Transcriptome and Co-Expression Network Analyses Identify the Molecular Signatures Underlying Drought Resistance in Yellowhorn

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Construction, and Sequencing
2.3. Transcriptome Assembly and Functional Annotation
2.4. Identification of Differentially Expressed Genes (DEGs)
2.5. WGCNA and Hub Genes Analysis
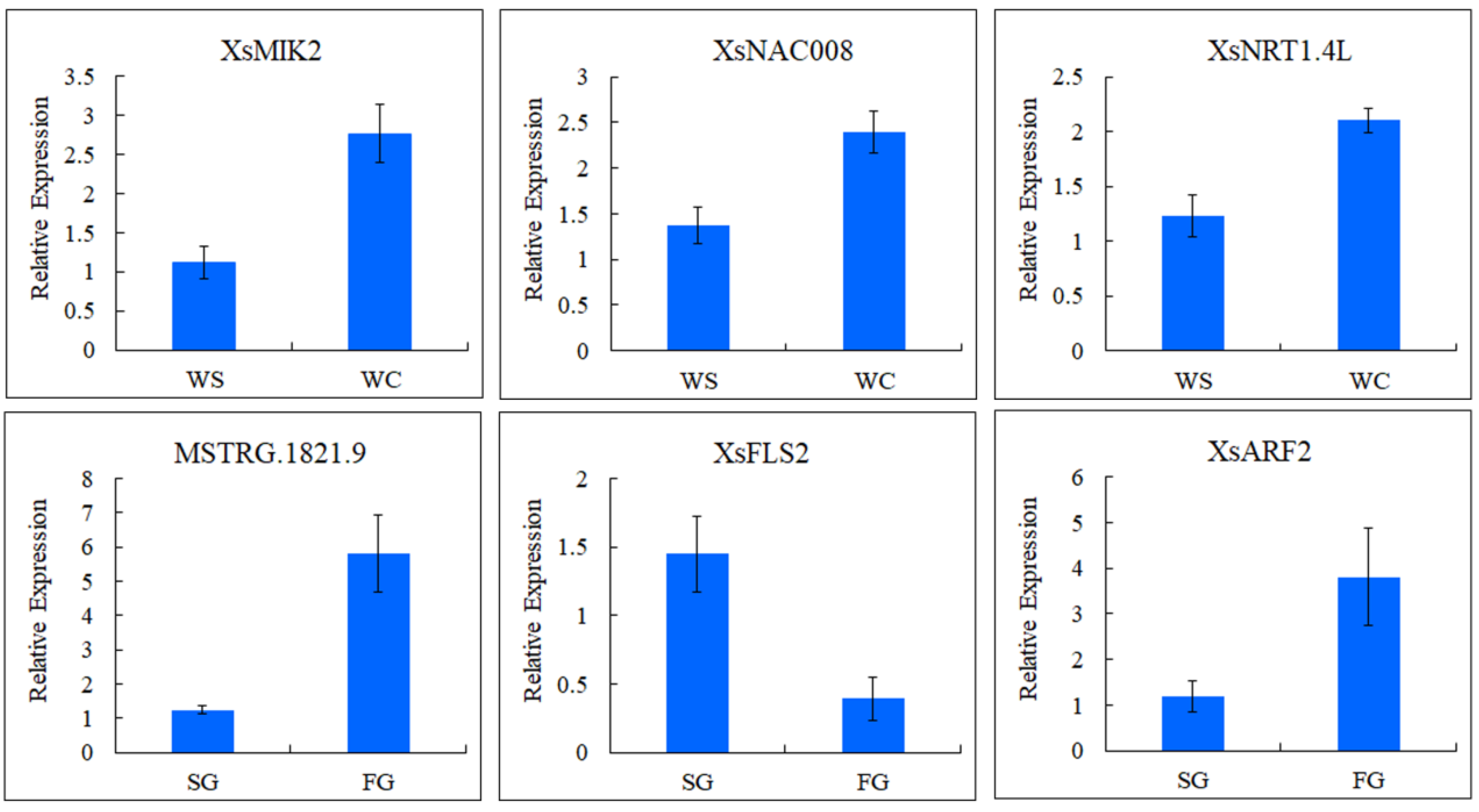
2.6. qRT-PCR Analysis
3. Results
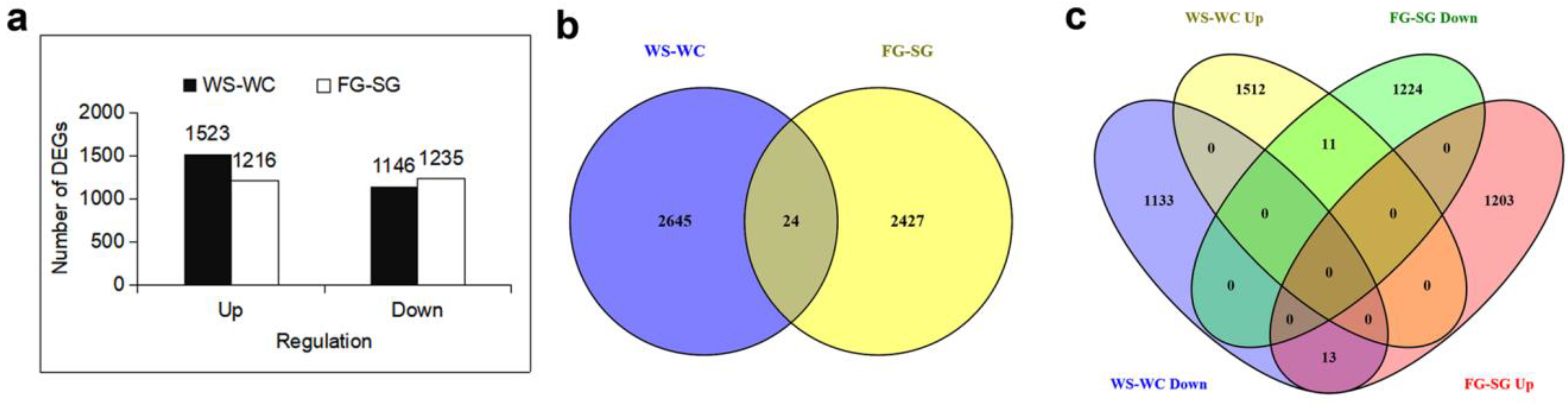
3.1. Identification of DEGs in the Four Types of Yellowhorn under Drought Conditions
3.2. GO Enrichment Analysis of DEGs
3.3. Pathway Enrichment Analysis of DEGs
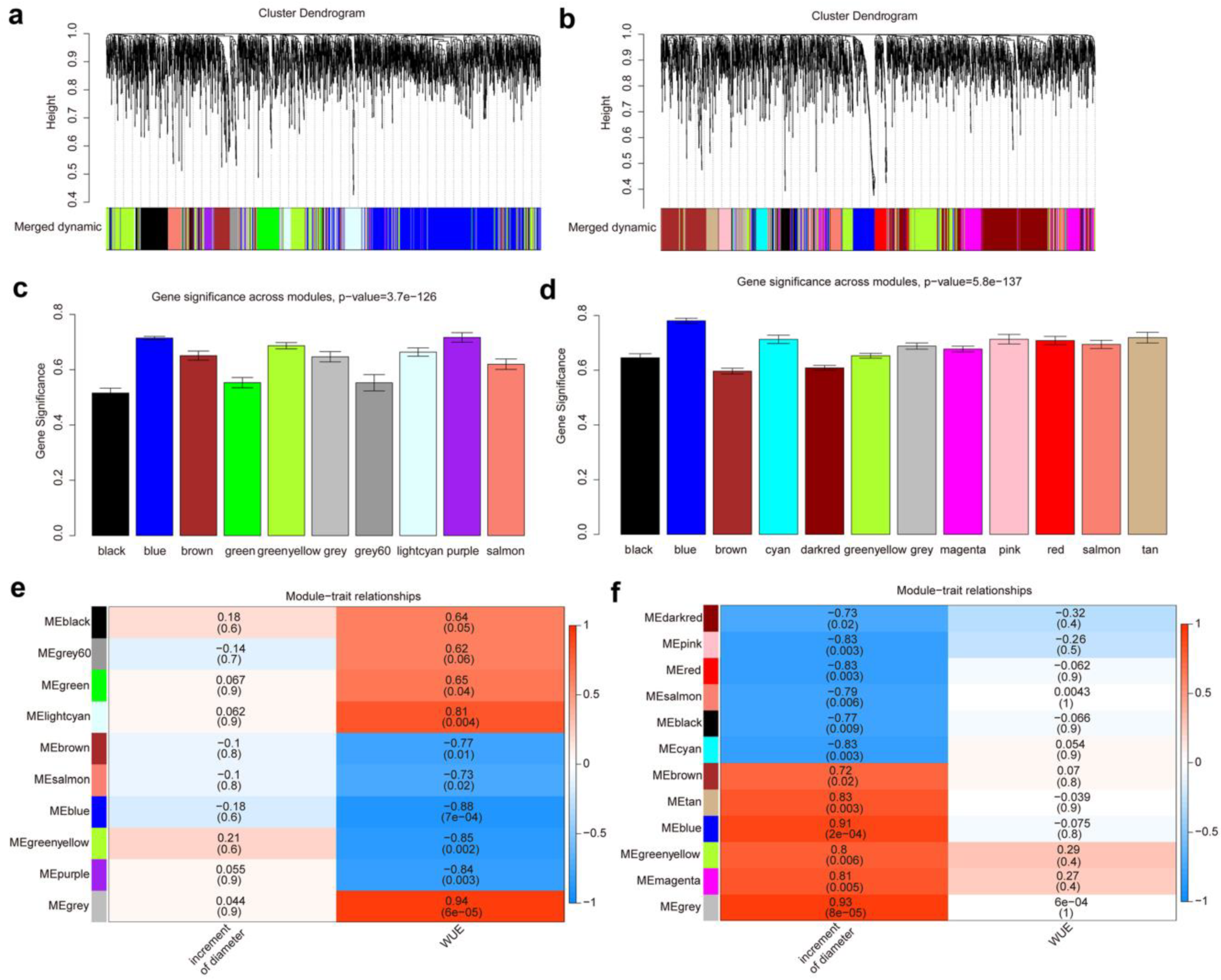
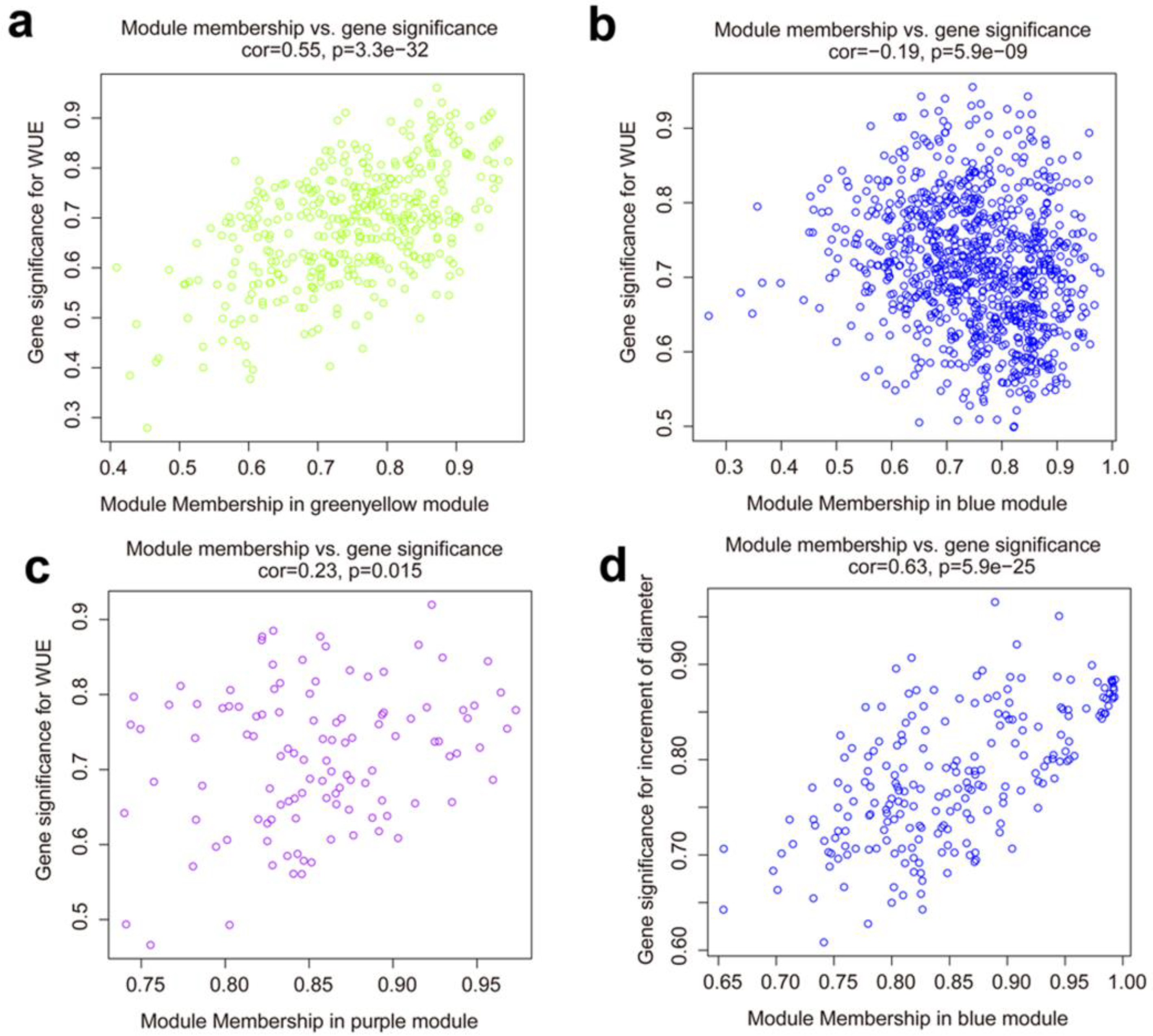
3.4. Co-Expression Network and Hub Genes Associated with the WUE and Growth under Drought Conditions
4. Discussion
4.1. Sugar and Nitrogen Metabolic and Biosynthetic Processes
4.2. Cell Wall- and Microtubule-Related Processes
4.3. Antioxidant Metabolic Processes
4.4. IAA Biosynthetic and Metabolic Processes
4.5. Phosphatidylinositol-Related Processes
4.6. Phosphorylation/Dephosphorylation Modification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Q.; Yang, L.; Ranjitkar, S.; Wang, J.J.; Wang, X.R.; Zhang, D.X.; Wang, Z.Y.; Huang, Y.Z.; Zhou, Y.M.; Deng, Z.X.; et al. Distribution and in situ conservation of a relic Chinese oil woody species yellowhorn Xanthoceras sorbifolium Bunge. Can. J. For. Res. 2017, 47, 1450–1456. [Google Scholar] [CrossRef]
- Yu, H.; Fan, S.; Bi, Q.; Wang, S.; Hu, X.; Chen, M.; Wang, L. Seed morphology, oil content and fatty acid composition variability assessment in yellow horn (Xanthoceras sorbifolium Bunge) germplasm for optimum biodiesel production. Ind. Crops Prod. 2017, 97, 425–430. [Google Scholar] [CrossRef]
- Venegas-Calerón, M.; Ruíz-Méndez, M.V.; Martínez-Force, E.; Garcés, R.; Salas, J.J. Characterization of Xanthoceras sorbifolium Bunge seeds: Lipids, proteins and saponins content. Ind. Crops Prod. 2017, 109, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Ruan, C.J.; Yan, R.; Wang, B.X.; Mopper, S.; Guan, W.K.; Zhang, J. The importance of yellow horn (Xanthoceras sorbifolium) for restoration of arid habitats and production of bioactive seed oils. Ecol. Eng. 2017, 99, 504–512. [Google Scholar] [CrossRef]
- Xie, Z.Y.; Zhang, W.H. Effects of drought and rewatering on growth and photosynthetic physioecological characteristics of Xanthoceras Sorbifolia. Chin. J. Appl. Ecol. 2018, 29, 1759–1767. [Google Scholar]
- Jin, H.; Zou, J.; Li, L.; Bai, X.; Zhu, T.; Li, J.; Xu, B.; Wang, Z. Physiological responses of yellowhorn seedlings to high temperatures under drought condition. Plant Biotechnol. Rep. 2019, 14, 111–120. [Google Scholar] [CrossRef]
- Basu, S.; Ramegowda, V.; Kumar, A.; Pereira, A. Plant adaptation to drought stress. F1000Research 2016, 5, 1554. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Chen, N.; Zhang, C.; Dong, X.; Zhao, C. Drought enhances the role of competition in mediating the relationship between tree growth and climate in semi-arid areas of northwest China. Forests 2019, 10, 804. [Google Scholar] [CrossRef] [Green Version]
- Honda, M.D.; Ishihara, K.L.; Pham, D.T.; Borthakur, D. Identification of drought-induced genes in giant leucaena (Leucaena leucocephala subsp. glabrata). Trees 2018, 32, 571–585. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar]
- Ma, J.; Li, R.Q.; Wang, H.G.; Li, D.X.; Wang, X.Y.; Zhang, Y.C.; Zhen, W.C.; Duane, H.J.; Yan, G.J.; Li, Y.M. Transcriptomics analyses reveal wheat responses to drought stress during reproductive stages under field conditions. Front. Plant Sci. 2017, 8, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, Y.X.; Hou, S.W. Research progress of protein phosphorylation under abiotic stresses in plants. Mol. Plant Breed. 2020, 18, 1176–1186. [Google Scholar]
- Mahmood, T.; Khalid, S.; Abdullah, M.; Ahmed, Z.; Shah, M.K.N.; Ghafoor, A.; Du, X.M. Insights into drought stress signaling in plants and the molecular genetic basis of cotton drought tolerance. Cells 2019, 9, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.R.; Feng, C.H. Effects of brassinolide on drought resistance of Xanthoceras Sorbifolia seedlings under water stress. Acta Physiol. Plant 2011, 33, 1293–1300. [Google Scholar] [CrossRef]
- Blum, A. Drought resistance, water-use efficiency, and yield potential-are they compatible, dissonant, or mutually exclusive? Aust. J. Agric. Res. 2005, 56, 1159–1168. [Google Scholar] [CrossRef]
- Raherison, E.S.M.; Giguère, I.; Caron, S.; Lamara, M.; Mackay, J.J. Modular organization of the white spruce (Picea glauca) transcriptome reveals functional organization and evolutionary signatures. New Phytol. 2015, 207, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Hasanuzzaman, M.; Shabala, L.; Brodribb, T.J.; Zhou, M.; Shabala, S. Understanding physiological and morphological traits contributing to drought tolerance in barley. J. Agron. Crop Sci. 2019, 205, 129–140. [Google Scholar] [CrossRef]
- Kuromori, T.; Mizoi, J.; Umezawa, T.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Drought stress signaling network. In Molecular Biology; Howell, S.H., Ed.; Springer: New York, NY, USA, 2014; pp. 383–409. [Google Scholar]
- Seki, M.; Umezawa, T.; Urano, K.; Shinozaki, K. Regulatory metabolic networks in drought stress responses. Curr. Opin. Plant Biol. 2007, 10, 296–302. [Google Scholar] [CrossRef]
- Das, A.; Rushton, P.; Rohila, J. Metabolomic profiling of soybeans (Glycine max L.) reveals the importance of sugar and nitrogen metabolism under drought and heat stress. Plants 2017, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Fàbregas, N.; Fernie, A.R. The metabolic response to drought. J. Exp. Bot. 2019, 70, 1077–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veres, S.; Zsombik, L.; Juhász, C. Regulation Effect of Different Water Supply to the Nitrogen and Carbon Metabolism. In Abiotic and Biotic Stress in Plants; de Oliveira, A.B., Ed.; IntechOpen: London, UK, 2019; p. 261. [Google Scholar]
- Zhang, G.; Meng, S.; Gong, J. The expected and unexpected roles of nitrate transporters in plant abiotic stress resistance and their regulation. Int. J. Mol. Sci. 2018, 19, 3535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.P.; Vicré-Gibouin, M.; Farrant, J.M.; Driouich, A. Adaptations of higher plant cell walls to water loss: Drought vs desiccation. Physiol. Plant. 2008, 134, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Tenhaken, R. Cell wall remodeling under abiotic stress. Front. Plant Sci. 2015, 5, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Liu, M. The microtubule cytoskeleton acts as a sensor for stress response signaling in plants. Mol. Biol. Rep. 2019, 46, 5603–5608. [Google Scholar] [CrossRef]
- Yu, S.G.; Kim, J.H.; Cho, A.H.; Oh, T.R.; Kim, W.T. Arabidopsis RING E3 ubiquitin ligase JUL1 participates in ABA-mediated microtubule depolymerization, stomatal closure, and tolerance response to drought stress. Plant J. 2020, 103, 824–842. [Google Scholar] [CrossRef]
- de Carvalho, M.H.C. Drought stress and reactive oxygen species. Plant Signal. Behav. 2008, 3, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Schuler, M. Functional genomics of p450s. Annu. Rev. Plant Biol. 2003, 54, 629–667. [Google Scholar] [CrossRef]
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef]
- Wang, L.; Hua, D.; He, J.; Duan, Y.; Chen, Z.; Hong, X.; Gong, Z. Auxin Response Factor2 (ARF2) and its regulated homeodomain gene HB33 mediate abscisic acid response in Arabidopsis. PLoS Genet. 2011, 7, e1002172. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Liu, Z.L.; Ning, K.; Wang, A.; Zeng, X.; Xu, J. Genomic and transcriptome analyses reveal that mapk- and phosphatidylinositol-signaling pathways mediate tolerance to 5-hydroxymethyl-2-furaldehyde for industrial yeast Saccharomyces cerevisiae. Sci. Rep. 2014, 4, 6556. [Google Scholar] [CrossRef]
- Munnik, T.; Vermeer, J.E.M. Osmotic stress-induced phosphoinositide and inositol phosphate signalling in plants. Plant Cell Environ. 2020, 33, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Kuang, R.; Chan, K.H.; Yeung, E.; Lim, B.L. Molecular and biochemical characterization of AtPAP15, a purple acid phosphatase with phytase activity, in Arabidopsis. Plant Physiol. 2009, 151, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, B.R.; Jeon, B.W.; Zhang, W.; Assmann, S.M. Open Stomata 1 (OST1) is limiting in abscisic acid responses of Arabidopsis guard cells. New Phytol. 2013, 200, 1049–1063. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Out (349) | All (3754) | p-Value | Q value | |
|---|---|---|---|---|---|
| WS-WC | Pantothenate and CoA biosynthesis | 7 | 20 | 0.0015 | 0.1641 |
| RNA degradation | 18 | 110 | 0.0117 | 0.4968 | |
| mRNA surveillance pathway | 19 | 120 | 0.0137 | 0.4968 | |
| Purine metabolism | 19 | 125 | 0.0205 | 0.5307 | |
| Alpha-linolenic acid metabolism | 8 | 39 | 0.0243 | 0.5307 | |
| Beta-alanine metabolism | 7 | 34 | 0.0336 | 0.6113 | |
| Ribosome | 32 | 256 | 0.0471 | 0.7333 | |
| FG-SG | Ribosome biogenesis in eukaryotes | 19 | 85 | 3.47 × 10−5 | 0.0036 |
| Spliceosome | 25 | 162 | 0.0012 | 0.0516 | |
| Inositol phosphate metabolism | 9 | 35 | 0.0015 | 0.0516 | |
| Phosphatidylinositol signaling system | 8 | 35 | 0.0059 | 0.1546 | |
| Sphingolipid metabolism | 7 | 30 | 0.0088 | 0.1835 | |
| Autophagy—other eukaryotes | 6 | 30 | 0.0311 | 0.5400 | |
| Overlap of WS-WC and FG-SG | Autophagy—other eukaryotes | 3 | 30 | 0.0009 | 0.0245 |
| Spliceosome | 5 | 162 | 0.0037 | 0.0481 |
| Module | Pathway | Out (54) | All (3754) | p-Value | Q value |
|---|---|---|---|---|---|
| Greenyellow | Ribosome | 13 | 256 | 4.50 × 10−5 | 0.0023 |
| Ubiquinone and other terpenoid-quinone biosynthesis | 2 | 22 | 0.0390 | 0.6290 | |
| Blue | SNARE interactions in vesicular transport | 2 | 41 | 0.0159 | 0.1889 |
| Base excision repair | 2 | 43 | 0.0175 | 0.1889 | |
| Riboflavin metabolism | 1 | 5 | 0.0238 | 0.1889 | |
| Ribosome | 4 | 256 | 0.0302 | 0.1889 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Cui, Y.; Wu, Z.; Zhao, Y.; Hu, X.; Bi, Q.; Yang, S.; Wang, L. Transcriptome and Co-Expression Network Analyses Identify the Molecular Signatures Underlying Drought Resistance in Yellowhorn. Forests 2020, 11, 840. https://doi.org/10.3390/f11080840
Liu X, Cui Y, Wu Z, Zhao Y, Hu X, Bi Q, Yang S, Wang L. Transcriptome and Co-Expression Network Analyses Identify the Molecular Signatures Underlying Drought Resistance in Yellowhorn. Forests. 2020; 11(8):840. https://doi.org/10.3390/f11080840
Chicago/Turabian StyleLiu, Xiaojuan, Yifan Cui, Zhiyan Wu, Yang Zhao, Xiaoyu Hu, Quanxin Bi, Suzhi Yang, and Libing Wang. 2020. "Transcriptome and Co-Expression Network Analyses Identify the Molecular Signatures Underlying Drought Resistance in Yellowhorn" Forests 11, no. 8: 840. https://doi.org/10.3390/f11080840