Gene Set Subtraction Reveals 633 Candidate Genes for Bamboo Culm Wall Thickening

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, DNA Extraction, and Whole Genome Resequencing
2.2. Sequence Alignment and Detection of Single Nucleotide Polymorphisms (SNPs) and Insertion-Deletion Mutations (InDels)
2.3. Genetic Annotation and Identification of Candidate Genes
3. Results
3.1. Genome-Wide Variation of Four Species of Bamboo
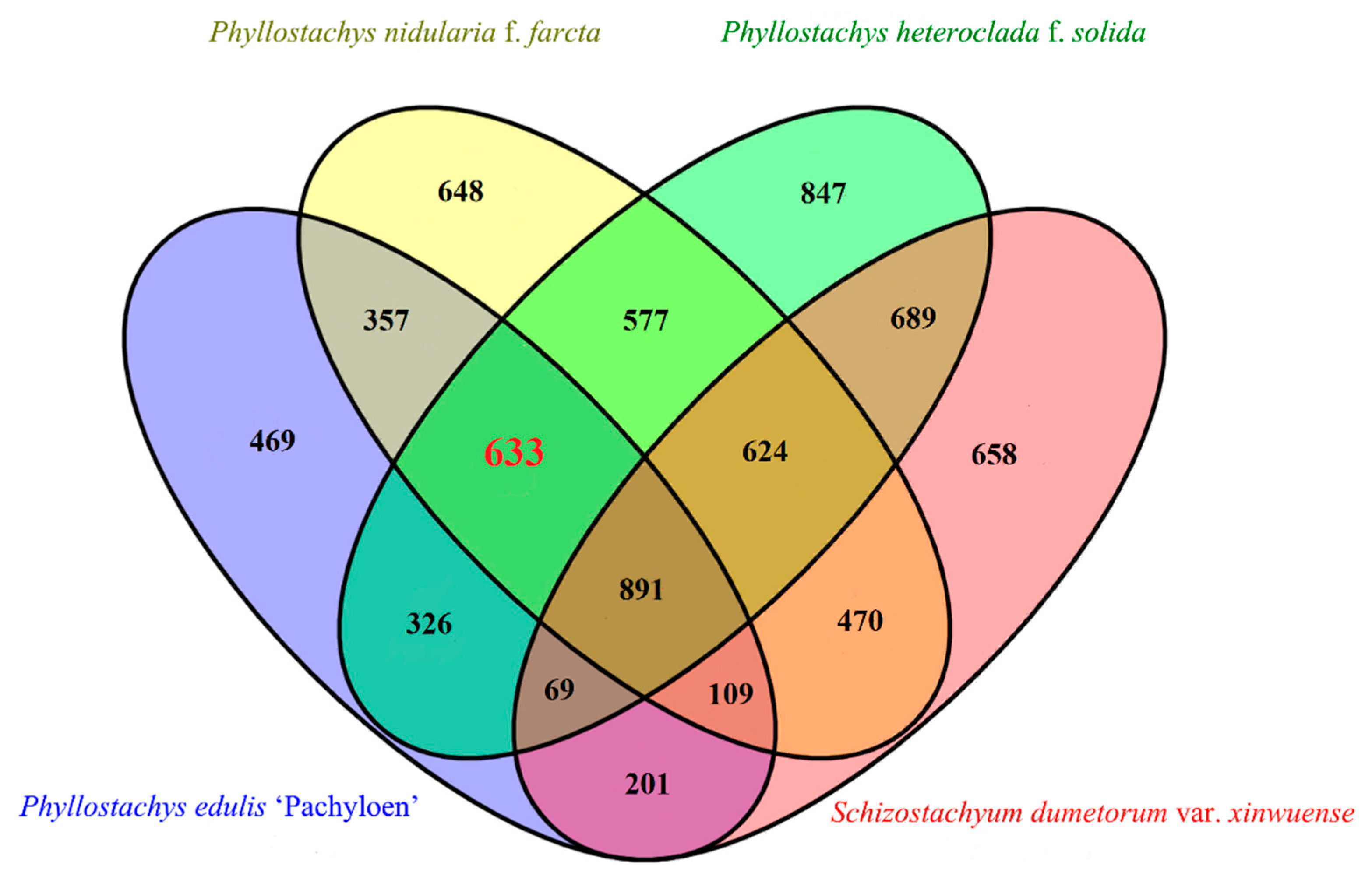
3.2. Determination of the Differential Gene Set
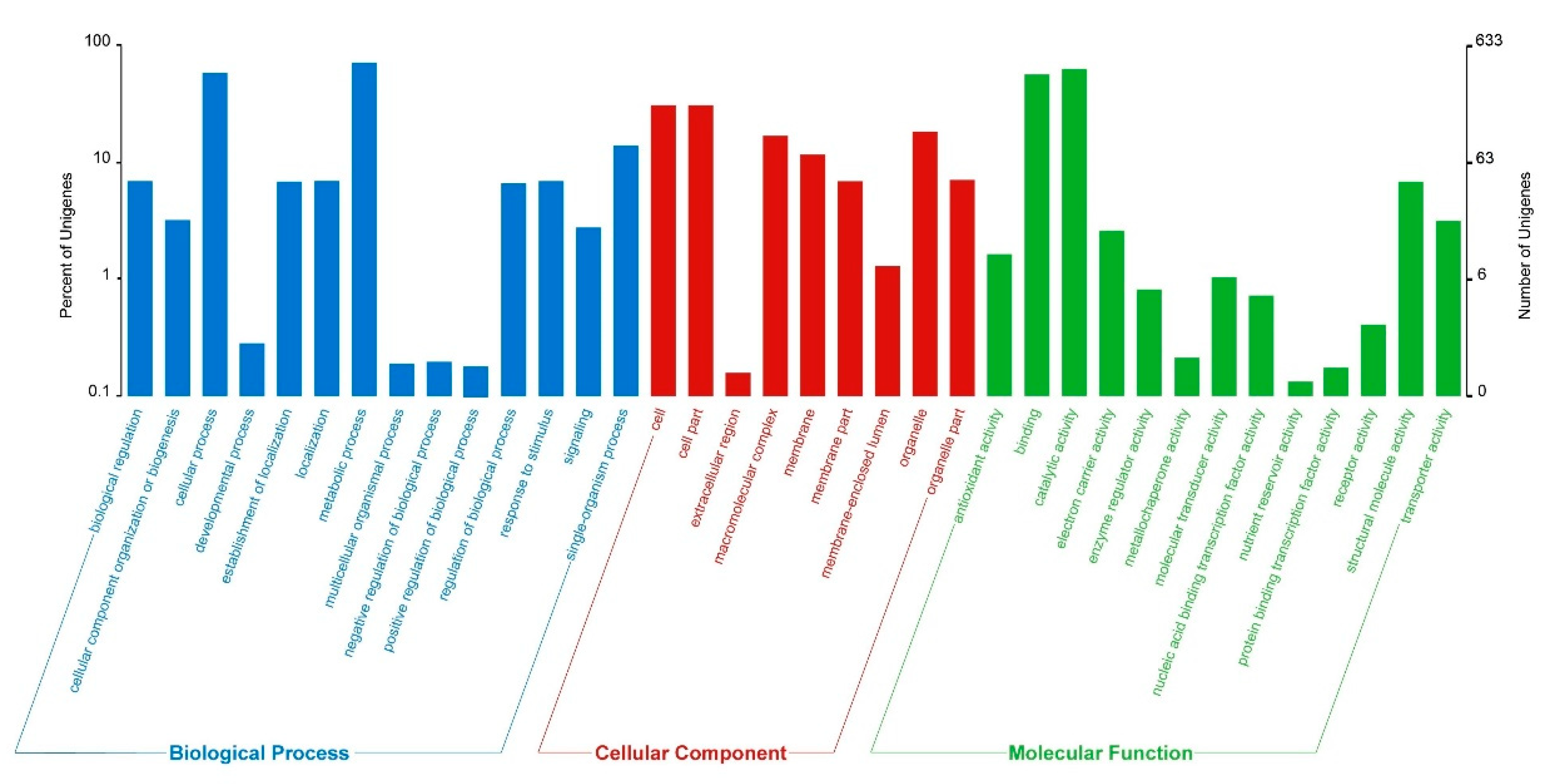
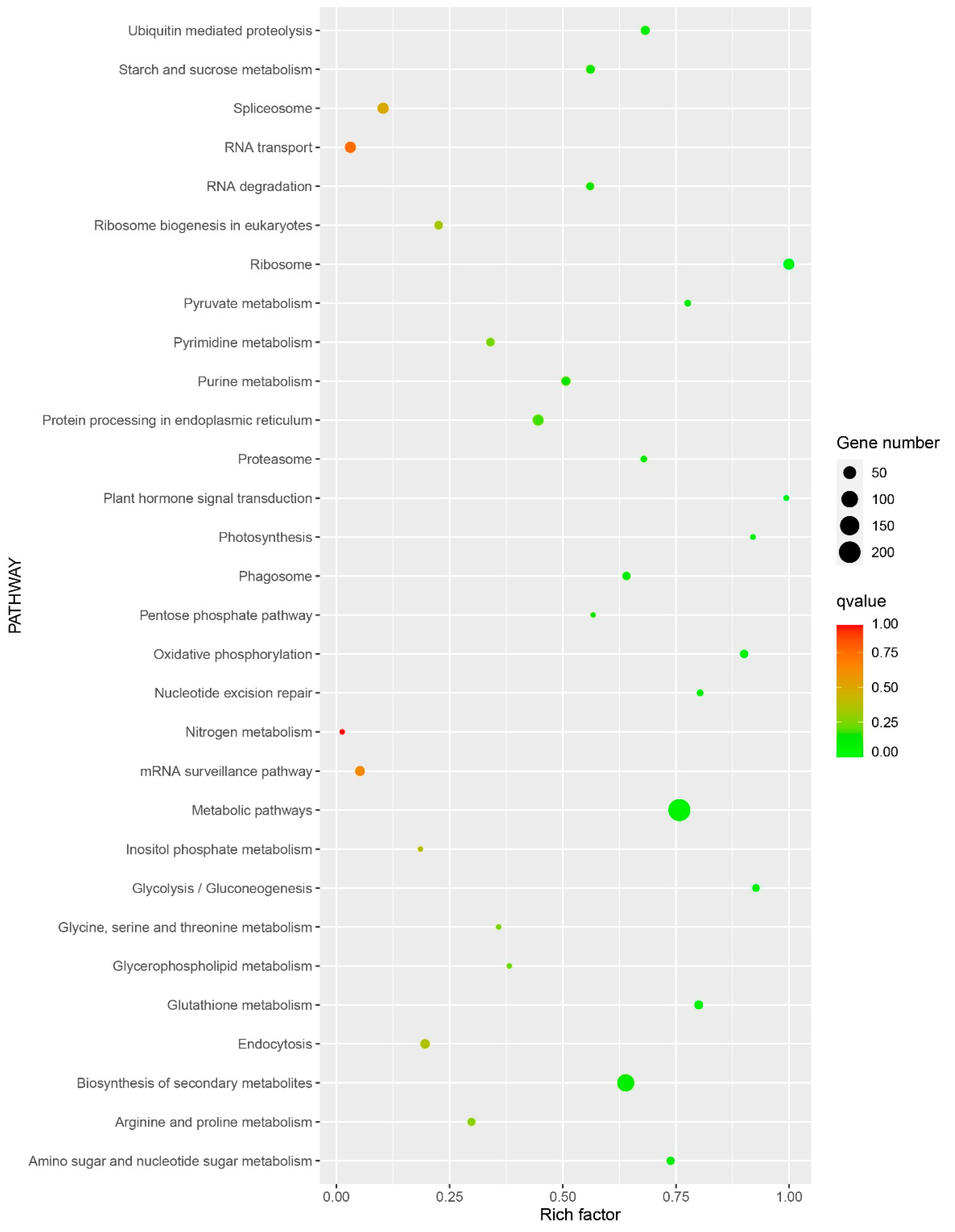
3.3. GO and KEGG Enrichment Analysis of Differentially Expressed Genes (DEGs)
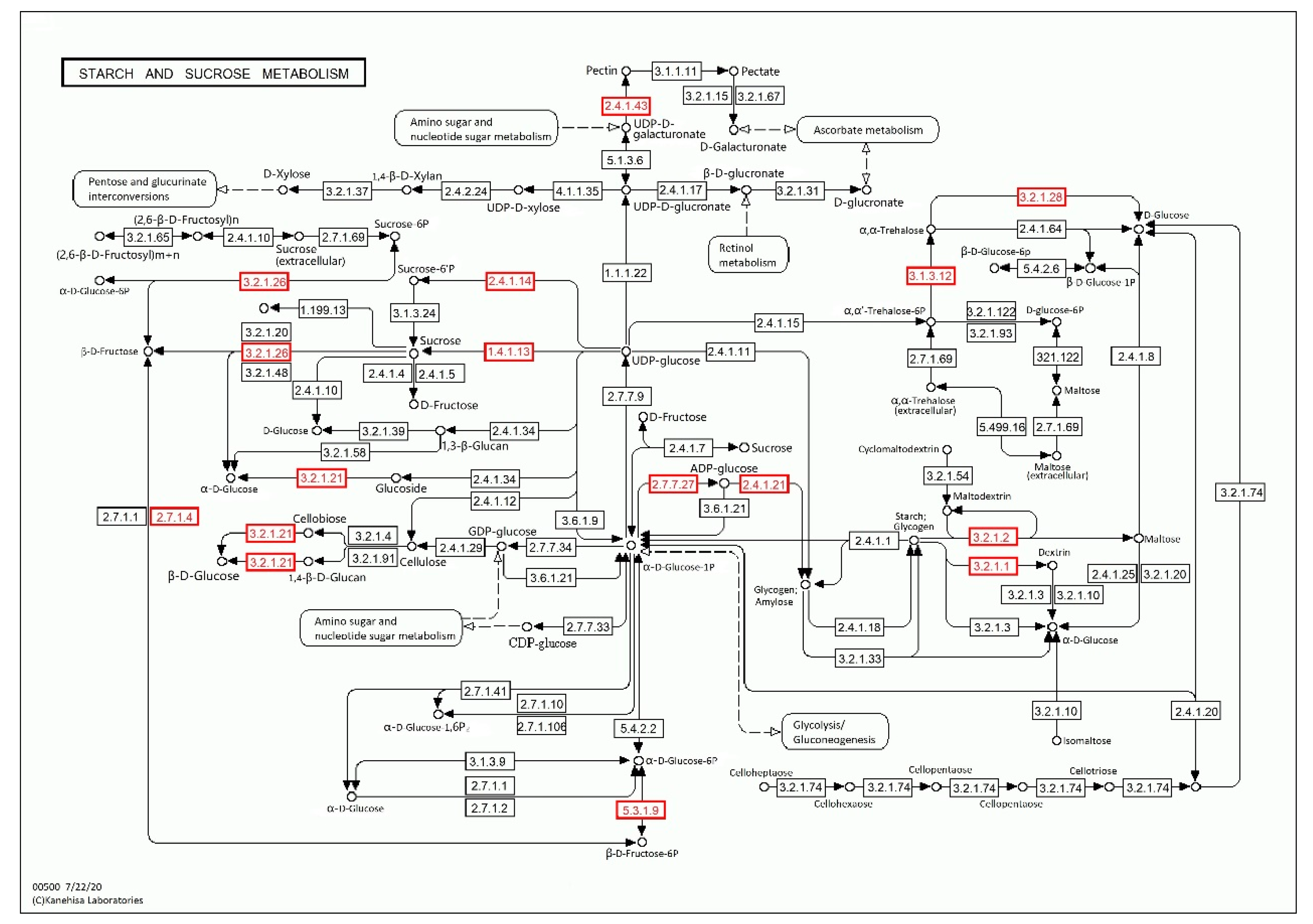
3.4. Analysis of Critical Pathways
4. Discussion
4.1. A New Perspective on Gene Set Subtraction
4.2. A Gene Set for Bamboo Culm Wall Thickness Regulation Containing 633 Candidate Genes
4.3. Reticulation Pathway for Bamboo Culm Wall Thickness Control
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GO | Gene Ontology |
| InDel | Insertion-Deletion |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| SNP | Single Nucleotide Polymorphism |
References
- Xie, J.; Hong, M. Effects of ecological factors on growth of Arundinaria spanostachya shoots in Liziping National Nature Reserve, China. Glob. Ecol. Conserv. 2020, 23. [Google Scholar] [CrossRef]
- Safial, A.Z.; Ramlah, L.; Azizi, B.; Binti, A.N.F.L.; Binti, E.N. Malay Wisdom in the Motifs of Bamboo Shoots. Shs Web Conf. 2018, 45, 02004. [Google Scholar]
- Soreng, R.J.; Peterson, P.M.; Romaschenko, K.; Davidse, G.; Zuloaga, F.O.; Judziewicz, E.J.; Filgueiras, T.S.; Davis, J.I.; Morrone, O. A worldwide phylogenetic classification of the Poaceae (Gramineae). J. Syst. Evol. 2015, 53, 117–137. [Google Scholar] [CrossRef]
- Huang, N.-J.; Li, J.-P.; Yang, G.-Y.; Yu, F. Two plastomes of Phyllostachys and reconstruction of phylogenic relationship amongst selected Phyllostachys species using genome skimming. Mitochondrial DNA Part B Resour. 2020, 5, 69–70. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, X.; Ding, Y.; Fei, Z.; Jiao, C.; Fan, M.; Yao, B.; Xin, P.; Chu, J.; Wei, Q. Cellular and molecular characterization of a thick-walled variant reveal a pivotal role of shoot apical meristem in transverse development of bamboo culm. J. Exp. Bot. 2019, 70, 3911–3926. [Google Scholar] [CrossRef] [PubMed]
- Miyashima, S.; Sebastian, J.; Lee, J.-Y.; Helariutta, Y. Stem cell function during plant vascular development. EMBO J. 2013, 32, 178–193. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Zhang, Y.; Zhou, J.; Wang, G.; Guo, Q. Transcriptome Reveals the Specificity ofPhyllostachys edulis’Pachyloen’ Shoots at Different Developmental Stages. Forests 2020, 11, 861. [Google Scholar] [CrossRef]
- Meraj, T.A.; Fu, J.; Raza, M.A.; Zhu, C.; Shen, Q.; Xu, D.; Wang, Q. Transcriptional Factors Regulate Plant Stress Responses Through Mediating Secondary Metabolism. Genes 2020, 11, 346. [Google Scholar] [CrossRef] [Green Version]
- Sharma, L.; Priya, M.; Kaushal, N.; Bhandhari, K.; Chaudhary, S.; Dhankher, O.P.; Prasad, P.V.V.; Siddique, K.H.M.; Nayyar, H. Plant growth-regulating molecules as thermoprotectants: Functional relevance and prospects for improving heat tolerance in food crops. J. Exp. Bot. 2020, 71, 569–594. [Google Scholar] [CrossRef]
- Toleco, M.R.; Naake, T.; Zhang, Y.; Heazlewood, J.L.; Fernie, A.R. Plant Mitochondrial Carriers: Molecular Gatekeepers That Help to Regulate Plant Central Carbon Metabolism. Plants 2020, 9, 117. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Yi, H.; Chen, X.; Wang, J. Post-Translational Modifications of Proteins Have Versatile Roles in Regulating Plant Immune Responses. Int. J. Mol. Sci. 2019, 20, 2807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trneny, O.; Vlk, D.; Mackova, E.; Matouskova, M.; Repkova, J.; Nedelnik, J.; Hofbauer, J.; Vejrazka, K.; Jakesova, H.; Jansa, J.; et al. Allelic Variants for Candidate Nitrogen Fixation Genes Revealed by Sequencing in Red Clover (Trifolium pratense L.). Int. J. Mol. Sci. 2019, 20, 5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Liu, L.; Safdar, L.B.; Xie, M.; Cheng, X.; Liu, Y.; Xiang, Y.; Tong, C.; Tu, J.; Huang, J.; et al. Characterization and Fine Mapping of a Yellow-Virescent Gene Regulating Chlorophyll Biosynthesis and Early Stage Chloroplast Development inBrassica napus. G3-Genes Genomes Genet. 2020, 10, 3201–3211. [Google Scholar] [CrossRef]
- Khanh Le, N.; Grondin, A.; Courtois, B.; Gantet, P. Next-Generation Sequencing Accelerates Crop Gene Discovery. Trends Plant Sci. 2019, 24, 263–274. [Google Scholar] [CrossRef]
- Lin, X.C.; Ruan, X.S.; Lou, Y.F.; Guo, X.Q.; Fang, W. Genetic similarity among cultivars of Phyllostachys pubescens. Plant Syst. Evol. 2009, 277, 67–73. [Google Scholar] [CrossRef]
- Darwis, A.; Iswanto, A.H.; Jeon, W.-S.; Kim, N.-H.; Wirjosentono, B.; Susilowati, A.; Hartono, R. Variation of Quantitative Anatomical Characteristics in the Culm of Belangke Bamboo (Gigantochloa pruriens). Bioresources 2020, 15, 6617–6626. [Google Scholar] [CrossRef]
- Zhang, B.; Guo, Y.; Liu, X.e.; Chen, H.; Yang, S.; Wang, Y.g. Mechanical Properties of the Fiber Cell Wall in Bambusa pervariabilis Bamboo and Analyses of Their Influencing Factors. Bioresources 2020, 15, 5316–5327. [Google Scholar] [CrossRef]
- Peng, Z.; Lu, Y.; Li, L.; Zhao, Q.; Feng, Q.; Gao, Z.; Lu, H.; Hu, T.; Yao, N.; Liu, K.; et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledo, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The Rate and Molecular Spectrum of Spontaneous Mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Moult, J. SNPs, protein structure, and disease. Hum. Mutat. 2001, 17, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, C.; Inoue, E.; Saito, T.; Ogata, N.; Kita, K.; Gonai, T.; Kasumi, M.; Ishii, R.; Sawamura, Y.; Takada, N.; et al. Transcriptome analysis of watercore in Pyrus pyrifolia by comparing pairs of susceptible and resistant F1 sibs. Sci. Hortic. 2020, 264. [Google Scholar] [CrossRef]
- Zhang, L.; Yin, X.; Zhang, J.; Wei, Y.; Huo, D.; Ma, C.; Chang, H.; Cai, K.; Shi, H. Comprehensive microbiome and metabolome analyses reveal the physiological mechanism of chlorotic Areca leaves. Tree Physiol. 2020. [Google Scholar] [CrossRef]
- Zhou, Q.; Fu, H.; Yang, D.; Ye, C.; Zhu, S.; Lin, J.; Ye, W.; Ji, G.; Ye, X.; Wu, X.; et al. Differential alternative polyadenylation contributes to the developmental divergence between two rice subspecies, japonica and indica. Plant J. 2019, 98, 260–276. [Google Scholar] [CrossRef]
- Sharma, N.; Singh, S.K.; Mahato, A.K.; Ravishankar, H.; Dubey, A.K.; Singh, N.K. Physiological and molecular basis of alternate bearing in perennial fruit crops. Sci. Hortic. 2019, 243, 214–225. [Google Scholar] [CrossRef]
- Yin, K.; Mylo, M.D.; Speck, T.; Wegst, U.G.K. Bamboo-inspired tubular scaffolds with functional gradients. J. Mech. Behav. Biomed. Mater. 2020, 110. [Google Scholar] [CrossRef]
- Luo, J.; Liu, R.; Zhang, S.; Lian, C.; Yang, F.; Fei, B. Comparative culm anatomy of metaxylem vessel pits in three different types of bamboo rhizome. IAWA J. 2020, 41, 141–158. [Google Scholar] [CrossRef]
- Wang, K.-L.; Wang, B.; Hu, R.; Zhao, X.; Li, H.; Zhou, G.; Song, L.; Wu, A.-m. Characterization of hemicelluloses in Phyllostachys edulis (moso bamboo) culm during xylogenesis. Carbohydr. Polym. 2019, 221, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Boardman, K.N. Comparative Photosynthesis of Sun and Shade Plants. Annu. Rev. Plant Physiol. 1977, 28, 355–377. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Chen, Z.J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 2007, 58, 377–406. [Google Scholar] [CrossRef] [Green Version]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.; Shi, T.; Wang, F.; Zhou, H.; Yang, J.; Wang, L.; Wang, S.; Su, Y.; Zhang, Z.; Qiao, Y.; et al. Interval mapping for red/green skin color in Asian pears using a modified QTL-seq method. Hortic. Res. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Liese, W.; Khl, M. Bamboo—The Plant and its Uses; Springer: Berlin, Germany, 2015. [Google Scholar]
- Guo, L.; Sun, X.; Li, Z.; Wang, Y.; Fei, Z.; Jiao, C.; Feng, J.; Cui, D.; Feng, X.; Ding, Y.; et al. Morphological dissection and cellular and transcriptome characterizations of bamboo pith cavity formation reveal a pivotal role of genes related to programmed cell death. Plant Biotechnol. J. 2019, 17, 982–997. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Region | SNP | InDel | |||||||
|---|---|---|---|---|---|---|---|---|---|
| P. edulis ‘Pachyloen’ | P. nidularia f. farcta | P. heteroclada f. solida | S. dumetorum var. xinwuense | P. edulis ‘Pachyloen’ | P. nidularia f. farcta | P. heteroclada f. solida | S. dumetorum var. xinwuense | ||
| Intergenic | 1,624,972 | 18,353,189 | 16,685,473 | 691,858 | 66,350 | 415,448 | 282,184 | 14,663 | |
| Intron | 32,534 | 795,600 | 763,191 | 295,724 | 2324 | 49,079 | 33,489 | 8934 | |
| Splice site acceptor | 119 | 1732 | 1710 | 979 | 9 | 175 | 129 | 67 | |
| Splice site donor | 128 | 1485 | 1499 | 1049 | 23 | 194 | 153 | 102 | |
| Splice site region | 1271 | 21,862 | 22,268 | 25,098 | 114 | 1663 | 1327 | 1015 | |
| Exon | Disruptive inframe insertion | - | - | - | - | 390 | 2102 | 1943 | 1002 |
| Disruptive inframe deletion | - | - | - | - | 252 | 2508 | 2280 | 1594 | |
| Synonymous | 13,070 | 131,717 | 137,171 | 195,782 | - | - | - | - | |
| Stop gained | 1131 | 13,622 | 14,290 | 17,597 | 20 | 157 | 200 | 72 | |
| Stop lost | 920 | 11,477 | 11,744 | 15,856 | 15 | 169 | 128 | 106 | |
| Stop retained | 163 | 2455 | 2496 | 3001 | - | - | - | - | |
| Synonymous | 13,070 | 131,717 | 137,171 | 195,782 | - | - | - | - | |
| Non-synonymous | 30,243 | 310,187 | 322,148 | 441,538 | - | - | - | - | |
| Multiple alleles | 308 | 14,294 | 16,683 | 5702 | 411 | 2352 | 2313 | 269 | |
| Homozygote | 294,832 | 14,912,672 | 11,846,991 | 1,158,569 | 33,127 | 398,617 | 234,711 | 22,002 | |
| Heterozygote | 1,423,820 | 4,800,212 | 6,178,104 | 519,040 | 38,799 | 88,095 | 98,721 | 9668 | |
| Total | 1,718,960 | 19,727,178 | 18,041,778 | 1,683,311 | 72,337 | 489,064 | 335,745 | 31,939 | |
| Gene ID | Function | Number of Amino Acid Changes |
|---|---|---|
| PH01001102G0470 | glycosyl hydrolases | 46 |
| PH01001643G0420 | kinase, pfkB family | 31 |
| PH01000125G0770 | sucrose synthase | 14 |
| PH01000173G0770 | alpha-amylase precursor | 6 |
| PH01005448G0030 | glycosyl transferase family 8 | 4 |
| PH01000171G0500 | beta-amylase | 4 |
| PH01004013G0130 | trehalose-6-phosphate synthase | 2 |
| PH01003937G0060 | glucose-1-phosphate adenylyltransferase large subunit, chloroplast precursor | 2 |
| PH01000269G0880 | starch synthase | 1 |
| PH01000362G0480 | trehalose precursor | 1 |
| PH01000453G0440 | trehalose synthase | 1 |
| PH01003516G0050 | glycosyl hydrolase family 3 protein | 1 |
| PH01000028G0260 | glycosyl hydrolases | 1 |
| Gene ID | Function | Number of Amino Acid Changes |
|---|---|---|
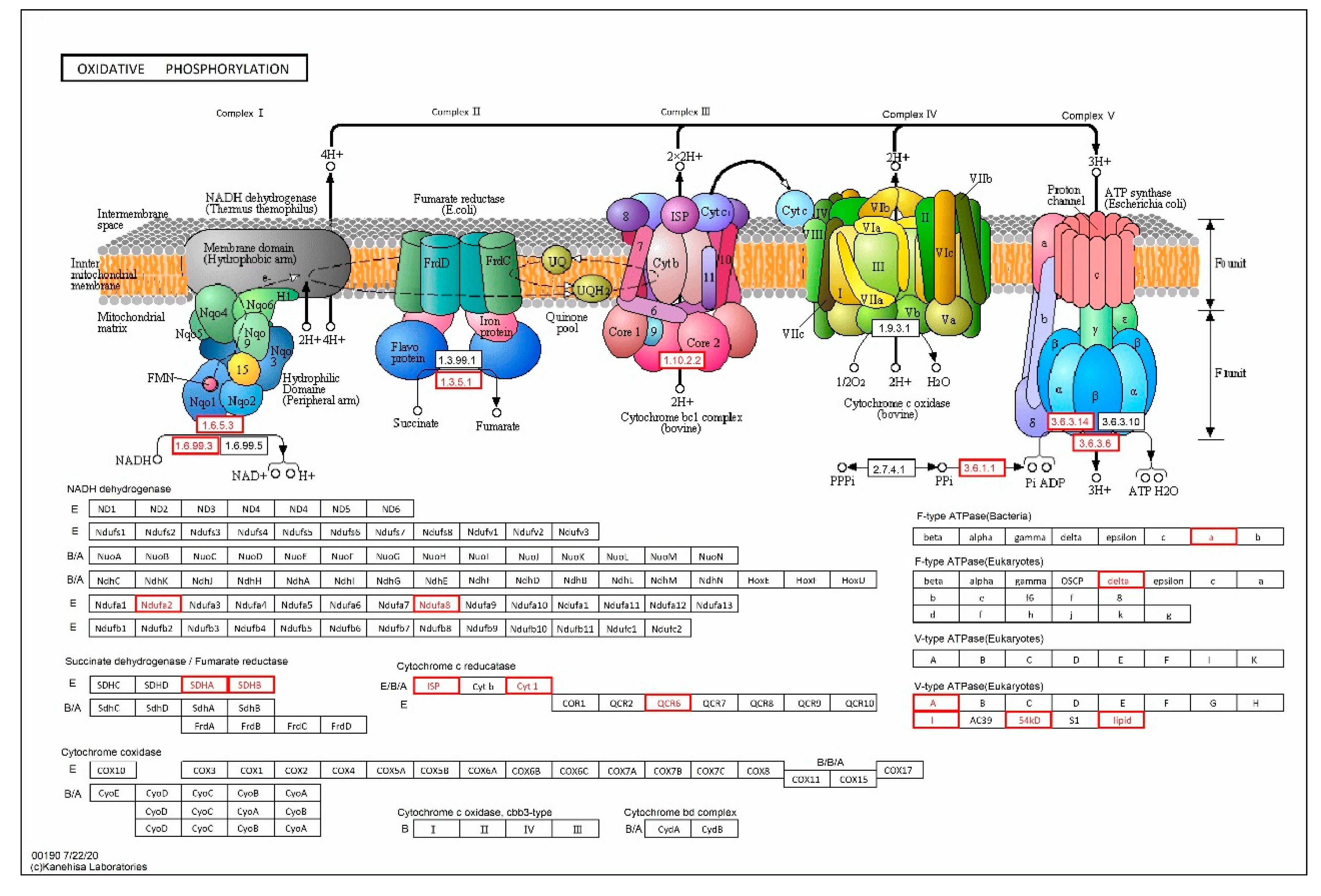
| PH01000003G0420 | succinate dehydrogenase iron-sulfur subunit 2, mitochondrial precursor | 15 |
| PH01001308G0210 | soluble inorganic pyrophosphatase | 13 |
| PH01000553G0250 | ATP synthase delta chain, mitochondrial precursor | 7 |
| PH01003807G0010 | plasma membrane ATPase | 7 |
| PH01002058G0310 | plasma membrane ATPase | 5 |
| PH01004724G0090 | inorganic H+ pyrophosphatase | 1 |
| PH01000202G0040 | protein kinase | 1 |
| PH01004931G0020 | soluble inorganic pyrophosphatase | 1 |
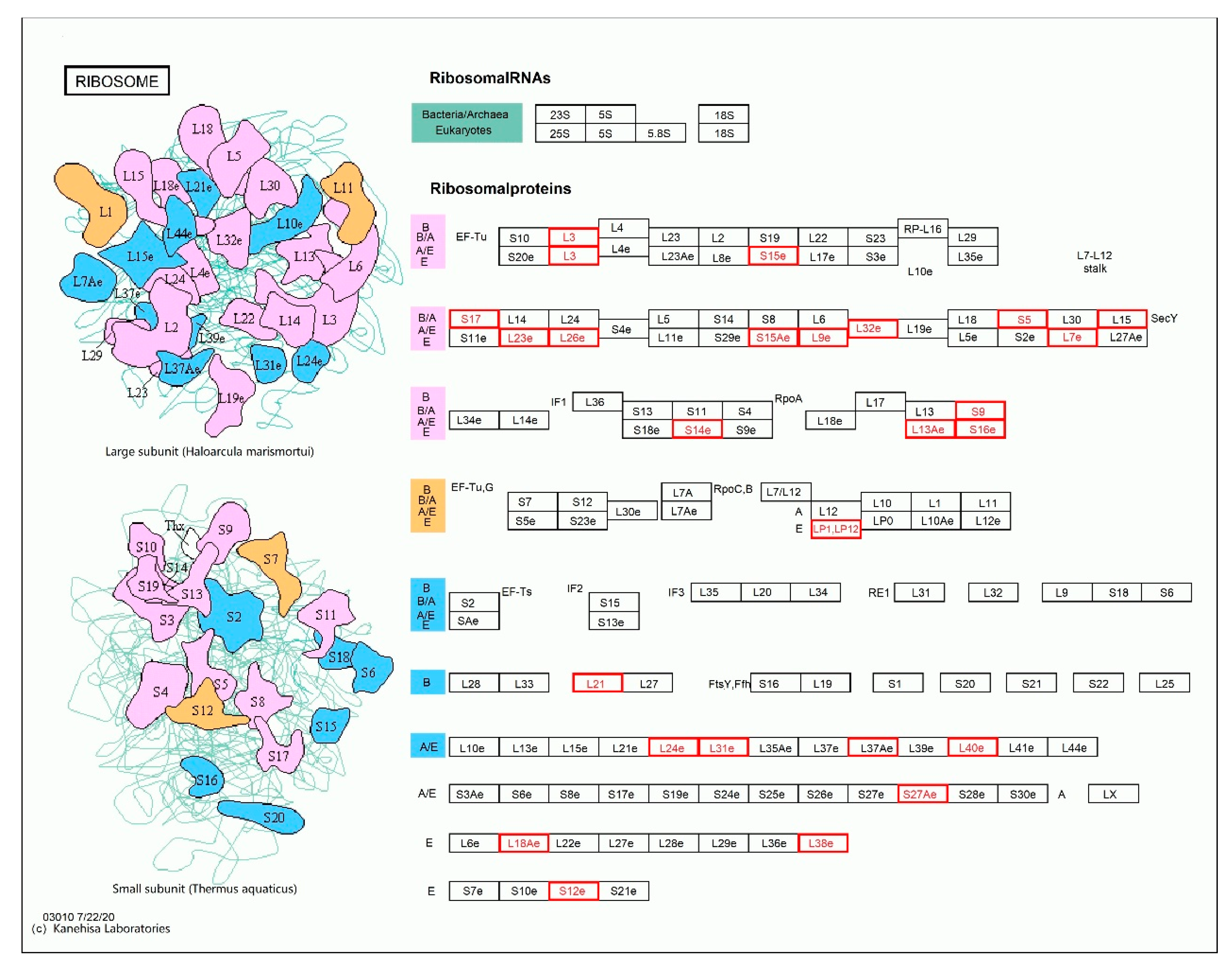
| Distribution | Gene ID | Function | Number of Amino Acid Changes | Type of Mutation in the Exon Region |
|---|---|---|---|---|
| Small subunit | PH01001546G0370 | 40S ribosomal protein S15 | 1 | - |
| PH01000291G1180 | phosphoglycerate mutase | 1 | - | |
| PH01000845G0690 | transportin-2 | 2 | - | |
| PH01005914G0060 | ribosomal protein | 2 | - | |
| PH01002868G0010 | ribosomal protein | 4 | - | |
| PH01002189G0040 | ribosomal protein L7Ae | 1 | - | |
| PH01000145G0580 | ribosomal protein L3 | 3 | Frameshift variant | |
| Large subunit | PH01000632G0190 | ribosomal protein L3 | 2 | Inframe deletion |
| PH01002363G0020 | ribosomal protein L3 | 2 | - | |
| PH01000734G0730 | ribosomal protein L24 | 1 | - | |
| PH01002396G0040 | 50S ribosomal protein L15 | 1 | Disruptive inframe insertion | |
| PH01000215G1180 | 60S ribosomal protein L31 | 1 | - | |
| PH01001171G0230 | expressed protein | 2 | Frameshift variant | |
| PH01000716G0300 | ribosomal protein L13 | 6 | Frameshift variant | |
| PH01000553G0230 | tyrosine protein kinase domain containing protein | 2 | Frameshift variant | |
| PH01001955G0010 | 50S ribosomal protein L21, chloroplast precursor | 2 | Frameshift variant | |
| PH01006046G0080 | 60S ribosomal protein L37a | 1 | - | |
| PH01002417G0020 | 60S ribosomal protein L38 | 1 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Zhou, J.; Yu, Z.; Li, J.; Cai, J.; Wang, G.; Guo, Q. Gene Set Subtraction Reveals 633 Candidate Genes for Bamboo Culm Wall Thickening. Forests 2020, 11, 1331. https://doi.org/10.3390/f11121331
Hu Y, Zhou J, Yu Z, Li J, Cai J, Wang G, Guo Q. Gene Set Subtraction Reveals 633 Candidate Genes for Bamboo Culm Wall Thickening. Forests. 2020; 11(12):1331. https://doi.org/10.3390/f11121331
Chicago/Turabian StyleHu, Yaping, Jie Zhou, Zhaoyan Yu, Jiajia Li, Jinfeng Cai, Guibin Wang, and Qirong Guo. 2020. "Gene Set Subtraction Reveals 633 Candidate Genes for Bamboo Culm Wall Thickening" Forests 11, no. 12: 1331. https://doi.org/10.3390/f11121331