Efficient Fingerprinting of the Tetraploid Salix psammophila Using SSR Markers

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction and SSR Analysis
2.3. Data Analysis and Core Markers Selection
3. Results
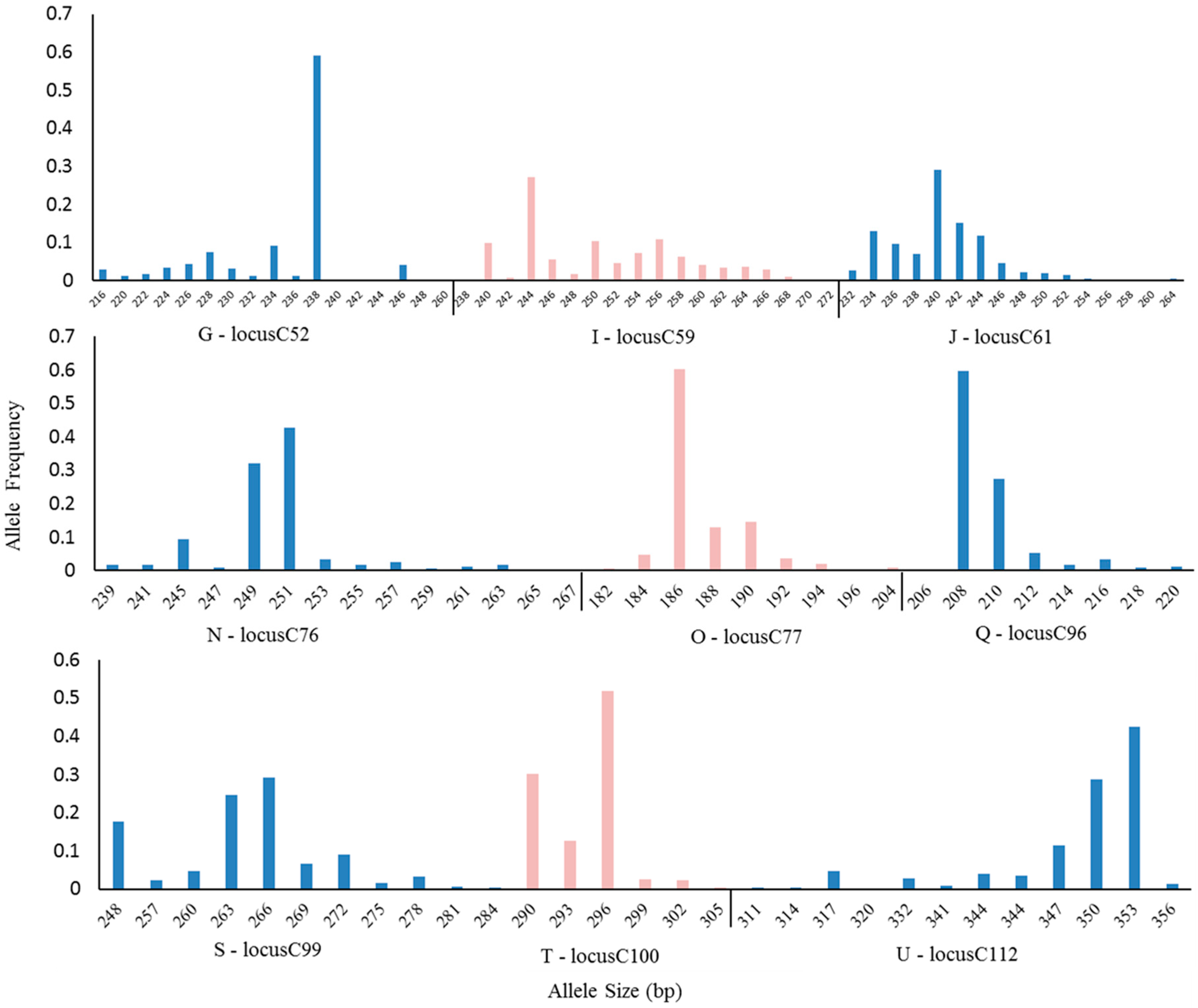
3.1. SSR Marker Polymorphism
3.2. DNA Fingerprinting Power
3.2.1. Single Marker Calculations
3.2.2. Screening of Marker Combinations
3.3. Construct Fingerprinting of S. psammophila
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Karp, A.; Hanley, S.J.; Trybush, S.O.; Macalpine, W.; Pei, M.; Sheild, L. Genetic improvement of willow for bioenergy and biofuels free access. J. Integr. Plant Biol. 2011, 53, 151–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madlung, A. Polyploidy and its effect on evolutionary success: Old questions revisited with new tools. Heredity 2013, 110, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Y.; Zhang, G. Study of adsorption characteristics of methylene blue onto activated carbon made by salix psammophila. Energy Procedia 2012, 16, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.B.; Yang, G.H.; Chen, J.C. Effects of pretreatment by xylanase on properties of salix psammophila kraft pulp. Adv. Mater. Res. 2012, 610–613, 3505–3510. [Google Scholar] [CrossRef]
- Qu, L.; Chen, J.C.; Yang, G.H.; Xue, Y. Effect of different process on the pulping properties of salix psammophila p-rc APMP. Adv. Mater. Res. 2012, 610–613, 581–585. [Google Scholar] [CrossRef]
- Baraket, G.; Abdallah, D.; Mustapha, S.B.; Tamarzizt, H.B.; Salhi-Hannachi, A. Combination of simple sequence repeat, s-locus polymorphism and phenotypic data for identification of tunisian plum species (prunus spp.). Biochem. Genet. 2019, 673–694. [Google Scholar] [CrossRef]
- Funk, J.L. Differences in plasticity between invasive and native plants from a low resource environment. J. Ecol. 2008, 96, 1162–1173. [Google Scholar] [CrossRef]
- Chapin, F.S.; Autumn, K.; Pugnaire, F. Evolution of suites of traits in response to environmental stress. Am. Nat. 1993, 142, S78–S92. [Google Scholar] [CrossRef]
- Jia, H.; Yang, H.; Sun, P.; Li, J.; Zhang, J.; Guo, Y.; Han, X.; Zhang, G.; Lu, M.; Hu, J. De novo transcriptome assembly, development of EST-SSR markers and population genetic analyses for the desert biomass willow, Salix psammophila. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, Q.; Xie, J.; Pan, Y.-B.; Zhou, F.; Guo, Y.; Chang, H.; Xu, H.; Zhang, W.; Zhang, C.; et al. SSR marker-assisted management of parental germplasm in sugarcane (saccharum sphybrids) breeding programs. Agronomy 2019, 9, 449. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Patel, D.A.; Parmar, H.; Dhaduk, H.L.; Sasidharan, N. Genetic diversity analysis in soybean (Glycine max (L.) Merrill.) using SSR markers. J. Pharmacogn. Phytochem. 2018, 7, 2380–2384. [Google Scholar]
- Fu, N.; Wang, P.-Y.; Liu, X.-D.; Shen, H.-L. Use of EST-SSR markers for evaluating genetic diversity and fingerprinting celery (apium graveolens l.) cultivars. Molecules 2014, 19, 1939–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.H.; Liang, K.N.; Zhou, Z.Z.; Ma, H.M. SSR genotyping-genetic diversity and fingerprinting of Teak (Tectona grandis) clones. J. Trop. For. Sci. 2016, 28, 48–58. [Google Scholar]
- Njung’e, V.; Deshpande, S.; Siambi, M.; Jones, R.; Silim, S.; De Villiers, S. SSR genetic diversity assessment of popular pigeonpea varieties in Malawi reveals unique fingerprints. Electron. J. Biotechnol. 2016, 21, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Peng, M.; Xu, L.-Y.; Wang, L.-Y.; Chen, S.-X.; Zou, Y.; Qi, G.-N.; Cheng, H. Fingerprinting 128 Chinese clonal tea cultivars using SSR markers provides new insights into their pedigree relationships. Tree Genet. Genomes 2015, 11, 90. [Google Scholar] [CrossRef]
- Jordens, R. Progress of plant variety protection based on the International Convention for the Protection of New Varieties of Plants (UPOV Convention). World Pat. Inf. 2005, 27, 232–243. [Google Scholar] [CrossRef]
- Agrimonti, C.; Vietina, M.; Pafundo, S.; Marmirolim, N. The use of food genomics to ensure the traceability of olive oil. Trends Food Sci. Technol. 2011, 22, 237–244. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, J.; Han, R.; Wang, Y.; Zhu, S. Fingerprinting and identification of closely related wheat (‘triticum aestivum’l.) cultivars using issr and fluorescence-labeled tp-m13-ssr markers. Aust. J. Crop. Sci. 2011, 5, 846. [Google Scholar]
- Zhang, Z.; Huang, K.; Yue, J.; Liu, M.; Qi, M.; Dong, J. DNA fingerprinting of malt varieties using tailed primer M13 microsatellite (TP-M13-SSR) markers. Shipin Kexue/Food Sci. 2018, 39, 183–188. [Google Scholar]
- Girichev, V.; Hanke, M.-V.; Peil, A.; Flachowsky, H. SSR fingerprinting of a German Rubus collection and pedigree based evaluation on trueness-to-type. Genet. Resour. Crop Evol. 2017, 64, 189–203. [Google Scholar] [CrossRef]
- Schlautman, B.; Bolivar-Medina, J.; Hodapp, S.; Zalapa, J. Cranberry SSR multiplexing panels for DNA horticultural fingerprinting and genetic studies. Sci. Hortic. 2017, 219, 280–286. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthi, B.K.; Naravaneni, R. SSR marker based DNA fingerprinting and diversity study in rice (Oryza sativa. L). Afr. J. Biotechnol. 2006, 5, 684–688. [Google Scholar]
- Hao, L.; Zhang, G.; Lu, D.; Hu, J.; Jia, H. Analysis of the genetic diversity and population structure of Salix psammophila based on phenotypic traits and simple sequence repeat markers. PeerJ 2019, 7, e6419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrall, P.H.; Young, A. AUTOTET: A program for analysis of autotetraploid genotypic data. J. Hered. 2000, 348–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, S.; Cernák, I.; Mousapour Gorji, A.; Géza Hegedűs, G.; Taller, J. PICcalc: An Online Program to Calculate Polymorphic Information Content for Molecular Genetic Studies. Biochem. Genet. 2012, 50, 670–672. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, H.; Wu, A.; Hou, Y.; An, Y.; Wei, C. Construction of fingerprinting for tea plant ( Camellia sinensis ) accessions using new genomic SSR markers. Mol. Breed. 2017, 37, 93. [Google Scholar] [CrossRef]
- Li, L.; Xu, X.; Zhang, G.; Zhang, X. Establishment of molecular identity cards for cucumis melo cultivars using ssr markers. HortScience 2018, 53, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Sun, Y.; Zhao, K.; Zhang, J.; Zhang, Y.; Li, X.; Xun, S.; Zhang, J.; Wang, S.; Li, Y. Development and application of EST-SSR markers for DNA fingerprinting and genetic diversity analysis of the main cultivars of black locust (robinia pseudoacacia L.) in China. Forests 2019, 10, 644. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wangle, Y.; Wangle, L.; Zhang, D. Molecular identification of mung bean accessions (Vigna radiata L.) from Northeast China using capillary electrophoresis with fluorescence-labeled SSR markers. Food Energy Secur. 2019, e182. [Google Scholar] [CrossRef] [Green Version]
- Freixas-Coutin, J.A.; An, S.; Postman, J.; Bassil, N.V.; Yates, B.; Shukla, M.; Saxena, P.K. Development of a reliable Corylus sreference database through the implementation of a DNA fingerprinting test. Planta 2019, 249, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.B.; Cordeiro, G.M.; Henry, R.J. Molecular genotyping of sugarcane clones with microsatellite markers. Maydica 2003, 48, 319–329. [Google Scholar]
- Tan, L.-Q.; Wang, L.-Y.; Xu, L.-Y.; Wu, L.-Y.; Peng, M.; Zhang, C.-C.; Wei, K.; Bai, P.-X.; Li, H.-L.; Cheng, H.; et al. SSR-based genetic mapping and QTL analysis for timing of spring bud flush, young shoot color, and mature leaf size in tea plant (Camellia sinensis). Tree Genet. Genomes 2016, 12, 1–13. [Google Scholar] [CrossRef]
- Siew, G.Y.; Wei Lun Ng, W.L.; Tan, S.W.; Alitheen, N.B.; Tan, S.G.; Yeap, S.K. Genetic variation and DNA fingerprinting of durian types in Malaysia using simple sequence repeat (SSR) markers. PeerJ 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Penghang Zhang, P.; Luo, J.; Liu, X.; Fan, S.; Liu, C.; Han, Y. The establishment of a DNA fingerprinting database for 73 varieties of Lactuca sativa capitate L. using SSR molecular markers. Hortic. Environ. Biotechnol. 2019, 60, 95–103. [Google Scholar] [CrossRef]
- Galli, Z.; Halász, G.; Kiss, E.; Heszky, L.; Dobránszki, J. Molecular identification of commercial apple cultivars with microsatellite markers. HortScience 2005, 40, 1974–1977. [Google Scholar] [CrossRef] [Green Version]
- Gökirmak, T.; Mehlenbacher, S.A.; Bassil, N.V. Characterization of European hazelnut (Corylus avellana) cultivars using SSR markers. Genet. Resour. Crop. Evol. 2009, 56, 147–172. [Google Scholar] [CrossRef]
- Gürcan, K.; Mehlenbacher, S.; Erdoğan, V. Genetic diversity in hazelnut (Corylus avellana L.) cultivars from Black Sea countries assessed using SSR markers. Plant Breed. 2010, 129, 422–434. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Code | A | Ai | G | G1 | Ho | He | M1/% | M2/% | PIC | PI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| c4 | A | 11 | 2.41 | 78 | 41 | 0.6 | 0.66 | 52.56 | 15.71 | 0.64 | 0.14 |
| c16 | B | 8 | 2.23 | 26 | 8 | 0.56 | 0.55 | 30.77 | 3.07 | 0.51 | 0.25 |
| c24 | C | 14 | 2.84 | 97 | 56 | 0.76 | 0.76 | 57.73 | 21.46 | 0.74 | 0.08 |
| c25 | D | 5 | 1.84 | 8 | 2 | 0.47 | 0.45 | 25.00 | 0.77 | 0.35 | 0.40 |
| c46 | E | 8 | 2.53 | 51 | 19 | 0.68 | 0.67 | 37.25 | 7.28 | 0.62 | 0.16 |
| c49 | F | 8 | 2.03 | 27 | 10 | 0.48 | 0.51 | 37.04 | 3.83 | 0.47 | 0.28 |
| c52 | G | 17 | 2.20 | 91 | 54 | 0.55 | 0.63 | 59.34 | 20.69 | 0.61 | 0.15 |
| c57 | H | 3 | 1.74 | 9 | 1 | 0.40 | 0.41 | 11.11 | 0.38 | 0.33 | 0.42 |
| c59 | I | 18 | 3.33 | 176 | 135 | 0.88 | 0.87 | 76.70 | 51.72 | 0.86 | 0.02 |
| c61 | J | 16 | 2.81 | 130 | 89 | 0.77 | 0.84 | 68.46 | 34.10 | 0.83 | 0.04 |
| c69 | K | 16 | 2.68 | 154 | 102 | 0.73 | 0.88 | 66.23 | 39.08 | 0.87 | 0.02 |
| c73 | L | 9 | 2.27 | 73 | 41 | 0.57 | 0.73 | 56.16 | 15.71 | 0.70 | 0.11 |
| c74 | M | 5 | 2.35 | 34 | 11 | 0.62 | 0.65 | 32.35 | 4.21 | 0.58 | 0.19 |
| c76 | N | 14 | 2.61 | 87 | 52 | 0.70 | 0.70 | 59.77 | 19.92 | 0.66 | 0.13 |
| c77 | O | 9 | 2.23 | 49 | 22 | 0.57 | 0.60 | 44.90 | 8.43 | 0.56 | 0.20 |
| c90 | P | 7 | 1.65 | 35 | 14 | 0.35 | 0.71 | 40.00 | 5.36 | 0.65 | 0.14 |
| c96 | Q | 8 | 2.07 | 36 | 13 | 0.53 | 0.56 | 36.11 | 4.98 | 0.51 | 0.25 |
| c97 | R | 16 | 2.07 | 92 | 60 | 0.49 | 0.65 | 65.22 | 22.99 | 0.63 | 0.13 |
| c99 | S | 11 | 2.29 | 97 | 51 | 0.60 | 0.81 | 52.58 | 19.54 | 0.78 | 0.06 |
| c100 | T | 6 | 2.31 | 31 | 13 | 0.63 | 0.62 | 41.94 | 4.98 | 0.56 | 0.20 |
| c112 | U | 12 | 2.5 | 76 | 39 | 0.66 | 0.72 | 51.32 | 14.94 | 0.68 | 0.12 |
| c115 | V | 8 | 2.23 | 47 | 15 | 0.57 | 0.68 | 31.91 | 5.75 | 0.62 | 0.16 |
| Mean | - | 10.41 | 2.33 | 68.36 | 38.55 | 0.60 | 0.67 | 47.02 | 14.77 | 0.63 | 0.17 |
| Total | - | 229 | - | 1504 | 848 | - | - | - | - | - | - |
| Nine-Marker Combination | GC1 | MC2/% | In Total of PIC |
|---|---|---|---|
| G+H+I+J+N+O+S+T+U | 261 | 100 | 5.88 |
| G+H+J+L+N+O+S+T+U | 261 | 100 | 5.72 |
| G+H+J+N+O+R+S+T+U | 261 | 100 | 5.65 |
| G+H+J+N+O+S+T+U+V | 261 | 100 | 5.64 |
| G+I+J+N+O+Q+S+T+U | 261 | 100 | 6.05 |
| G+J+L+N+O+Q+S+T+U | 261 | 100 | 5.89 |
| G+J+N+O+Q+R+S+T+U | 261 | 100 | 5.82 |
| G+J+N+O+Q+S+T+U+V | 261 | 100 | 5.81 |
| A+G+H+I+J+N+S+T+U | 259 | 99.23 | 5.96 |
| A+G+H+J+L+N+S+T+U | 259 | 99.23 | 5.79 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, L.; Zhai, Y.; Zhang, G.; Lu, D.; Huang, H. Efficient Fingerprinting of the Tetraploid Salix psammophila Using SSR Markers. Forests 2020, 11, 176. https://doi.org/10.3390/f11020176
Hao L, Zhai Y, Zhang G, Lu D, Huang H. Efficient Fingerprinting of the Tetraploid Salix psammophila Using SSR Markers. Forests. 2020; 11(2):176. https://doi.org/10.3390/f11020176
Chicago/Turabian StyleHao, Lei, Yongguang Zhai, Guosheng Zhang, Dongye Lu, and Haiguang Huang. 2020. "Efficient Fingerprinting of the Tetraploid Salix psammophila Using SSR Markers" Forests 11, no. 2: 176. https://doi.org/10.3390/f11020176