Transcriptomic Responses of Dove Tree (Davidia involucrata Baill.) to Heat Stress at the Seedling Stage


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Heat Treatment
2.2. RNA Extraction, cDNA Library Preparation and Transcriptome Sequencing
2.3. Transcriptome Assembly and Functional Annotation
2.4. Differential Expression Analysis
2.5. qRT-PCR Analysis
3. Results
3.1. Illumina Sequencing and Transcriptome Assembly
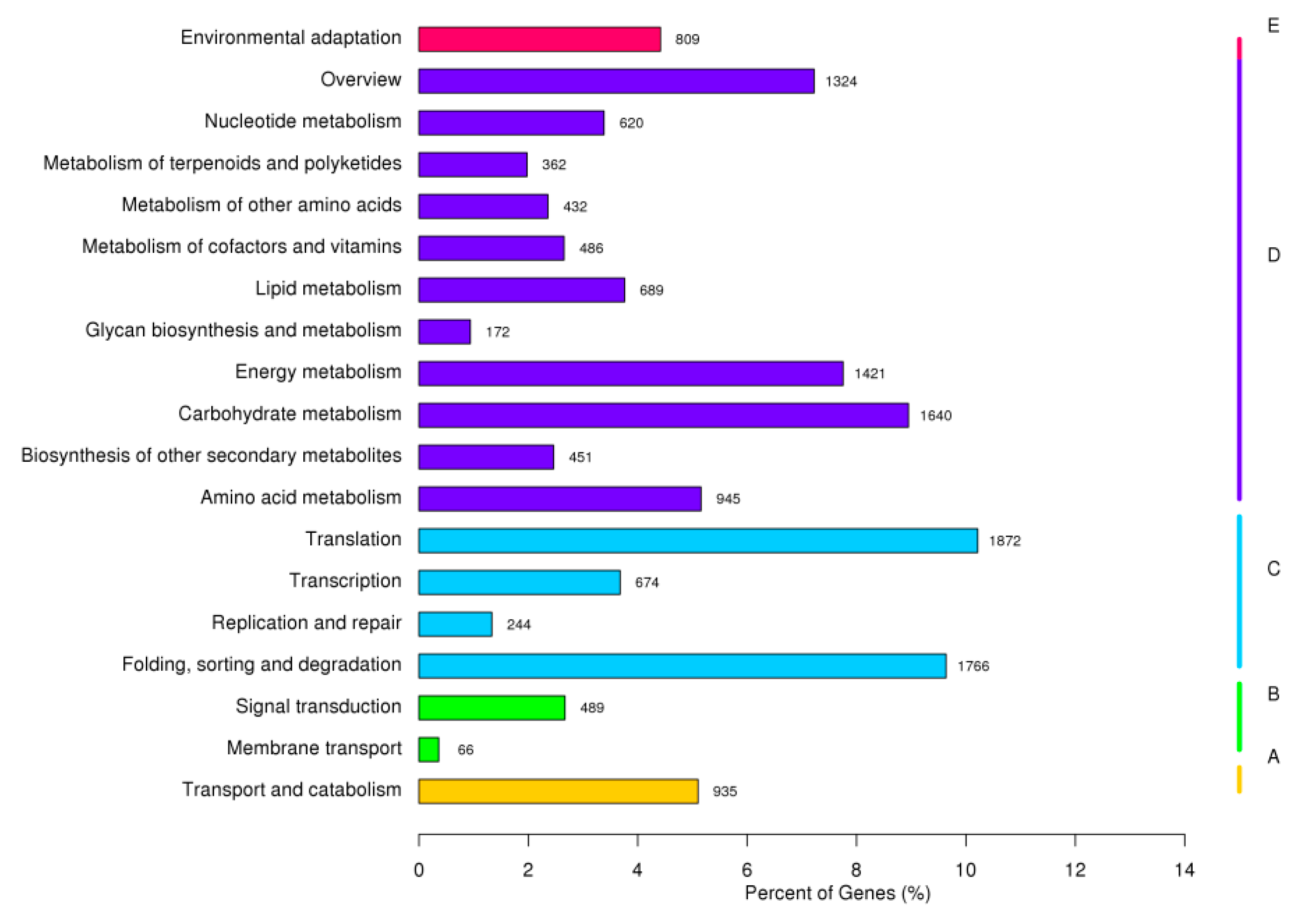
3.2. Functional Annotation and Classification of D. involucrata Unigenes
3.3. Identification of DEGs
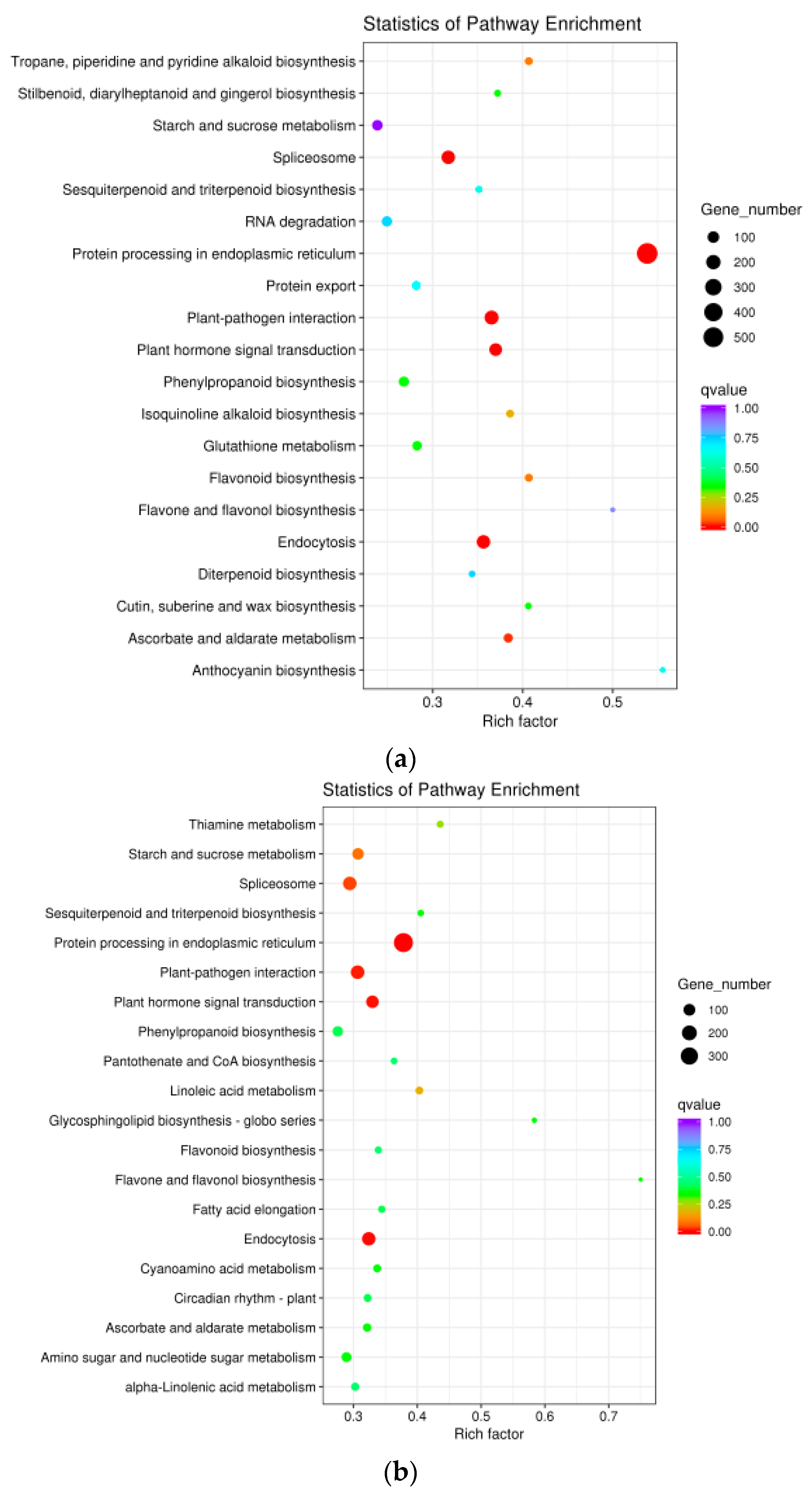
3.4. GO and KEGG Enrichment Analyses of Heat-Responsive Genes
3.5. Heat Shock Transcription Factors Responding to Heat Stress
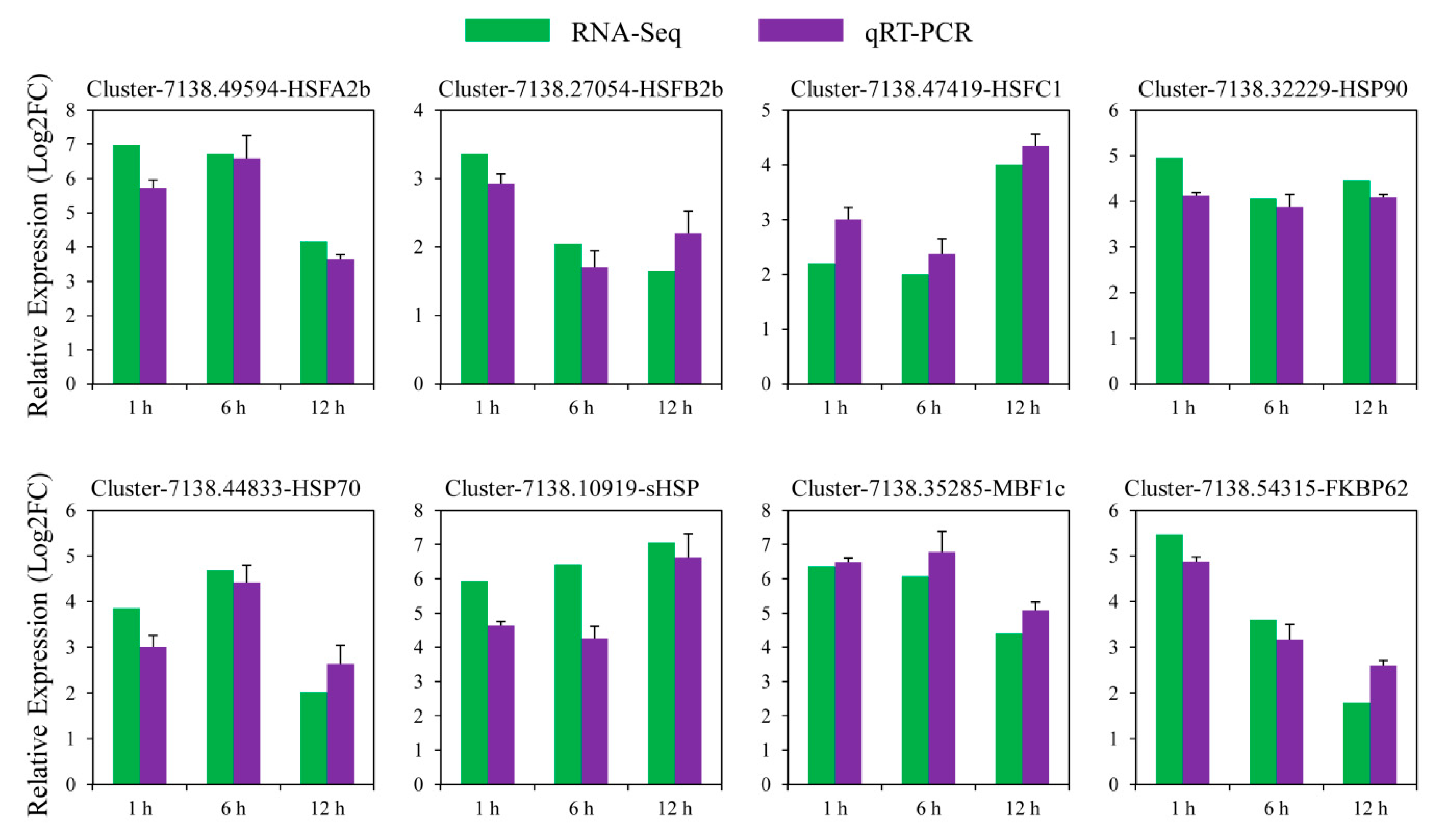
3.6. Verification of D. involucrata DEGs by qRT-PCR
4. Discussion
4.1. Heat Stress Treatment Used for Transcriptome Analysis in D. involucrata
4.2. Heat Stress Affected Expression of Genes Associated with Protein Processing in the ER
4.3. Influence of Heat Stress on Plant Hormone Signal Transduction
4.4. Hsfs May Play a Pivotal Role During Heat Stress
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, Y.X.; Chen, L.; Juan, L.; Li, Y.; Chen, F. Suppression subtractive hybridization cloning of cDNAs of differentially expressed genes in dovetree (Davidia involucrata) bracts. Plant Mol. Biol. Rep. 2002, 20, 231–238. [Google Scholar] [CrossRef]
- He, Z.C.; Li, J.Q.; Wang, H.C. Karyomorphology of Davidia involucrata and Camptotheca acuminata, with special reference to their systematic positions. Bot. J. Linn. Soc. 2004, 144, 193–198. [Google Scholar] [CrossRef]
- Sun, J.F.; Gong, Y.B.; Renner, S.S.; Huang, S.Q. Multifunctional bracts in the dove tree Davidia involucrata (Nyssaceae: Cornales): Rain protection and pollinator attraction. Am. Nat. 2008, 171, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Shimozu, Y.; Kimura, Y.; Esumi, A.; Aoyama, H.; Kuroda, T.; Sakagami, H.; Hatano, T. Ellagitannins of Davidia involucrata. I. structure of davicratinic acid A and effects of davidia tannins on drug-resistant bacteria and human oral squamous cell carcinomas. Molecules 2017, 22, 470. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.Q.; Dong, Y.F.; Herrando-Moraira, S.; Matsui, T.; Ohashi, H.; He, L.Y.; Nakao, K.; Tanaka, N.; Tomita, M.; Li, X.S.; et al. Potential effects of climate change on geographic distribution of the Tertiary relict tree species Davidia involucrata in China. Sci. Rep. 2017, 7, 43822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Zhou, B.; Lian, X. Natural distribution of Davidia involucrata and introduction analysis. J. Beijing For. Univ. 1995, 17, 25–30. [Google Scholar]
- Chen, Y.; Su, Z.X. Research on the protection of Davidia involucrata populations, a rare and endangered plant endemic to China. Acta Ecol. Sin. 2011, 31, 5466–5474. [Google Scholar]
- Li, Y.Q.; Lei, N.F.; Xu, Y.; Chen, F. Effects of high temperature stress on physiological and biochemical indicators of Davidia involucrata leaves. J. Sichuan Univ. 2009, 46, 809–813. [Google Scholar]
- Zhang, C.C.; Zhou, Q.; Ren, S.H.; Zou, J.; Li, Y.Y. Study on high temperature stress test of Davidia involucrata Baill. Hortic. Seed 2017, 5, 45–47. [Google Scholar]
- Driedonks, N.; Rieu, I.; Vriezen, W.H. Breeding for plant heat tolerance at vegetative and reproductive stages. Plant Reprod. 2016, 29, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Mesihovic, A.; Iannacone, R.; Firon, N.; Fragkostefanakis, S. Heat stress regimes for the investigation of pollen thermotolerance in crop plants. Plant Reprod. 2016, 29, 93–105. [Google Scholar] [CrossRef]
- Vierling, E. The roles of heat shock proteins in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1991, 42, 579–620. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Röth, S.; Schleiff, E.; Scharf, K.D. Prospects of engineering thermotolerance in crops through modulation of heat stress transcription factor and heat shock protein networks. Plant Cell Environ. 2015, 38, 1881–1895. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Simm, S.; Paul, P.; Bublak, D.; Scharf, K.D.; Schleiff, E. Chaperone network composition in Solanum lycopersicum explored by transcriptome profiling and microarray meta-analysis. Plant Cell Environ. 2015, 38, 693–709. [Google Scholar] [CrossRef]
- Nover, L.; Bharti, K.; Döring, P.; Mishra, S.K.; Ganguli, A.; Scharf, K.D. Arabidopsis and the heat stress transcription factor world: How many heat stress transcription factors do we need? Cell Stress Chaperones 2001, 6, 177–189. [Google Scholar] [CrossRef]
- Scharf, K.D.; Berberich, T.; Ebersberger, I.; Nover, L. The plant heat stress transcription factor (Hsf) family: Structure, function and evolution. Biochim. Biophys. Acta 2012, 1819, 104–119. [Google Scholar] [CrossRef]
- Nover, L.; Scharf, K.D.; Gagliardi, D.; Vergne, P.; Czarnecka-Verner, E.; Gurley, W.B. The Hsf world: Classification and properties of plant heat stress transcription factors. Cell Stress Chaperones 1996, 1, 215–223. [Google Scholar] [CrossRef]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef]
- von Koskull-Döring, P.; Scharf, K.D.; Nover, L. The diversity of plant heat stress transcription factors. Trends Plant Sci. 2007, 12, 452–457. [Google Scholar] [CrossRef]
- Scharf, K.D.; Rose, S.; Zott, W.; Schöffl, F.; Nover, L. Three tomato genes code for heat stress transcription factors with a region of remarkable homology to the DNA-binding domain of the yeast HSF. EMBO J. 1990, 9, 4495–4501. [Google Scholar] [CrossRef]
- Scharf, K.D.; Rose, S.; Thierfelder, J.; Nover, L. Two cDNAs for tomato heat stress transcription factors. Plant Physiol. 1993, 102, 1355–1356. [Google Scholar] [CrossRef]
- Treuter, E.; Nover, L.; Ohme, K.; Scharf, K.D. Promoter specificity and deletion analysis of three heat stress transcription factors of tomato. Mol. Gen. Genet. 1993, 240, 113–125. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Simm, S.; El-Shershaby, A.; Hu, Y.; Bublak, D.; Mesihovic, A.; Darm, K.; Mishra, S.K.; Tschiersch, B.; Theres, K.; et al. The repressor and co-activator HsfB1 regulates the major heat stress transcription factors in tomato. Plant Cell Environ. 2019, 42, 874–890. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Mesihovic, A.; Simm, S.; Paupière, M.J.; Hu, Y.; Paul, P.; Mishra, S.K.; Tschiersch, B.; Theres, K.; Bovy, A.; et al. HsfA2 controls the activity of developmentally and stress-regulated heat stress protection mechanisms in tomato male reproductive tissues. Plant Physiol. 2016, 170, 2461–2477. [Google Scholar] [CrossRef]
- Berz, J.; Simm, S.; Schuster, S.; Scharf, K.D.; Schleiff, E.; Ebersberger, I. HEATSTER: A database and web server for identification and classification of heat stress transcription factors in plants. Bioinform. Biol. Insights 2019, 13, 1177932218821365. [Google Scholar] [CrossRef]
- Suzuki, N.; Bajad, S.; Shuman, J.; Shulaev, V.; Mittler, R. The transcriptional co-activator MBF1c is a key regulator of thermotolerance in Arabidopsis thaliana. J. Biol. Chem. 2008, 283, 9269–9275. [Google Scholar] [CrossRef]
- Meiri, D.; Breiman, A. Arabidopsis ROF1 (FKBP62) modulates thermotolerance by interacting with HSP90.1 and affecting the accumulation of HsfA2-regulated sHSPs. Plant J. 2009, 59, 387–399. [Google Scholar] [CrossRef]
- Zhang, C.; Li, G.; Chen, T.; Feng, B.; Fu, W.; Yan, J.; Islam, M.R.; Jin, Q.; Tao, L.; Fu, G. Heat stress induces spikelet sterility in rice at anthesis through inhibition of pollen tube elongation interfering with auxin homeostasis in pollinated pistils. Rice 2018, 11, 14. [Google Scholar] [CrossRef]
- Yan, J.; Yu, L.; Xuan, J.; Lu, Y.; Lu, S.; Zhu, W. De novo transcriptome sequencing and gene expression profiling of spinach (Spinacia oleracea L.) leaves under heat stress. Sci. Rep. 2016, 6, 19473. [Google Scholar] [CrossRef]
- Hwang, J.E.; Kim, Y.J.; Shin, M.H.; Hyun, H.J.; Bohnert, H.J.; Park, H.C. A comprehensive analysis of the Korean fir (Abies koreana) genes expressed under heat stress using transcriptome analysis. Sci. Rep. 2018, 8, 10233. [Google Scholar] [CrossRef]
- Wang, K.; Liu, Y.; Tian, J.; Huang, K.; Shi, T.; Dai, X.; Zhang, W. Transcriptional profiling and identification of heat-responsive genes in perennial ryegrass by RNA-sequencing. Front. Plant Sci. 2017, 8, 1032. [Google Scholar] [CrossRef]
- Qian, Y.; Ren, Q.; Zhang, J.; Chen, L. Transcriptomic analysis of the maize (Zea mays L.) inbred line B73 response to heat stress at the seedling stage. Gene 2019, 692, 68–78. [Google Scholar] [CrossRef]
- Wu, L.; Taohua, Z.; Gui, W.; Xu, L.; Li, J.; Ding, Y. Five pectinase gene expressions highly responding to heat stress in rice floral organs revealed by RNA-seq analysis. Biochem. Biophys. Res. Commun. 2015, 463, 407–413. [Google Scholar] [CrossRef]
- Keller, M.; Consortium, S.; Simm, S. The coupling of transcriptome and proteome adaptation during development and heat stress response of tomato pollen. BMC Genom. 2018, 19, 447. [Google Scholar] [CrossRef]
- Li, M.; Dong, X.; Peng, J.; Xu, W.; Ren, R.; Liu, J.; Cao, F.; Liu, Z. De novo transcriptome sequencing and gene expression analysis reveal potential mechanisms of seed abortion in dove tree (Davidia involucrata Baill.). BMC Plant Biol. 2016, 16, 82. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wan, X.L.; Zhou, Q.; Wang, Y.Y.; Wang, W.E.; Bao, M.Z.; Zhang, J.W. Identification of heat-responsive genes in carnation (Dianthus caryophyllus L.) by RNA-seq. Front. Plant Sci. 2015, 6, 519. [Google Scholar] [CrossRef]
- Wahid, A.; Gelani, S.; Ashraf, M.; Foolad, M.R. Heat tolerance in plants: An overview. Environ. Exp. Bot. 2007, 61, 199–223. [Google Scholar] [CrossRef]
- Xu, X.; Peng, G.; Wu, C.; Han, Q. Global warming induces female cuttings of Populus cathayana to allocate more biomass, C and N to aboveground organs than do male cuttings. Aust. J. Bot. 2010, 58, 519–526. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, J.; Liu, Q.; He, H.; Xu, X.; Dong, T. Sexual differences in growth and defence of Populus yunnanensis under drought stress. Can. J. Forest Res. 2019, 49, 491–499. [Google Scholar] [CrossRef]
- Kranner, I.; Minibayeva, F.V.; Beckett, R.P.; Seal, C.E. What is stress? Concepts, definitions and applications in seed science. New Phytol. 2010, 188, 655–673. [Google Scholar] [CrossRef]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Bokszczanin, K.L.; Fragkostefanakis, S. Perspectives on deciphering mechanisms underlying plant heat stress response and thermotolerance. Front. Plant Sci. 2013, 4, 315. [Google Scholar] [CrossRef]
- Zhang, Y.; Mian, M.A.; Chekhovskiy, K.; So, S.; Kupfer, D.; Lai, H.; Roe, B.A. Differential gene expression in Festuca under heat stress conditions. J. Exp. Bot. 2005, 56, 897–907. [Google Scholar] [CrossRef]
- Vidya, S.M.; Kumar, H.S.V.; Bhatt, R.M.; Laxman, R.H.; Ravishankar, K.V. Transcriptional profiling and genes involved in acquired thermotolerance in Banana: A non-model crop. Sci. Rep. 2018, 8, 10683. [Google Scholar] [CrossRef]
- Fragkostefanakis, S.; Mesihovic, A.; Hu, Y.; Schleiff, E. Unfolded protein response in pollen development and heat stress tolerance. Plant Reprod. 2016, 29, 81–91. [Google Scholar] [CrossRef]
- Li, B.; Gao, K.; Ren, H.; Tang, W. Molecular mechanisms governing plant responses to high temperatures. J. Integr. Plant Biol. 2018, 60, 757–779. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, H. Differential accumulation of proteins in leaves and roots associated with heat tolerance in two Kentucky bluegrass genotypes differing in heat tolerance. Acta Physiol. Plant. 2016, 38, 213. [Google Scholar] [CrossRef]
- Cui, F.; Liu, L.; Zhao, Q.; Zhang, Z.; Li, Q.; Lin, B.; Wu, Y.; Tang, S.; Xie, Q. Arabidopsis ubiquitin conjugase UBC32 is an ERAD component that functions in brassinosteroid-mediated salt stress tolerance. Plant Cell 2012, 24, 233–244. [Google Scholar] [CrossRef]
- Kirst, M.E.; Meyer, D.J.; Gibbon, B.C.; Jung, R.; Boston, R.S. Identification and characterization of endoplasmic reticulum-associated degradation proteins differentially affected by endoplasmic reticulum stress. Plant Physiol. 2005, 138, 218–231. [Google Scholar] [CrossRef]
- Qian, D.; Chen, G.; Tian, L.; Qu, L.Q. OsDER1 is an ER-associated protein degradation factor that responds to ER Stress. Plant Physiol. 2018, 178, 402–412. [Google Scholar] [CrossRef]
- Kostova, Z.; Tsai, Y.C.; Weissman, A.M. Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Semin. Cell Dev. Biol. 2007, 18, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhang, Y.; Qi, J.; Chi, Y.; Fan, B.; Yu, J.Q.; Chen, Z. E3 ubiquitin ligase CHIP and NBR1-mediated selective autophagy protect additively against proteotoxicity in plant stress responses. PLoS Genet. 2014, 10, e1004116. [Google Scholar] [CrossRef]
- Yan, J.; Wang, J.; Li, Q.; Hwang, J.R.; Patterson, C.; Zhang, H. AtCHIP, a U-box-containing E3 ubiquitin ligase, plays a critical role in temperature stress tolerance in Arabidopsis. Plant Physiol. 2003, 132, 861–869. [Google Scholar] [CrossRef]
- Jaillais, Y.; Chory, J. Unraveling the paradoxes of plant hormone signaling integration. Nat. Struct. Mol. Biol. 2010, 17, 642–645. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Vain, T.; Viotti, C.; Doyle, S.M.; Tarkowská, D.; Novák, O.; Zipfel, C.; Sitbon, F.; Robert, S.; Hofius, D. Vacuole integrity maintained by DUF300 proteins is required for brassinosteroid signaling regulation. Mol. Plant 2018, 11, 553–567. [Google Scholar] [CrossRef]
- Ku, Y.S.; Sintaha, M.; Cheung, M.Y.; Lam, H.M. Plant hormone signaling crosstalks between biotic and abiotic stress responses. Int. J. Mol. Sci. 2018, 19, 3206. [Google Scholar] [CrossRef]
- Wani, S.H.; Kumar, V.; Shriram, V.; Sah, S.K. Phytohormones and their metabolic engineering for abiotic stress tolerance in crop plants. Crop J. 2016, 4, 162–176. [Google Scholar] [CrossRef] [Green Version]
- Nie, S.; Huang, S.; Wang, S.; Mao, Y.; Liu, J.; Ma, R.; Wang, X. Enhanced brassinosteroid signaling intensity via SlBRI1 overexpression negatively regulates drought resistance in a manner opposite of that via exogenous BR application in tomato. Plant Physiol. Biochem. 2019, 138, 36–47. [Google Scholar] [CrossRef]
- Peleg, Z.; Blumwald, E. Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 2011, 14, 290–295. [Google Scholar] [CrossRef]
- Jiroutova, P.; Oklestkova, J.; Strnad, M. Crosstalk between brassinosteroids and ethylene during plant growth and under abiotic stress conditions. Int. J. Mol. Sci. 2018, 19, 3283. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, P.; Li, C.; Xia, G. The moss jasmonate ZIM-domain protein PnJAZ1 confers salinity tolerance via crosstalk with the abscisic acid signalling pathway. Plant Sci. 2019, 280, 1–11. [Google Scholar] [CrossRef]
- Yang, Y.G.; Lv, W.T.; Li, M.J.; Wang, B.; Sun, D.M.; Deng, X. Maize membrane-bound transcription factor Zmbzip17 is a key regulator in the cross-talk of ER quality control and ABA signaling. Plant Cell Physiol. 2013, 54, 2020–2033. [Google Scholar] [CrossRef]
- Chen, Y.; Aung, K.; Rolčík, J.; Walicki, K.; Friml, J.; Brandizzi, F. Inter-regulation of the unfolded protein response and auxin signaling. Plant J. 2014, 77, 97–107. [Google Scholar] [CrossRef]
- Simm, S.; Scharf, K.D.; Jegadeesan, S.; Chiusano, M.L.; Firon, N.; Schleiff, E. Survey of genes involved in biosynthesis, transport, and signaling of phytohormones with focus on Solanum lycopersicum. Bioinform. Biol. Insights 2016, 10, 185–207. [Google Scholar] [CrossRef]
- Liu, H.C.; Charng, Y.Y. Common and distinct functions of Arabidopsis class A1 and A2 heat shock factors in diverse abiotic stress responses and development. Plant Physiol. 2013, 163, 276–290. [Google Scholar] [CrossRef]
- Mishra, S.K.; Tripp, J.; Winkelhaus, S.; Tschiersch, B.; Theres, K.; Nover, L.; Scharf, K.D. In the complex family of heat stress transcription factors, HsfA1 has a unique role as master regulator of thermotolerance in tomato. Genes Dev. 2002, 16, 1555–1567. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.C.; Liao, H.T.; Charng, Y.Y. The role of class A1 heat shock factors (HSFA1s) in response to heat and other stresses in Arabidopsis. Plant Cell Environ. 2011, 34, 738–751. [Google Scholar] [CrossRef]
- Yoshida, T.; Ohama, N.; Nakajima, J.; Kidokoro, S.; Mizoi, J.; Nakashima, K.; Maruyama, K.; Kim, J.M.; Seki, M.; Todaka, D.; et al. Arabidopsis HsfA1 transcription factors function as the main positive regulators in heat shock-responsive gene expression. Mol. Genet. Genom. 2011, 286, 321–332. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length Range | Transcripts | Unigenes |
|---|---|---|
| 200–500 bp | 158,998 | 63,485 |
| 0.5–1k bp | 82,691 | 37,693 |
| 1–2k bp | 85,562 | 21,967 |
| >2k bp | 78,547 | 15,778 |
| Total | 405,798 | 138,923 |
| Min length (bp) | 201 | 201 |
| Mean length (bp) | 1212 | 941 |
| Max length (bp) | 17,633 | 17,633 |
| N50 (bp) | 2111 | 1493 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Vetukuri, R.R.; Xu, W.; Xu, X. Transcriptomic Responses of Dove Tree (Davidia involucrata Baill.) to Heat Stress at the Seedling Stage. Forests 2019, 10, 656. https://doi.org/10.3390/f10080656
Liu Q, Vetukuri RR, Xu W, Xu X. Transcriptomic Responses of Dove Tree (Davidia involucrata Baill.) to Heat Stress at the Seedling Stage. Forests. 2019; 10(8):656. https://doi.org/10.3390/f10080656
Chicago/Turabian StyleLiu, Qinsong, Ramesh R. Vetukuri, Wenjuan Xu, and Xiao Xu. 2019. "Transcriptomic Responses of Dove Tree (Davidia involucrata Baill.) to Heat Stress at the Seedling Stage" Forests 10, no. 8: 656. https://doi.org/10.3390/f10080656
APA StyleLiu, Q., Vetukuri, R. R., Xu, W., & Xu, X. (2019). Transcriptomic Responses of Dove Tree (Davidia involucrata Baill.) to Heat Stress at the Seedling Stage. Forests, 10(8), 656. https://doi.org/10.3390/f10080656