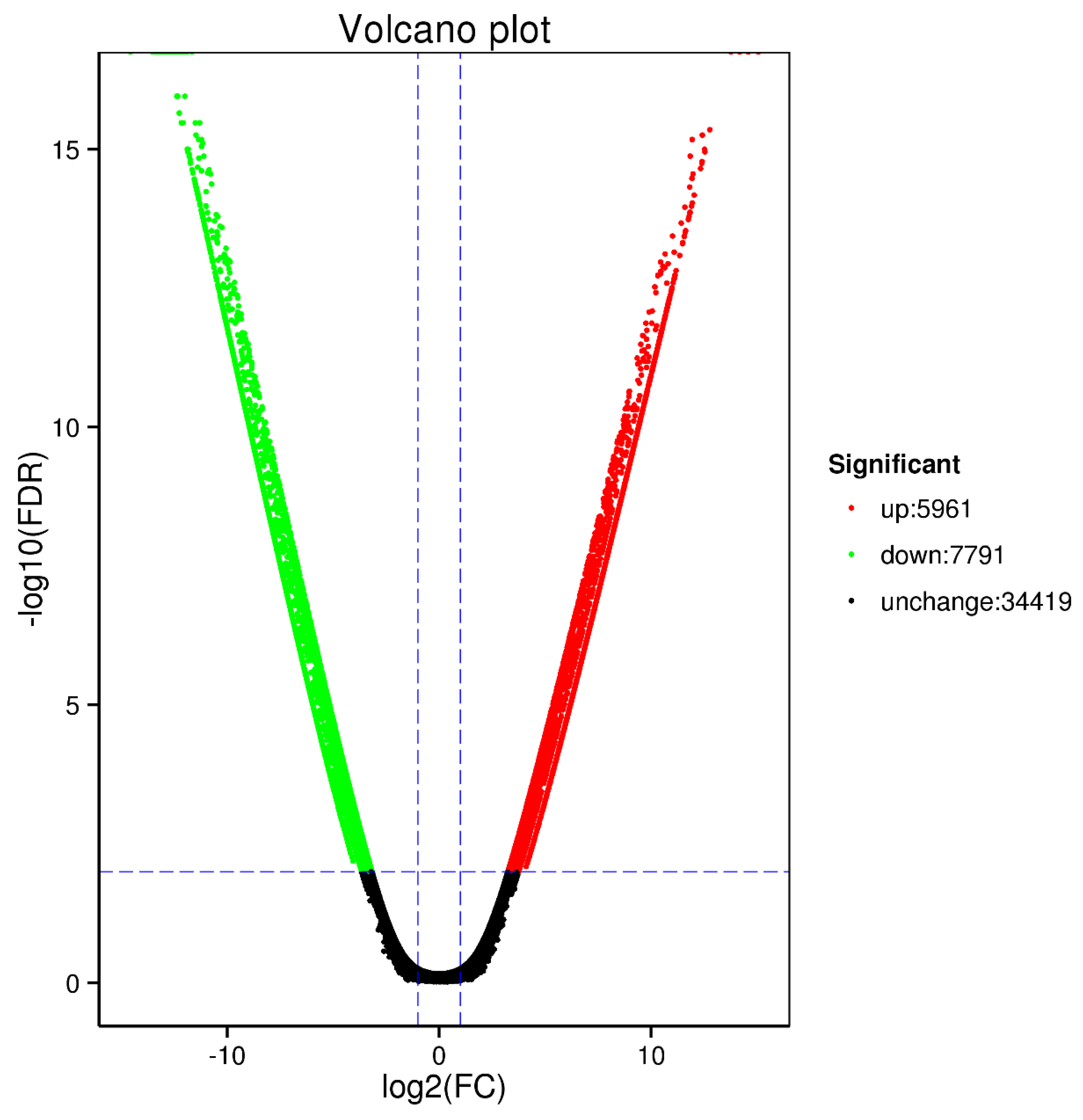

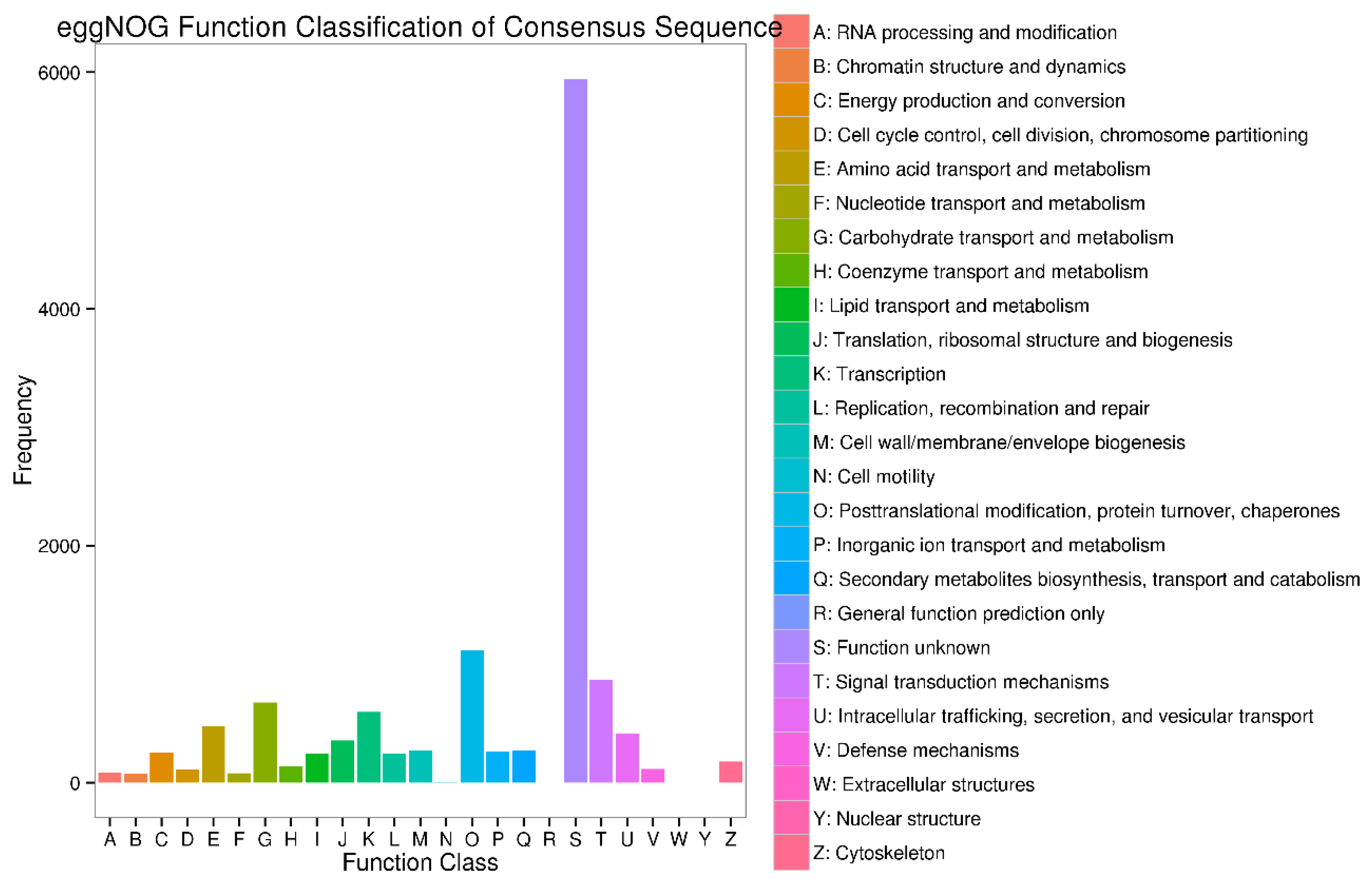
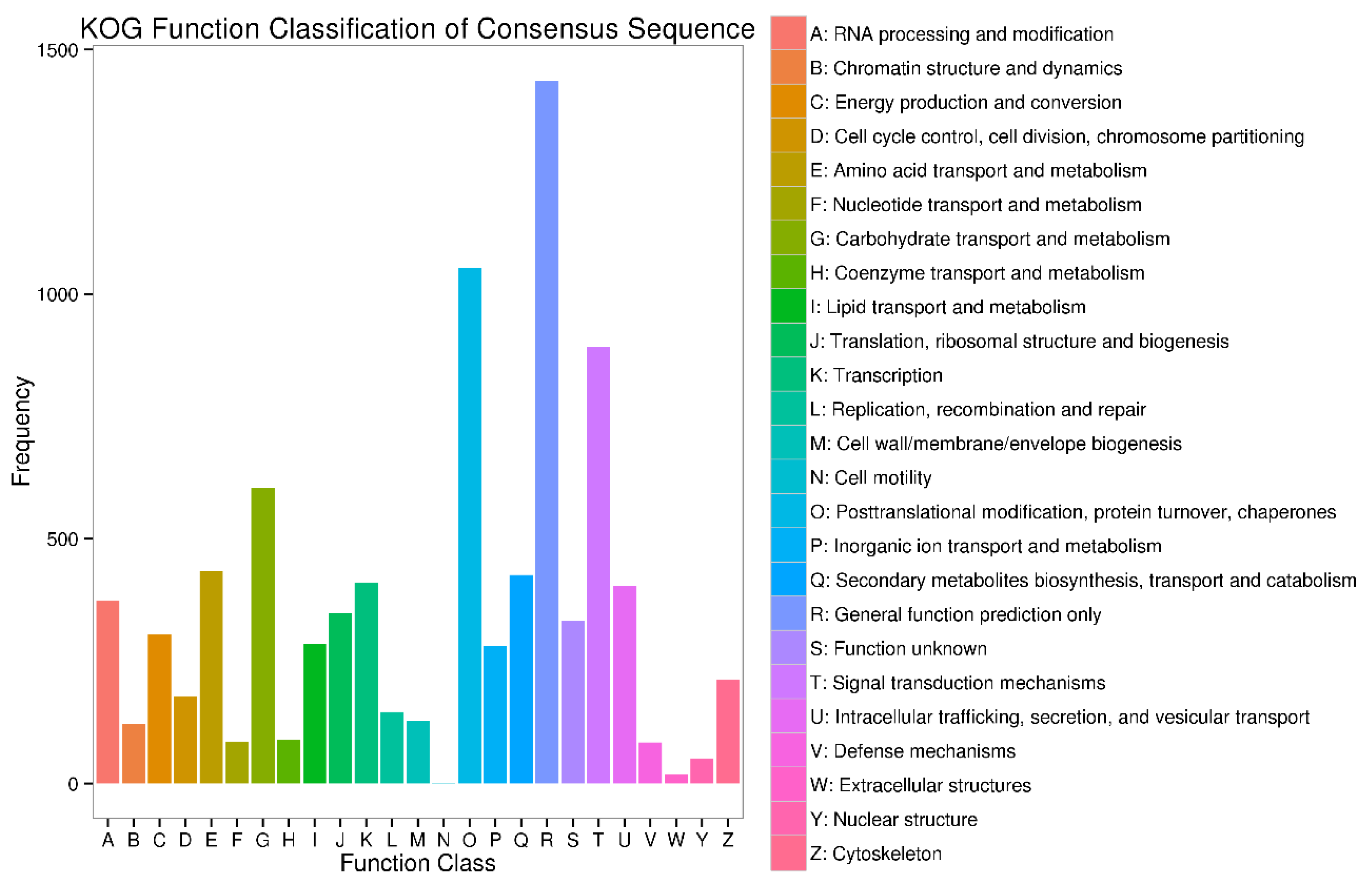
Appendix B
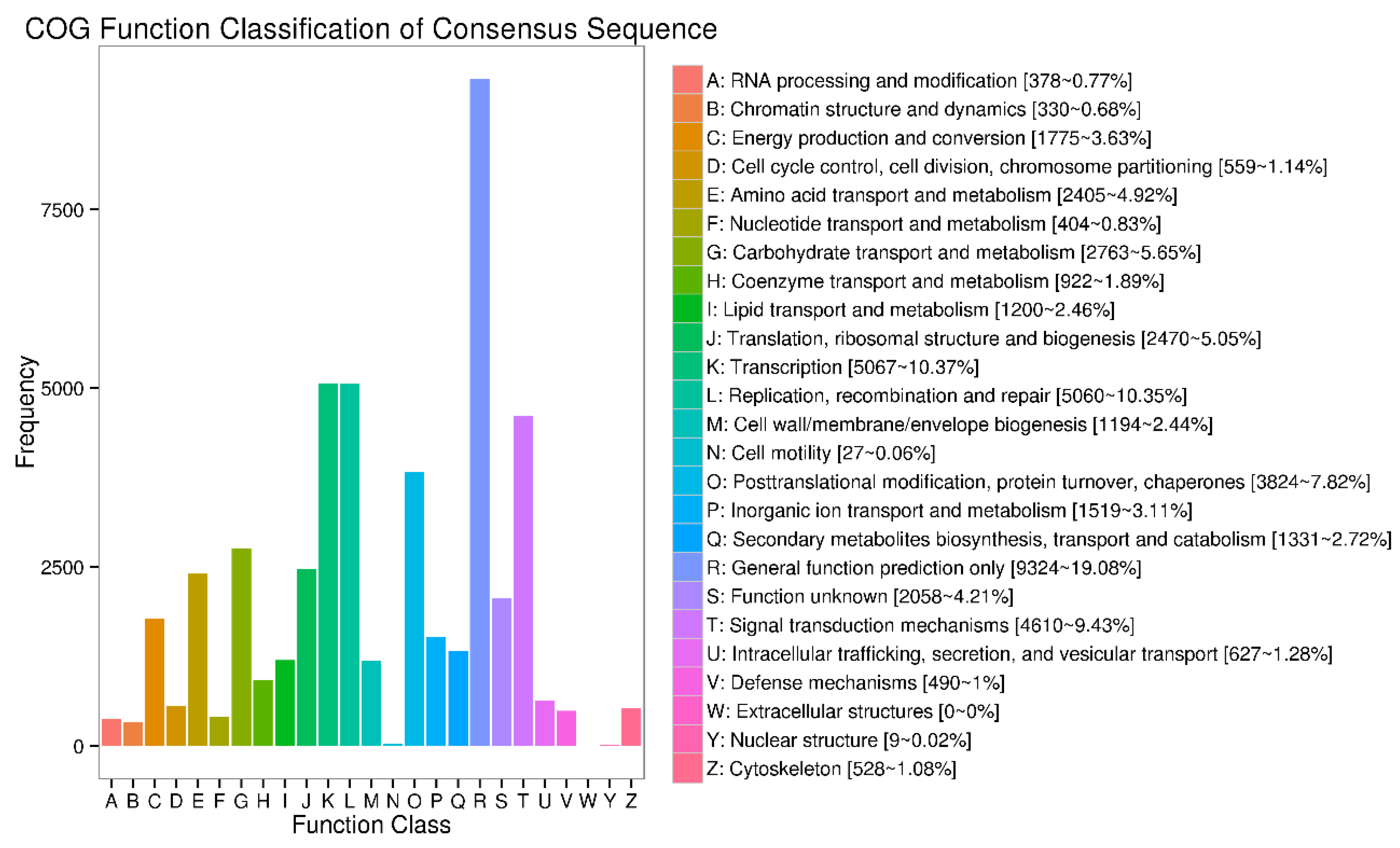

References
- Clark, A., III; Daniels, R.F. In Wood quality of slash pine and its effect on lumber, paper, and other products. In Proceedings of the Slash Pine Symposium, Jekyll Island, GA, USA, 23–25 April 2002. [Google Scholar]
- Neale, D.B.; Wheeler, N.C. The Conifers: Genomes, Variation and Evolution; Springer: Berlin/Heidelberg, Germany, 2019; p. 271. [Google Scholar]
- Zhang, S.; Jiang, J.; Luan, Q. Index selection for growth and construction wood properties in Pinus elliottii open-pollinated families in southern China. South. For. A J. For. Sci. 2018, 80, 209–216. [Google Scholar]
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.; Keeling, C.I.; Brand, D.; Vandervalk, B.P.; et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinform. (Oxf. Engl.) 2013, 29, 1492–1497. [Google Scholar] [CrossRef]
- Jackman, S.D.; Warren, R.L.; Gibb, E.A.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Raymond, A.; Pleasance, S.; Coope, R.; Wildung, M.R.; et al. Organellar Genomes of White Spruce (Picea glauca): Assembly and Annotation. Genome Biol. Evol. 2015, 8, 29–41. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Neale, D.B.; McGuire, P.E.; Wheeler, N.C.; Stevens, K.A.; Crepeau, M.W.; Cardeno, C.; Zimin, A.V.; Puiu, D.; Pertea, G.M.; Sezen, U.U.; et al. The Douglas-Fir Genome Sequence Reveals Specialization of the Photosynthetic Apparatus in Pinaceae. G3 (BethesdaMd.) 2017, 7, 3157–3167. [Google Scholar] [CrossRef] [Green Version]
- Leitch, I.; Johnston, E.; Pellicer, J.; Hidalgo, O.; Bennett, M. Plant DNA C-values database. Available online: https://cvalues.science.kew.org/ (accessed on 7 April 2019).
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marcais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and assembly of the 22-gb loblolly pine genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef]
- Stevens, K.A.; Wegrzyn, J.L.; Zimin, A.; Puiu, D.; Crepeau, M.; Cardeno, C.; Paul, R.; Gonzalez-Ibeas, D.; Koriabine, M.; Holtz-Morris, A.E.; et al. Sequence of the Sugar Pine Megagenome. Genetics 2016, 204, 1613–1626. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Acosta, J.J.; Fahrenkrog, A.M.; Neves, L.G.; Resende, M.F.; Dervinis, C.; Davis, J.M.; Holliday, J.A.; Kirst, M. Exome Resequencing Reveals Evolutionary History, Genomic Diversity, and Targets of Selection in the Conifers Pinus taeda and Pinus elliottii. Genome Biol. Evol. 2019, 11, 508–520. [Google Scholar] [CrossRef]
- De Oliveira Junkes, C.F.; de Araújo Júnior, A.T.; de Lima, J.C.; de Costa, F.; Füller, T.; de Almeida, M.R.; Neis, F.A.; da Silva Rodrigues-Corrêa, K.C.; Fett, J.P.; Fett-Neto, A.G. Resin tapping transcriptome in adult slash pine (Pinus elliottii var. elliottii). Ind. Crop. Prod. 2019, 139, 111545. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinform. (Oxf. Engl.) 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Chen, S.; Yang, P.; Jiang, F.; Wei, Y.; Ma, Z.; Kang, L. De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PloS one. 2010, 5, e15633. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinform. (Oxf. Engl.) 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Pundir, S.; Martin, M.J.; O’Donovan, C. UniProt Protein Knowledgebase. Methods Mol. Biol. (Clifton N.J.) 2017, 1558, 41–55. [Google Scholar] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evol. Int. J. Org. Evol. 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Sammeth, M.; Foissac, S.; Guigó, R. A general definition and nomenclature for alternative splicing events. PLoS Comput. Biol. 2008, 4, e1000147. [Google Scholar] [CrossRef]
- Gernandt, D.S.; López, G.G.; García, S.O.; Liston, A. Phylogeny and classification of Pinus. Taxon 2005, 54, 29–42. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. TIG 2002, 18, 486. [Google Scholar] [CrossRef]
- Muranty, H.; Jorge, V.; Bastien, C.; Lepoittevin, C.; Bouffier, L.; Sanchez, L. Potential for marker-assisted selection for forest tree breeding: Lessons from 20 years of MAS in crops. Tree Genet. Genomes 2014, 10, 1491–1510. [Google Scholar] [CrossRef]
- Cappa, E.P.; El-Kassaby, Y.A.; Garcia, M.N.; Acuna, C.; Borralho, N.M.; Grattapaglia, D.; Marcucci Poltri, S.N. Impacts of population structure and analytical models in genome-wide association studies of complex traits in forest trees: A case study in Eucalyptus globulus. PLoS ONE 2013, 8, e81267. [Google Scholar] [CrossRef]
- David, M.R. Ecology and Biogeography of Pinus; Cambridge University Press: New York, NY, USA, 1998; pp. 69–91. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoform | COG | GO | KEGG | KOG | Pfam | Swiss-Prot | Egg NOG | NR | All |
|---|---|---|---|---|---|---|---|---|---|
| Isoform Number | 32,755 | 48,720 | 31,939 | 46,177 | 64,413 | 56,543 | 73,004 | 76,173 | 76,411 |
| Percentage (%) | 40.77 | 60.64 | 39.76 | 57.48 | 80.18 | 70.38 | 90.87 | 94.81 | 95.11 |
| Genus | Pinus/Picea spp. | Data Source | Number of Unigenes/Genes | Total Length(bp) | Mean Length(bp) |
|---|---|---|---|---|---|
| Pinus | Pinus contorta | PlantGDB | 13,570 | 13,018,301 | 959.34 |
| Pinus | Pinus banksiana | PlantGDB | 13,040 | 12,504,634 | 958.94 |
| Pinus | Pinus taeda | Treegenesdb(v1.01) | 84,525 | 66,216,274 | 783.39 |
| Pinus | Pinus patula | NCBI | 52,735 | 72,146,457 | 1368.09 |
| Pinus | Pinus elliottii | This study | 80,339 | 190,161,398 | 2366.99 |
| Pinus | Pinus massoniana | NCBI | 194,821 | 106,618,461 | 547.26 |
| Pinus | Pinus canariensis | NCBI | 47,792 | 43,957,406 | 919.76 |
| Pinus | Pinus pinaster | PlantGDB | 15,648 | 8,925,915 | 570.42 |
| Pinus | Pinus albicaulis | NCBI | 129,522 | 116,155,243 | 896.8 |
| Pinus | Pinus monticola | NCBI | 54,661 | 46,069,734 | 842.83 |
| Pinus | Pinus flexilis | NCBI | 14,279 | 16,275,848 | 1139.85 |
| Picea | Picea glauca | NCBI | 354,861 | 349,225,041 | 984.12 |
| Species | Orthologous Pairs | Peaks of Ks Values | Number of Genes under Positive Selection | Ratio |
|---|---|---|---|---|
| Picea glauca | 2752 | 0.148301 | 54 | 1.96% |
| Pinus albicaulis | 4173 | 0.072611 | 106 | 2.54% |
| Pinus banksiana | 2758 | 0.010958 | 296 | 10.73% |
| Pinus canariensis | 4870 | 0.030305 | 302 | 6.20% |
| Pinus contorta | 3011 | 0.008745 | 251 | 8.34% |
| Pinus flexilis | 2245 | 0.075452 | 58 | 2.58% |
| Pinus massoniana | 5907 | 0.027807 | 398 | 6.74% |
| Pinus monticola | 5047 | 0.070412 | 108 | 2.14% |
| Pinus patula | 5416 | 0.005033 | 393 | 7.26% |
| Pinus pinaster | 2496 | 0.028913 | 187 | 7.49% |
| Pinus taeda | 3695 | 0.012831 | 402 | 10.88% |
| P. patula | P. banksiana | P. contorta | P. taeda | P. massoniana | P. pinaster | P. canariensis | P. monticola | P. flexilis | P. albicaulis | Picea glauca | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Response to plant phytohormone | |||||||||||
| Salicylic acid | 2 | 2 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Abscisic acid | 2 | 3 | 0 | 2 | 1 | 3 | 0 | 0 | 0 | 0 | 0 |
| Gibberellin | 2 | 1 | 1 | 4 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| Jasmonic acid | 2 | 1 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Auxin | 1 | 2 | 1 | 3 | 3 | 3 | 1 | 0 | 0 | 0 | 0 |
| Meristem activity | |||||||||||
| meristem initiation | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| meristem growth | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| meristem determinacy | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| vegetative to reproductive phase transition of meristem | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| defense to biological and abiotic factors | |||||||||||
| defense response to fungus | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| defense response to bacterium | 2 | 2 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| response to salt stress | 2 | 3 | 1 | 3 | 0 | 2 | 2 | 0 | 0 | 1 | 0 |
| response to osmotic stress | 1 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| response to oxidative stress | 1 | 1 | 2 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| response to endoplasmic reticulum stress | 4 | 1 | 2 | 0 | 1 | 0 | 3 | 0 | 0 | 0 | 0 |
| antifungal | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| response to cold | 2 | 0 | 2 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| response to water deprivation | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| response to light | |||||||||||
| response to blue light | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| response to high light intensity | 3 | 1 | 1 | 1 | 2 | 0 | 2 | 0 | 0 | 0 | 0 |
| response to red light | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| response to far red light | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| red, far-red light phototransduction | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diao, S.; Ding, X.; Luan, Q.; Jiang, J. A Complete Transcriptional Landscape Analysis of Pinus elliottii Engelm. Using Third-Generation Sequencing and Comparative Analysis in the Pinus Phylogeny. Forests 2019, 10, 942. https://doi.org/10.3390/f10110942
Diao S, Ding X, Luan Q, Jiang J. A Complete Transcriptional Landscape Analysis of Pinus elliottii Engelm. Using Third-Generation Sequencing and Comparative Analysis in the Pinus Phylogeny. Forests. 2019; 10(11):942. https://doi.org/10.3390/f10110942
Chicago/Turabian StyleDiao, Shu, Xianying Ding, Qifu Luan, and Jingmin Jiang. 2019. "A Complete Transcriptional Landscape Analysis of Pinus elliottii Engelm. Using Third-Generation Sequencing and Comparative Analysis in the Pinus Phylogeny" Forests 10, no. 11: 942. https://doi.org/10.3390/f10110942
APA StyleDiao, S., Ding, X., Luan, Q., & Jiang, J. (2019). A Complete Transcriptional Landscape Analysis of Pinus elliottii Engelm. Using Third-Generation Sequencing and Comparative Analysis in the Pinus Phylogeny. Forests, 10(11), 942. https://doi.org/10.3390/f10110942