Measuring the Heat of Interaction between Lignocellulosic Materials and Water



Abstract
:1. Introduction
1.1. Wood-Water Interactions
1.2. Nomenclature
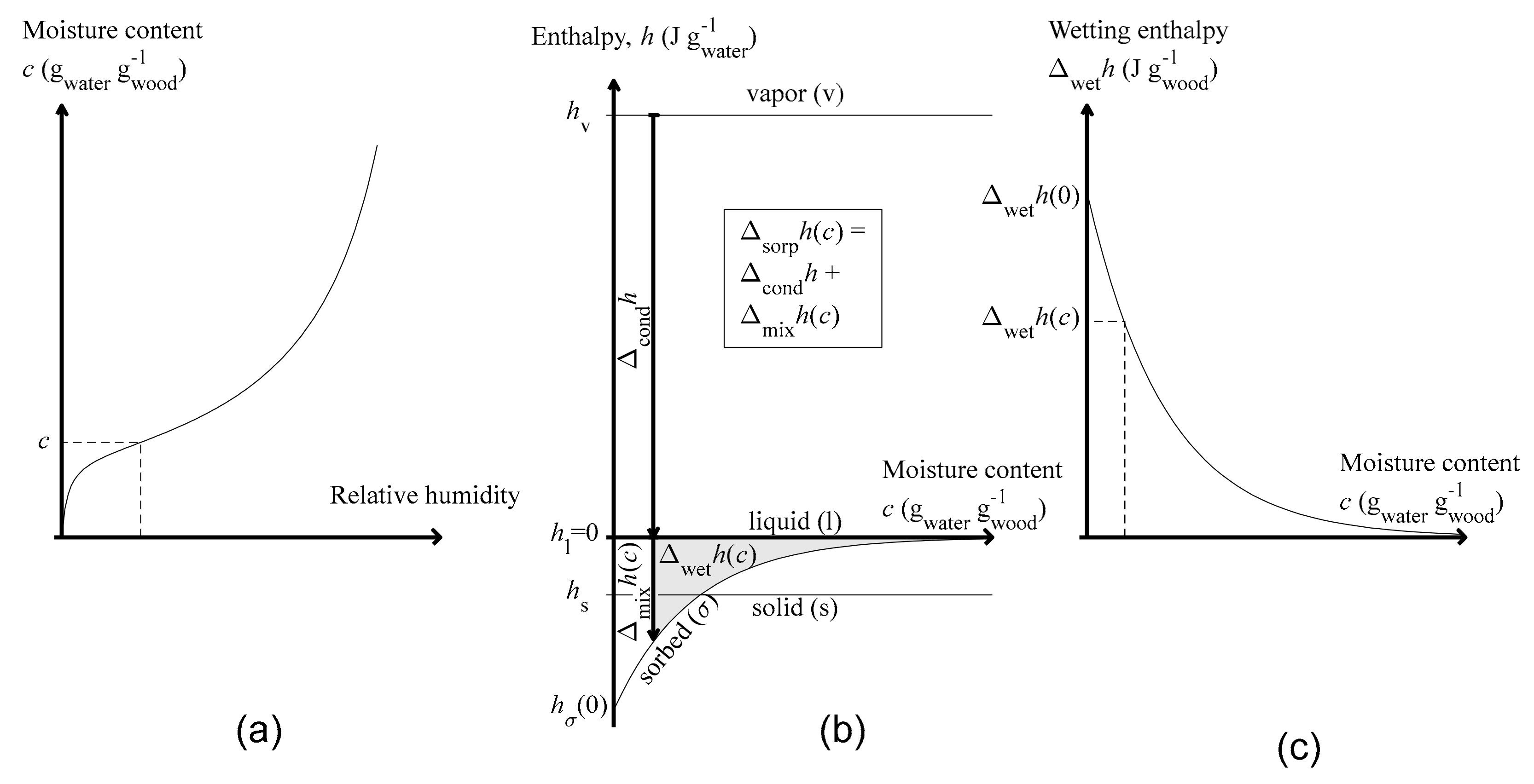
1.3. Perspective on Sorption
- The larger entity does not have a defined molecular mass, at least not if it is a material, like wood, so we need to work with specific units (J g−1) when the denominator refers to the material.
- The enthalpy when a vapor molecule binds to a solid is not a constant, but it depends on how many molecules have previously been sorbed on the solid, i.e., the moisture content. Chemically, the sorption process may be similar to the ligand-protein binding process (both are typically hydrogen bonding), but while all the protein molecules and their binding sites are identical, vapor can be bound in accessible porous materials in many different ways.
- Chemical thermodynamics states that processes spontaneously take place when they result in a decrease in the Gibbs energy. This “driving force” has contributions from two terms: enthalpy and entropy. The enthalpic contributions reflect the strength of the binding. For ligand-protein binding processes in an aqueous environment, there can also be large entropical contributions, from the loss of conformational degrees of freedom in the molecules involved and that water molecules are expelled from the binding site [16]. Such phenomena can also take place during sorption on, for example, bio-molecules. Some sorption processes can then be said to be entropy driven, see, for example, reference [15]. This phenomenon is limited for vapor sorption on relatively rigid solids, like wood.
1.4. Sorption Isotherms
1.5. Sorption and Mixing
1.6. Wetting
1.7. Clausius-Clapeyron Equation
- The sorption isosteres (ln(φ) as a function of 1/T) should be linear. In practice they are often more or less non-linear, possibly because of measurement errors or the use of non-optimal fitting equations.
- The enthalpy is assumed to be constant with temperature, which it strictly is not.
- The Clausius–Clapeyron assumes that the sorption is reversible, which it probably is not because of hysteresis.
1.8. Literature Data
2. Materials
3. Methods
3.1. Isosteric Method
3.2. Solution Calorimetry
3.3. Sorption Calorimetry
3.4. RH Perfusion Calorimetry
4. Results
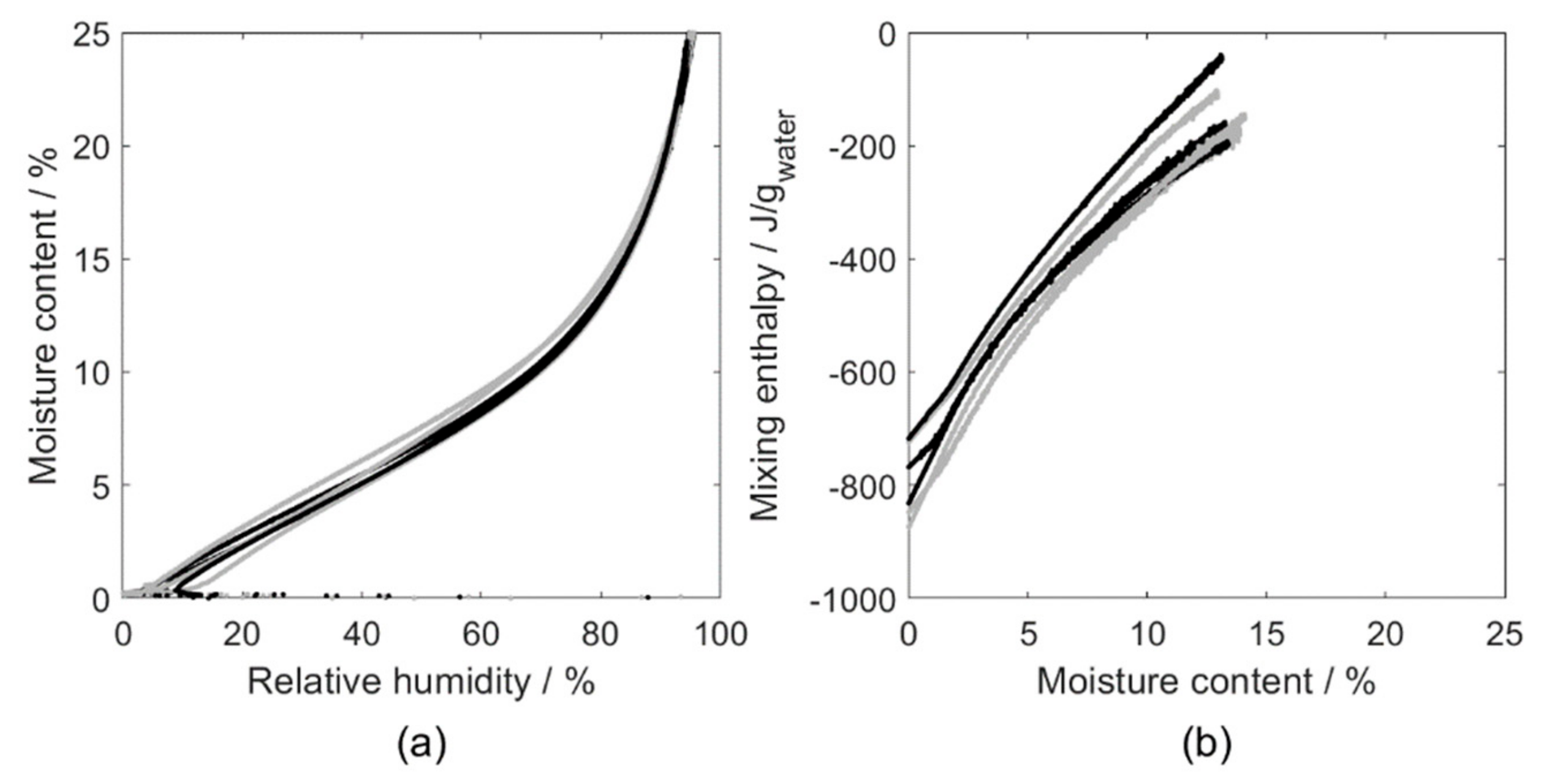
4.1. Isosteric Method
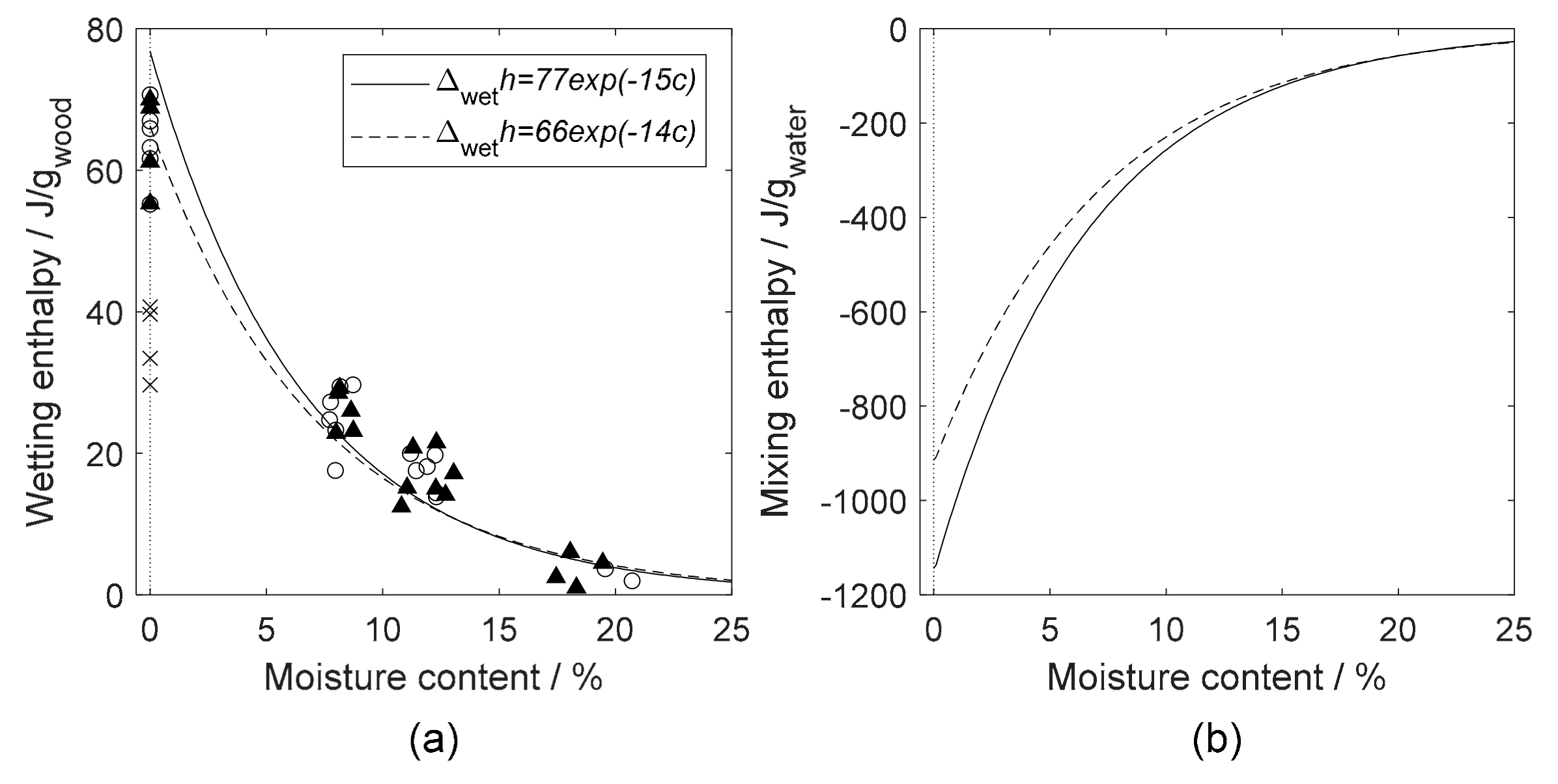
4.2. Heat of Wetting
4.3. Sorption Calorimetry
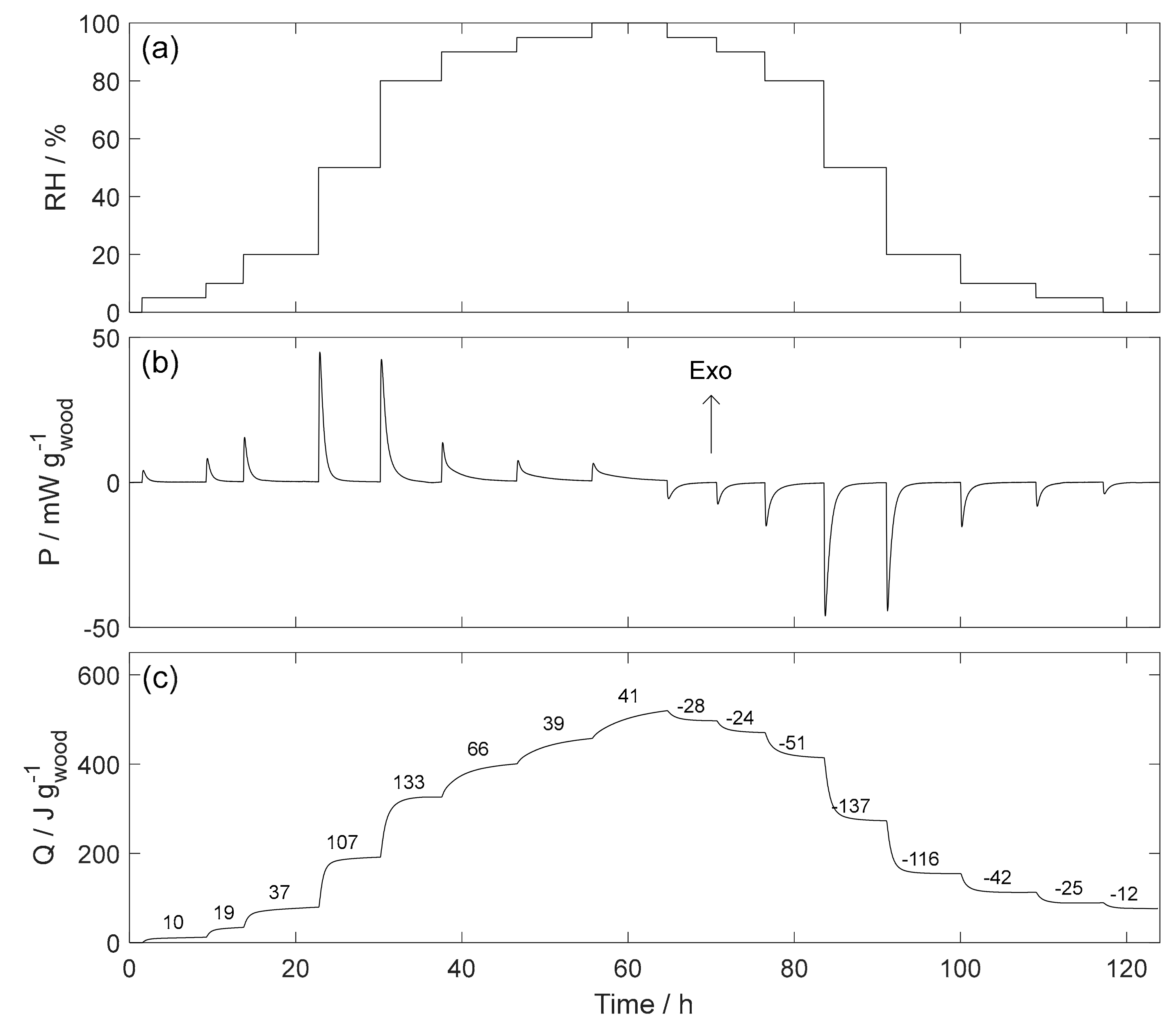
4.4. RH Perfusion Calorimetry
5. Discussion
5.1. Isosteric Method
5.2. Solution Calorimetry
5.3. Sorption Calorimetry
5.4. RH Perfusion Calorimetry
- In this type of measurement, the results of a blank run (without sample) should be subtracted from the sample result, as there will also be sorption and the corresponding heats on the internal surfaces of the vial (this was not done in the present measurements).
- This method should probably not be used with small steps in RH, like the 0-5-10% RH that we used, as this generates small peaks and small moisture content changes, and large uncertainties in the results.
- The RH that is generated by the equipment needs to be calibrated over the whole RH range, especially at low RHs, where the mass flow controllers that the RH generation is based on can be less reliable. We made a one-point calibration at 75% RH (that gave 0.9% RH higher value, which is good).
- The drying of the samplse may need to be improved; it is not clear what the initial state of the sample was after the initial drying at a set value of 0% RH at 25 °C. Possibly, the sample should be externally dried, for example, in a vacuum oven, and then quickly transferred to the calorimetric vial.
5.5. General
- Different terminology is used. For example, are the terms differential heat and integral heat [3] sometimes used for what we have called mixing enthalpy and wetting enthalpy, respectively; differential heat of swelling [9] is our mixing enthalpy; and, the term isosteric heat is typically the same as our mixing enthalpy, but used if the isosteric method has been used.
- Different symbols are used; for example are Q, H, ∆H, and ∆h used for enthalpy change. We prefer to use Q for a measured heat and ∆h for a calculated specific enthalpy change.
- Different sign conventions are used. We have followed the thermochemical convention that looks at processes from the standpoint of the system, so heat production (loss of heat by the system) gives negative enthalpy. However, it is common to instead show, e.g., mixing enthalpies as positive.
- A minor issue is the use of calories in the older literature, but as long as the units are written out, the multiplication by 4.184 J cal−1 converts heats and enthalpies from calories to joules (the SI-unit).
- All of the sorption and mixing enthalpies can be based either on gram or on mole, as it is the water that they refer to; wetting enthalpies for wood can only be given per mass of wood, as wood has no molar mass. If an enthalpy has units of J g−1, it is thus not possible to know whether it is J gwood−1 (a wetting enthalpy) or J gwater−1 (a mixing or sorption enthalpy), so we encourage the use of sub-indices to indicate this.
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Volbehr, B. Untersuchungen über die Quellung der Holzfaser; Druck von Schmidt and Klaunig: Kiel, Germany, 1896. [Google Scholar]
- Glass, S.V.; Zelinka, S.L.; Johnson, J.A. Investigation of Historic Equilibrium Moisture Content Data from the Forest Products Laboratory; FPL-GTR-229; USDA Forest Service: Washington, DC, USA, 2014; p. 37. [Google Scholar]
- Skaar, C. Wood-Water Relations; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1988; ISBN 3642736858. [Google Scholar]
- Engelund, E.T.; Thygesen, L.G.; Svensson, S.; Hill, C.S. A critical discussion of the physics of wood–water interactions. Wood Sci. Technol. 2013, 47, 141–161. [Google Scholar] [CrossRef]
- Berthold, J.; Desbrières, J.; Rinaudo, M.; Salmén, L. Types of adsorbed water in relation to the ionic groups and their counter-ions for some cellulose derivatives. Polymer 1994, 35, 5729–5736. [Google Scholar] [CrossRef]
- Ping, Z.H.; Nguyen, Q.T.; Chen, S.M.; Zhou, J.Q.; Ding, Y.D. States of water in different hydrophilic polymers—DSC and FTIR studies. Polymer 2001, 42, 8461–8467. [Google Scholar] [CrossRef]
- Thygesen, L.G.; Engelund, E.T.; Hoffmeyer, P. Water sorption in wood and modified wood at high values of relative humidity. Part I: Results for untreated, acetylated, and furfurylated Norway spruce. Holzforschung 2010, 64, 315–323. [Google Scholar] [CrossRef]
- Rautkari, L.; Hill, C.; Curling, S.; Jalaludin, Z.; Ormondroyd, G. What is the role of the accessibility of wood hydroxyl groups in controlling moisture content? J. Mater. Sci. 2013, 48, 6352–6356. [Google Scholar] [CrossRef]
- Stamm, A.J.; Loghborough, K.W. Thermodynamics of the Swelling of Wood. J. Phys. Chem. C 1935, 39, 121–132. [Google Scholar] [CrossRef]
- Weichert, L. Untersuchungen über das Sorption-und Quellungsverhalten von Fichte, Buche und Buchen-Preßvollholz bei Temperaturen zwischen 20° und 100°C. Eur. J. Wood Wood Prod. 1963, 21, 290–300. [Google Scholar] [CrossRef]
- Simpson, W.T. Predicting Equilibrium Moisture Content of Wood by Mathematical Models. Wood Fiber Sci. 1973, 5, 41–49. [Google Scholar]
- Rees, W.H.; Hearle, J.W.S. Moisture in Textiles. Heat of Absorption; Textile Institute: Manchester, UK; Butterworths Scientific Publications: London, UK, 1960. [Google Scholar]
- Cohen, E.R.; Cvitas, T.; Frey, J.; Holmstrom, B.; Kuchitsu, K.; Marquardt, R.; Mills, I.; Pavese, F.; Quack, M.; Stohner, J.; et al. Quantities, Units and Symbols in Physical Chemistry, 3rd ed.; IUPAC and RSC Publishing: Cambridge, UK, 2007; ISBN 978-0-85404-433-7. [Google Scholar]
- Gál, S. Über die Ausdrucksweisen der Konzentration des Wasserdampfes bei Wasserdampf-Sorptionsmessungen. HCA 1972, 55, 1752–1757. [Google Scholar] [CrossRef]
- Vuignier, K.; Schappler, J.; Veuthey, J.-L.; Carrupt, P.-A.; Martel, S. Drug-protein binding: A critical review of analytical tools. Anal. Bioanal. Chem. 2010, 398, 53–66. [Google Scholar] [CrossRef]
- Freire, E. Isothermal titration calorimetry: Controlling binding forces in lead optimization. Drug Discov. Today Technol. 2004, 1, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Markova, N.; Sparr, E.; Wadsö, L.; Wennerström, H. A Calorimetric Study of Phospholipid Hydration. Simultaneous Monitoring of Enthalpy and Free Energy. J. Phys. Chem. B 2000, 104, 8053–8060. [Google Scholar] [CrossRef]
- Marabi, A.; Mayor, G.; Raemy, A.; Bauwens, I.; Claude, J.; Burbidge, A.; Wallach, R.; Saguy, I.S. Solution calorimetry: A novel perspective into the dissolution process of food powders. Food Res. Int. 2007, 40, 1286–1298. [Google Scholar] [CrossRef]
- Simon, C.; Esteban, L.; Palacios, P.D.; Garcia Fernández, F.; García-Iruela, A. Thermodynamic properties of the water sorption isotherms of wood of limba (Terminalia superba Engl. and Diels), obeche (Triplochiton scleroxylon K. Schum.), radiata pine (Pinus radiata D. Don) and chestnut (Castanea sativa Mill.). Ind. Crop. Prod. 2016, 94, 122–131. [Google Scholar] [CrossRef]
- Sirota, N. Certain Problems of Polymorphism (II) Generalized Clausius-Clapeyron Equation and Ostwald’s Step Rule. Cryst. Res. Technol. 1987, 22, 1343–1381. [Google Scholar] [CrossRef]
- Bahar, R.; Azzouz, S.; Remond, R.; Ouertani, S.; Elaieb, M.T.; El Cafci, M.A. Moisture sorption isotherms and thermodynamic properties of Oak wood (Quercus robur and Quercus canariensis): Optimization of the processing parameters. Heat Mass Transf. 2016, 46, 304. [Google Scholar] [CrossRef]
- Dieste, A.; Krause, A.; Militz, H. Modification of Fagus sylvatica (L.) with 1, 3-dimethylol-4, 5-dihydroxyethylene urea (DMDHEU): Part 1. Estimation of heat adsorption by the isosteric method (Hailwood-Horrobin model) and by solution calorimetry. Holzforschung 2008, 62, 577. [Google Scholar] [CrossRef]
- Mizoue, L.S.; Tellinghuisen, J. Calorimetric vs. van’t Hoff binding enthalpies from isothermal titration calorimetry: Ba2+-crown ether complexation. Biophys. Chem. 2004, 110, 15–24. [Google Scholar] [CrossRef]
- Kocherbitov, V. Salt-saturated salt solution as a standard system for sorption calorimetry. Thermochim. Acta 2004, 421, 105–110. [Google Scholar] [CrossRef]
- Albèr, C.; Engblom, J.; Falkman, P.; Kocherbitov, V. Hydration of hyaluronan: Effects on structural and thermodynamic properties. J. Phys. Chem. B 2015, 119, 4211–4219. [Google Scholar] [CrossRef]
- Benson, S.W. Some Observations on the Structures of Liquid Alcohols and Their Heats of Vaporization. J. Am. Chem. Soc. 1996, 118, 10645–10649. [Google Scholar] [CrossRef]
- Wadsö, L.; Markova, N. A double twin isothermal microcalorimeter. Thermochim. Acta 2000, 360, 101–107. [Google Scholar] [CrossRef]
- Wadsö, L.; Markova, N. A method to simultaneously determine sorption isotherms and sorption enthalpies with a double twin microcalorimeter. Rev. Sci. Instrum. 2002, 73, 2743. [Google Scholar] [CrossRef]
- Wadso, L. Operational issues in isothermal calorimetry. Cem. Concr. Res. 2010, 40, 1129–1137. [Google Scholar] [CrossRef]
- Miller, D.P.; Lechuga-Ballesteros, D. Rapid Assessment of the Structural Relaxation Behavior of Amorphous Pharmaceutical Solids: Effect of Residual Water on Molecular Mobility. Pharm. Res. 2006, 23, 2291–2305. [Google Scholar] [CrossRef] [PubMed]
- Ostrovskii, V.E.; Gostev, B.V. Heat effects and rates and molecular mechanisms of water sorption by perfluorinated polymer materials bearing functional groups. J. Therm. Anal. 1996, 46, 397–416. [Google Scholar] [CrossRef]
- Zelinka, S.L.; Glass, S.V.; Thybring, E.E. Myth versus reality: Do parabolic sorption isotherm models reflect actual wood–water thermodynamics? Wood Sci. Technol. 2018, 52, 1701–1706. [Google Scholar] [CrossRef]
- Willems, W. Equilibrium thermodynamics of wood moisture revisited: Presentation of a simplified theory. Holzforschung 2016, 70, 963–970. [Google Scholar] [CrossRef]
- Willems, W. Hydrostatic pressure and temperature dependence of wood moisture sorption isotherms. Wood Sci. Technol. 2014, 48, 483–498. [Google Scholar] [CrossRef]
- Glass, S.V.; Boardman, C.R.; Thybring, E.E.; Zelinka, S.L. Quantifying and reducing errors in equilibrium moisture content measurements with dynamic vapor sorption (DVS) experiments. Wood Sci. Technol. 2018, 52, 909–927. [Google Scholar] [CrossRef] [Green Version]
- Glass, S.V.; Boardman, C.R.; Zelinka, S.L. Short hold times in dynamic vapor sorption measurements mischaracterize the equilibrium moisture content of wood. Wood Sci. Technol. 2016, 51, 243–260. [Google Scholar] [CrossRef]
- Kajita, H. Heat of wetting of wood in water. I. Kyoto Pref. Univ. Bull. 1977, 20, 49–61. [Google Scholar]
- Kelsey, K.E.; Clarke, L.N. the heat of sorption of water in wood. Aust. J. Appl. Sci. 1955, 7, 161–175. [Google Scholar]
- Hearmon, R.F.S.; Burcham, J.N. Specific Heat and Heat of Wetting of Wood. Nature 1955, 176, 978. [Google Scholar] [CrossRef]
- Kajita, H. Heat of Wetting of the Constituents of Wood in Water. J. Soc. Mat. Sei. Jpn. 1977, 26, 417–423. [Google Scholar] [CrossRef]
- Hearle, J.W.S.; Peters, R.H. Moisture in Textiles; Textile Institute: Manchester, UK; Butterworths Scientific Publications: London, UK, 1960. [Google Scholar]
- Wadsö, L. Principles of a microcalorimetric technique for the study of mould activity as a function of relative humidity. J. Therm. Anal. 1997, 49, 1053–1060. [Google Scholar] [CrossRef]
- Wadsö, L. Describing non-Fickian water-vapour sorption in wood. J. Mater. Sci. 1994, 29, 2367–2372. [Google Scholar] [CrossRef]
- Kocherbitov, V.; Wadsö, L. A desorption calorimetric method for use at high water activities. Thermochim. Acta 2004, 411, 31–36. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | General Property | Water Related Property | Units |
|---|---|---|---|
| c | Vapor content | Moisture content | gvapor gsolid−1, gwater gwood−1 |
| ∆condh | Condensation enthalpy | J gvapor−1, J gwater−1 | |
| ∆mixh | Mixing enthalpy | J gvapor−1, J gwater−1 | |
| ∆sorph | Sorption enthalpy | J gvapor−1, J gwater−1 | |
| ∆weth | Wetting enthalpy | J gsolid−1, J gwood−1 | |
| ∆vaph | Vaporization enthalpy | J gvapor−1, J gwater−1 | |
| m | Mass | g | |
| φ | Relative vapor pressure | Relative humidity | Pa Pa−1 |
| Word | Meaning | ||
| Absorption | Uptake of vapor or liquid by a solid | ||
| Adsorption | Sorption of vapor on a surface | ||
| Condensation | Phase change from vapor to liquid | ||
| Desorption | Loss of vapor by a solid | ||
| Mixing | Uptake of liquid by a solid | ||
| Sorption | Exchange of vapor between solid and gas phases | ||
| Vaporization | Phase change from liquid to vapor | ||
| Wetting | Uptake of liquid by a solid until saturation | ||
| Isosteric Method | Solution Calorimetry | Sorption Calorimetry | RH Perfusion Calorimetry | |
|---|---|---|---|---|
| What is measured? | Sorption isotherms at different temperatures | The temperature difference when a sample is wetted | The thermal powers of vaporization and sorption when a sample is humidified | The heats produced when the RH around a sample is changed |
| Calculation procedure | The Clausius-Clapeyron equation (Equation (5)) | The heat produced when wood is wetted (saturated) with water (Equation (2)) | Three equations (Equations (9)–(11)) are used | Integration of each peak and combination with moisture content change from isotherm (Equation (12)) |
| What enthalpy is calculated? | Sorption/mixing enthalpy | Wetting enthalpy | Sorption/mixing enthalpy | Sorption/mixing enthalpy |
| Equipment | Sorption balance | Solution calorimeter | Sorption calorimeter | Isothermal calorimeter and RH-perfusion cell |
| Advantages | Sorption balances are rather common instruments | Solution calorimeters are rather common instruments | Rather quick measurement (1–2 days); gives both isotherm and enthalpy | Can be used for any RH-step; can be used in both absorption and desorption |
| Disadvantages | It is not clear if the theoretical assumptions are fulfilled; the isotherm data needs to be of high quality; measurements takes a long time (month) | Results need to be differentiated to give mixing enthalpy; many samples need to be measured | Very uncommon instrument; some uncertainties about the interpretation of the results | Rather common instrument, but the RH perfusion system is not common; relatively large uncertainties in several input factors |
| RH-Step | c/gwater gwood−1 | Q J gwood−1 | ∆sorph/J gwater−1 | ∆mixh/J gwater−1 | |||
|---|---|---|---|---|---|---|---|
| Initial | Final | Difference | Calc. | Lit. | |||
| 20→50% | 0.042 | 0.085 | 0.043 | 121 | −2800 | −360 | −490 |
| 50→80% | 0.085 | 0.148 | 0.063 | 135 | −2150 | 300 | −240 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nopens, M.; Wadsö, L.; Ortmann, C.; Fröba, M.; Krause, A. Measuring the Heat of Interaction between Lignocellulosic Materials and Water. Forests 2019, 10, 674. https://doi.org/10.3390/f10080674
Nopens M, Wadsö L, Ortmann C, Fröba M, Krause A. Measuring the Heat of Interaction between Lignocellulosic Materials and Water. Forests. 2019; 10(8):674. https://doi.org/10.3390/f10080674
Chicago/Turabian StyleNopens, Martin, Lars Wadsö, Christian Ortmann, Michael Fröba, and Andreas Krause. 2019. "Measuring the Heat of Interaction between Lignocellulosic Materials and Water" Forests 10, no. 8: 674. https://doi.org/10.3390/f10080674
APA StyleNopens, M., Wadsö, L., Ortmann, C., Fröba, M., & Krause, A. (2019). Measuring the Heat of Interaction between Lignocellulosic Materials and Water. Forests, 10(8), 674. https://doi.org/10.3390/f10080674