
Soil Microbiome Composition along the Natural Norway Spruce Forest Life Cycle

 , and
, and
Abstract
:1. Introduction
- (1)
- Microbial communities will differ along the gradient of time since the last disturbance. The communities of regenerating sites will be less contributed by ectomycorrhizal fungi due to a loss of their tree partners but more represented by copiotrophic saprobes and other nitrophilic groups compared to the old-growth forests, where these microbial groups will be restricted by low pH and low N availability. We expect that the changes will be the most evident in the youngest phases of forest regeneration and the dynamics will be flattened with the consolidation of the closed forest.
- (2)
- Fungal and bacterial communities will be shaped by different factors. The time since the last disturbance (i.e., the advancement of succession towards the old-growth forest) will be the key factor for the fungal community, especially for the ectomycorrhizal fungi dependent on their host trees, while bacteria will be largely determined by the soil pH and the C:N ratio.
- (3)
- The temporary change in the character of vegetation due to disturbance will be manifested in the soil by a polygenetic wave, i.e., changes in the soil most strongly and immediately affect the upper horizons; towards the depth the effect will be delayed and flattened. Accordingly, the effect of time since the last disturbance on the microbiome composition will be the most evident in organic horizons and will decrease with soil depth.
2. Materials and Methods
2.1. Study Sites and Soil Sampling
2.2. Soil Chemistry Analyses
2.3. Soil Microbial Communities
2.4. Statistical Analysis
3. Results
3.1. Soil Morphology and Classification
3.2. Soil Chemistry
3.3. Soil Microbial Communities
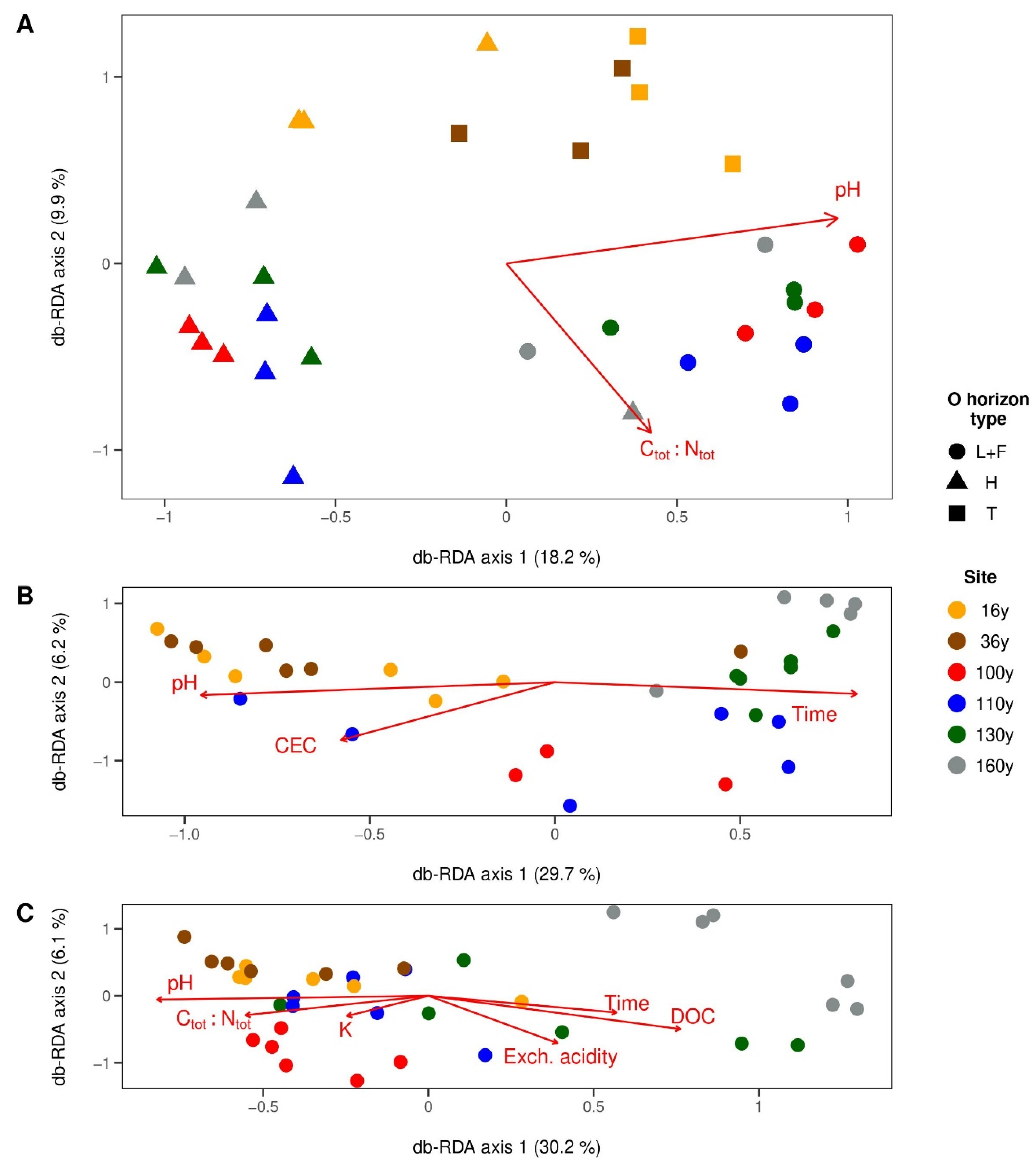
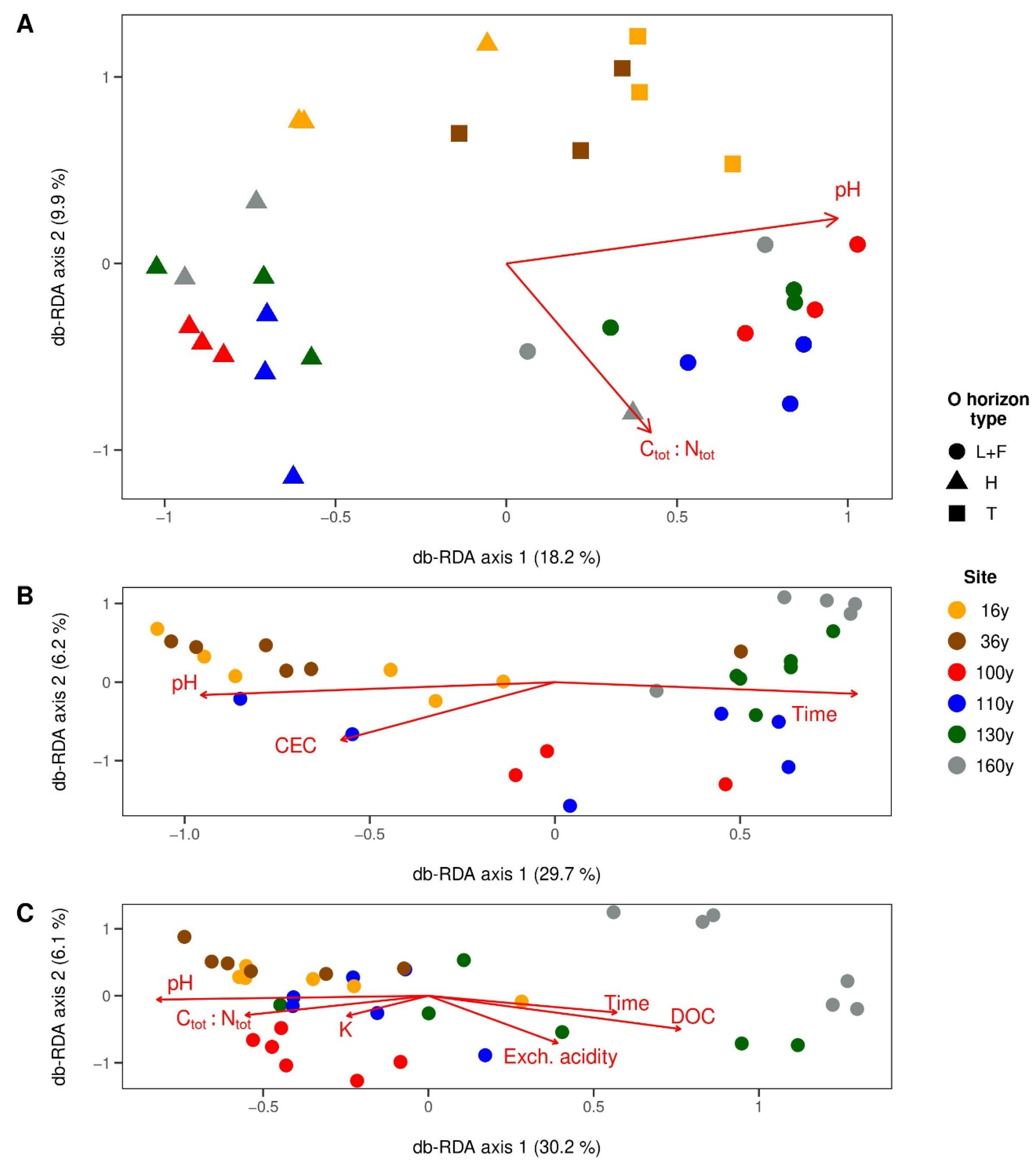
3.4. Factors Shaping Microbial Communities in Respective Horizons
3.4.1. Surface Organic Horizons
3.4.2. Upper Mineral Horizons
3.4.3. Spodic Horizons
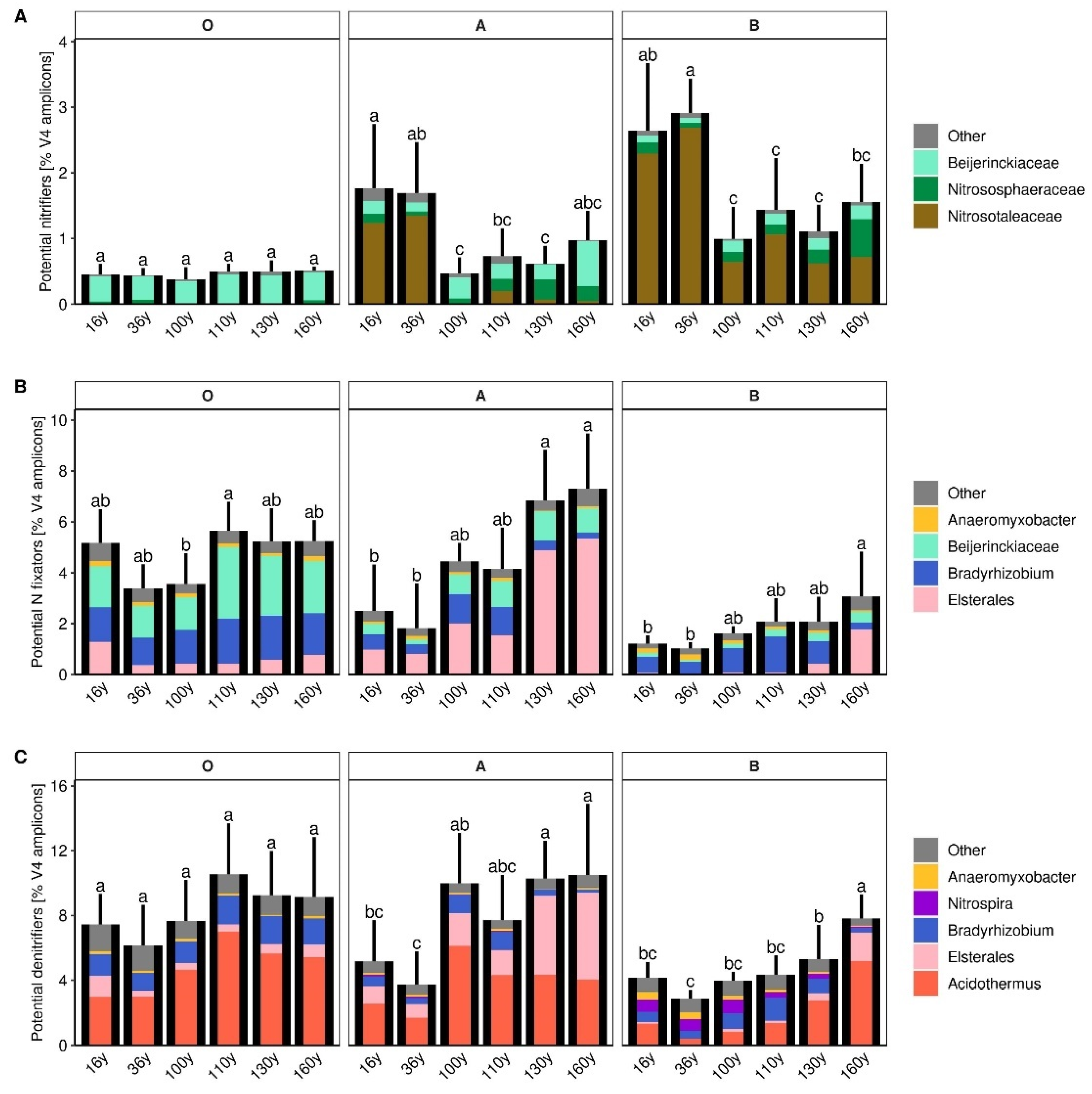
3.5. Microbial Groups Potentially Participating in Nitrification, N Fixation and Nitrate Respiration
4. Discussion
4.1. Vegetation and Soil Chemistry along the Chronosequence
4.2. General Patterns in the Soil Microbiome Composition
4.3. Soil Bacterial Communities
4.4. Soil Fungal Communities
4.5. The Effect of Disturbance within the Soil Profile
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sabatini, F.M.; Burrascano, S.; Keeton, W.S.; Levers, C.; Lindner, M.; Pötzschner, F.; Verkerk, P.J.; Bauhus, J.; Buchwald, E.; Chaskovsky, O.; et al. Where are Europe’s last primary forests? Divers. Distrib. 2018, 24, 1426–1439. [Google Scholar] [CrossRef] [Green Version]
- Nowakowska, J.A.; Hsiang, T.; Patynek, P.; Stereńczak, K.; Olejarski, I.; Oszako, T. Health assessment and genetic structure of monumental Norway spruce trees during a bark beetle (Ips typographus L.) outbreak in the Bialowieza Forest District, Poland. Forests 2020, 11, 647. [Google Scholar] [CrossRef]
- Čada, V.; Morrissey, R.C.; Michalová, Z.; Bače, R.; Janda, P.; Svoboda, M. Frequent severe natural disturbances and non-equilibrium landscape dynamics shaped the mountain spruce forest in central Europe. For. Ecol. Manag. 2016, 363, 169–178. [Google Scholar] [CrossRef]
- Carter, V.A.; Moravcová, A.; Chiverrell, R.C.; Clear, J.L.; Finsinger, W.; Dreslerová, D.; Halsall, K.; Kuneš, P. Holocene-scale fire dynamics of central European temperate spruce-beech forests. Quat. Sci. Rev. 2018, 191, 15–30. [Google Scholar] [CrossRef]
- Bobek, P.; Svobodová-Svitavská, H.; Pokorný, P.; Šamonil, P.; Kuneš, P.; Kozáková, R.; Abraham, V.; Klinerová, T.; Švarcová, M.G.; Jamrichová, E.; et al. Divergent fire history trajectories in Central European temperate forests revealed a pronounced influence of broadleaved trees on fire dynamics. Quat. Sci. Rev. 2019, 222, 105865. [Google Scholar] [CrossRef]
- Panayotov, M.; Kulakowski, D.; Laranjeiro Dos Santos, L.; Bebi, P. Wind disturbances shape old Norway spruce-dominated forest in Bulgaria. For. Ecol. Manag. 2011, 262, 470–481. [Google Scholar] [CrossRef]
- Trotsiuk, V.; Svoboda, M.; Janda, P.; Mikolas, M.; Bace, R.; Rejzek, J.; Samonil, P.; Chaskovskyy, O.; Korol, M.; Myklush, S. A mixed severity disturbance regime in the primary Picea abies (L.) Karst. forests of the Ukrainian Carpathians. For. Ecol. Manag. 2014, 334, 144–153. [Google Scholar] [CrossRef]
- Janda, P.; Trotsiuk, V.; Mikoláš, M.; Bače, R.; Nagel, T.A.; Seidl, R.; Seedre, M.; Morrissey, R.C.; Kucbel, S.; Jaloviar, P.; et al. The historical disturbance regime of mountain Norway spruce forests in the Western Carpathians and its influence on current forest structure and composition. For. Ecol. Manag. 2017, 388, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Panayotov, M.; Bebi, P.; Tsvetanov, N.; Alexandrov, N.; Laranjeiro, L.; Kulakowski, D. The disturbance regime of Norway spruce forests in Bulgaria. Can. J. For. Res. 2015, 45, 1143–1153. [Google Scholar] [CrossRef]
- Phillips, J.D.; Šamonil, P. Biogeomorphological Domination of Forest Landscapes: An example from the Šumava Mountains, Czech Republic. Geomorphology 2021, 383, 107698. [Google Scholar] [CrossRef]
- Oulehle, F.; Wright, R.F.; Svoboda, M.; Bače, R.; Matějka, K.; Kaňa, J.; Hruška, J.; Couture, R.-M.; Kopáček, J. Effects of Bark Beetle Disturbance on Soil Nutrient Retention and Lake Chemistry in Glacial Catchment. Ecosystems 2019, 22, 725–741. [Google Scholar] [CrossRef]
- Jabiol, B.; Zanella, A.; Ponge, J.F.; Sartori, G.; Englisch, M.; van Delft, B.; de Waal, R.; Le Bayon, R.C. A proposal for including humus forms in the World Reference Base for Soil Resources (WRB-FAO). Geoderma 2013, 192, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Šamonil, P.; Phillips, J.D.; Pawlik, Ł. Indirect biogeomorphic and soil evolutionary effects of spruce bark beetle. Glob. Planet. Chang. 2020, 195, 103317. [Google Scholar] [CrossRef]
- Sauer, D.; Sponagel, H.; Sommer, M.; Giani, L.; Jahn, R.; Stahr, K. Podzol: Soil of the year 2007. A review on its genesis, occurrence, and functions. J. Plant Nutr. Soil Sci. 2007, 170, 581–597. [Google Scholar] [CrossRef]
- Phillips, J.D.; Marion, D.A. Pedological memory in forest soil development. For. Ecol. Manag. 2004, 188, 363–380. [Google Scholar] [CrossRef] [Green Version]
- Barrett, L.R.; Schaetzl, R.J. Regressive pedogenesis following a century of deforestation: Evidence for depodzolization. Soil Sci. 1998, 163, 482–497. [Google Scholar] [CrossRef]
- Šamonil, P.; Daněk, P.; Schaetzl, R.J.; Vašíčková, I.; Valtera, M. Soil mixing and genesis as affected by tree uprooting in three temperate forests. Eur. J. Soil Sci. 2015, 66, 589–603. [Google Scholar] [CrossRef]
- Augusto, L.; Ranger, J.; Binkley, D.; Rothe, A. Impact of several common tree species of European temperate forests on soil fertility. Ann. For. Sci. 2002, 59, 233–253. [Google Scholar] [CrossRef] [Green Version]
- Urbanová, M.; Šnajdr, J.; Baldrian, P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 2015, 84, 53–64. [Google Scholar] [CrossRef]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Drenkhan, R.; Pritsch, K.; Buegger, F.; Padari, A.; Hagh-Doust, N.; Mikryukov, V.; Gohar, D.; et al. Regional-Scale In-Depth Analysis of Soil Fungal Diversity Reveals Strong pH and Plant Species Effects in Northern Europe. Front. Microbiol. 2020, 11, 1–31. [Google Scholar] [CrossRef]
- Bruns, T.D.; Bidartondo, M.I.; Taylor, D.L. Host specificity in ectomycorrhizal communities: What do exceptions tell us? Integr. Comp. Biol. 2002, 42, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Choma, M.; Tahovská, K.; Kaštovská, E.; Bárta, J.; Růžek, M.; Oulehle, F. Bacteria but not fungi respond to soil acidification rapidly and consistently in both a spruce and beech forest. FEMS Microbiol. Ecol. 2020, 96. [Google Scholar] [CrossRef] [PubMed]
- Bahnmann, B.; Mašínová, T.; Halvorsen, R.; Davey, M.L.; Sedlák, P.; Tomšovský, M.; Baldrian, P. Effects of oak, beech and spruce on the distribution and community structure of fungi in litter and soils across a temperate forest. Soil Biol. Biochem. 2018, 119, 162–173. [Google Scholar] [CrossRef]
- Nacke, H.; Goldmann, K.; Schöning, I.; Pfeiffer, B.; Kaiser, K.; Villamizar, G.A.C.; Schrumpf, M.; Buscot, F.; Daniel, R.; Wubet, T. Fine spatial scale variation of soil microbial communities under European beech and Norway spruce. Front. Microbiol. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choma, M.; Rappe-George, M.O.; Bárta, J.; Čapek, P.; Kaštovská, E.; Gärdenäs, A.I.; Šantrůčková, H. Recovery of the ectomycorrhizal community after termination of long-term nitrogen fertilisation of a boreal Norway spruce forest. Fungal Ecol. 2017, 29, 116–122. [Google Scholar] [CrossRef]
- Tahovská, K.; Choma, M.; Kaštovská, E.; Oulehle, F.; Bárta, J.; Šantrůčková, H.; Moldan, F. Positive response of soil microbes to long-term nitrogen input in spruce forest: Results from Gårdsjön whole-catchment N-addition experiment. Soil Biol. Biochem. 2020, 143, 107732. [Google Scholar] [CrossRef]
- Högberg, P.; Näsholm, T.; Franklin, O.; Högberg, M.N. Tamm Review: On the nature of the nitrogen limitation to plant growth in Fennoscandian boreal forests. For. Ecol. Manag. 2017, 403, 161–185. [Google Scholar] [CrossRef] [Green Version]
- Šantrůčková, H.; Vaněk, D.; Krištůfková, M. Decomposition rate and nutrient release from plant litter of Norway spruce forest in the Bohemian Forest. Biologia 2006, 61, S499–S508. [Google Scholar] [CrossRef]
- Kaňa, J.; Kopáček, J.; Tahovská, K.; Šantrůčková, H. Tree dieback and related changes in nitrogen dynamics modify the concentrations and proportions of cations on soil sorption complex. Ecol. Indic. 2019, 97, 319–328. [Google Scholar] [CrossRef]
- Fiala, K.; Tůma, I.; Holub, P.; Jandák, J. The role of Calamagrostis communities in preventing soil acidification and base cation losses in a deforested mountain area affected by acid deposition. Plant Soil 2005, 268, 35–49. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedersoo, L.; Bahram, M.; Polme, S.; Koljalg, U.; Yorou, N.S.; Wijesundera, R.; Villarreal Ruiz, L.; Vasco-Palacios, A.M.; Pham Quang, T.; Suija, A.; et al. Global diversity and geography of soil fungi. Science 2014, 346, 1078. [Google Scholar] [CrossRef] [Green Version]
- Glassman, S.I.; Wang, I.J.; Bruns, T.D. Environmental filtering by pH and soil nutrients drives community assembly in fungi at fine spatial scales. Mol. Ecol. 2017, 26, 6960–6973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousk, J.; Brookes, P.C.; Bååth, E. Investigating the mechanisms for the opposing pH relationships of fungal and bacterial growth in soil. Soil Biol. Biochem. 2010, 42, 926–934. [Google Scholar] [CrossRef]
- Uroz, S.; Courty, P.E.; Pierrat, J.C.; Peter, M.; Buée, M.; Turpault, M.P.; Garbaye, J.; Frey-Klett, P. Functional Profiling and Distribution of the Forest Soil Bacterial Communities Along the Soil Mycorrhizosphere Continuum. Microb. Ecol. 2013, 66, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Baldrian, P. Forest microbiome: Diversity, complexity and dynamics. FEMS Microbiol. Rev. 2017, 41, 109–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Štursová, M.; Šnajdr, J.; Cajthaml, T.; Bárta, J.; Šantrůčková, H.; Baldrian, P. When the forest dies: The response of forest soil fungi to a bark beetle-induced tree dieback. ISME J. 2014, 8, 1920–1931. [Google Scholar] [CrossRef] [Green Version]
- Kyaschenko, J.; Clemmensen, K.E.; Hagenbo, A.; Karltun, E.; Lindahl, B.D. Shift in fungal communities and associated enzyme activities along an age gradient of managed Pinus sylvestris stands. ISME J. 2017, 11, 863–874. [Google Scholar] [CrossRef]
- Spake, R.; Ezard, T.H.G.G.; Martin, P.A.; Newton, A.C.; Doncaster, C.P. A meta-analysis of functional group responses to forest recovery outside of the tropics. Conserv. Biol. 2015, 29, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Hagenbo, A.; Kyaschenko, J.; Clemmensen, K.E.; Lindahl, B.D.; Fransson, P. Fungal community shifts underpin declining mycelial production and turnover across a Pinus sylvestris chronosequence. J. Ecol. 2018, 106, 490–501. [Google Scholar] [CrossRef]
- Rudawska, M.; Wilgan, R.; Janowski, D.; Iwański, M.; Leski, T. Shifts in taxonomical and functional structure of ectomycorrhizal fungal community of Scots pine (Pinus sylvestris L.) underpinned by partner tree ageing. Pedobiologia 2018, 71, 20–30. [Google Scholar] [CrossRef]
- Kohout, P.; Charvátová, M.; Štursová, M.; Mašínová, T.; Tomšovský, M.; Baldrian, P. Clearcutting alters decomposition processes and initiates complex restructuring of fungal communities in soil and tree roots. ISME J. 2018, 12, 692–703. [Google Scholar] [CrossRef]
- Veselá, P.; Vašutová, M.; Edwards-Jonášová, M.; Cudlín, P. Soil fungal community in Norway spruce forests under bark beetle attack. Forests 2019, 10, 109. [Google Scholar] [CrossRef] [Green Version]
- Vašutová, M.; Edwards-Jonášová, M.; Veselá, P.; Effenberková, L.; Fleischer, P.; Cudlín, P. Management regime is the most important factor influencing ectomycorrhizal species community in Norway spruce forests after windthrow. Mycorrhiza 2018, 28, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Carrara, J.E.; Walter, C.A.; Hawkins, J.S.; Peterjohn, W.T.; Averill, C.; Brzostek, E.R. Interactions among plants, bacteria, and fungi reduce extracellular enzyme activities under long-term N fertilization. Glob. Chang. Biol. 2018, 24, 2721–2734. [Google Scholar] [CrossRef] [PubMed]
- Rosling, A.; Landeweert, R.; Lindahl, B.D.; Larsson, K.H.; Kuyper, T.W.; Taylor, A.F.S.; Finlay, R.D. Vertical distribution of ectomycorrhizal fungal taxa in a podzol soil profile. New Phytol. 2003, 159, 775–783. [Google Scholar] [CrossRef]
- Tedersoo, L.; Kõljalg, U.; Hallenberg, N.; Larsson, K.H. Fine scale distribution of ectomycorrhizal fungi and roots across substrate layers including coarse woody debris in a mixed forest. New Phytol. 2003, 159, 153–165. [Google Scholar] [CrossRef]
- Giurgiu, V.; Donita, N.; Bândiu, C.; Radu, S.; Cenusa, R.; Dissescu, R.; Stoiculescu, C.; Biris, I.-A. Les Forêts Vierges de Roumanie; Forêt Wallonne: Louvain-la-Nueve, Belgium, 2001. [Google Scholar]
- Kubát, K.; Hrouda, L.; Chrtek, J.; Kaplan, Z.; Kirschner, J.; Štěpánek, J. Klíč ke květeně České Republiky; Academia: Prague, Czech Republic, 2002; ISBN 80-200-0836-5. [Google Scholar]
- Svoboda, M.; Janda, P.; Bače, R.; Fraver, S.; Nagel, T.A.; Rejzek, J.; Mikoláš, M.; Douda, J.; Boublík, K.; Šamonil, P.; et al. Landscape-level variability in historical disturbance in primary Picea abies mountain forests of the Eastern Carpathians, Romania. J. Veg. Sci. 2014, 25, 386–401. [Google Scholar] [CrossRef]
- Valtera, M.; Šamonil, P.; Boublík, K. Soil variability in naturally disturbed Norway spruce forests in the Carpathians: Bridging spatial scales. For. Ecol. Manag. 2013, 310, 134–146. [Google Scholar] [CrossRef]
- IUSS Working Group WRB. World Reference Base for Soil Resources 2006, First Update 2007; Michéli, E., Schad, P., Spaargaren, O., Eds.; Food and Agriculture Organization: Rome, Italy, 2007. [Google Scholar]
- Valtera, M.; Šamonil, P.; Svoboda, M.; Janda, P. Effects of topography and forest stand dynamics on soil morphology in three natural Picea abies mountain forests. Plant Soil 2015, 392, 57–69. [Google Scholar] [CrossRef]
- Valtera, M.; Šamonil, P. Soil organic carbon stocks and related soil properties in a primary Picea abies (L.) Karst. volcanic-mountain forest. Catena 2018, 165, 217–227. [Google Scholar] [CrossRef]
- IUSS Working Group WRB. World Reference Base for Soil RESOURCES 2014. International Soil Classification System for Naming Soils and Creating Legends for SOIL maps; Food and Agriculture Organization: Rome, Italy, 2014; ISBN 9789251083697. [Google Scholar]
- Schoeneberger, P.J.; Wysocki, D.A.; Benham, E.C. Field Book for Describing and Sampling Soils; National Soil Survey Center, Natural Resources Conservation Service, U.S. Department of Agriculture: Lincoln, NE, USA, 2012; ISBN 9780160915420.
- Klinka, K.; Krestov, P.; Fons, J.; Chourmouzis, C. Towards a Taxonomic Classification of Humus Forms: Third Approximation; Forestry Sciences Department: Vancouver, BC, Canada, 1997. [Google Scholar]
- Zbíral, J. Analýza Půd I.; Ústřední kontrolní a zkušební ústav zemědělský: Brno, Czech Republic, 2002. [Google Scholar]
- Zbíral, J. Analýza Půd II.; Ústřední kontrolní a zkušební ústav zemědělský: Brno, Czech Republic, 2003. [Google Scholar]
- Zbíral, J.; Honsa, I.; Malý, S.; Čižmár, D. Analýza Půd III.; Ústřední kontrolní a zkušební ústav zemědělský: Brno, Czech Republic, 2004. [Google Scholar]
- Gillman, G.P.; Sumpter, M.E. Modification of the compulsive exchange method for measuring exchange characteristics of soils. Aust. J. Soil Res. 1986, 17, 61–66. [Google Scholar] [CrossRef]
- Bremner, J.M. Nitrogen-total. In Methods of Soil Analysis. Part 3. Chemical Methods. Number 5 in Soil Science Society of America Book Series; Sparks, D.L., Page, A.L., Helmke, P.A., Loeppert, R.H., Soltanpour, P.N., Tabatabai, M.A., Johnston, C.T., Sumner, M.E., Eds.; Soil Science Society of America, Inc. and American Society of Agronomy: Madison, WI, USA, 1996; pp. 1085–1121. [Google Scholar]
- Bray, R.H.; Kurtz, L.T. Determination of total, organic and available forms of phosphorus in soils. Soil Sci. 1945, 59, 39–45. [Google Scholar] [CrossRef]
- Boschetti, G.; Quintero, C. Extracción del P dosponible por el método Bray y Kuztz no. 1 In Tecnologías en análisis de suelos. In Tecnologías en Análisis de Suelos; Marbán, L., Ratto, S.E., Eds.; Associación Argentina de la Ciencia del Suelo: Buenos Aires, Argentina, 2005; pp. 159–168. [Google Scholar]
- Urich, T.; Lanzen, A.; Qi, J.; Huson, D.H.; Schleper, C.; Schuster, S.C. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 2008, 3, e2527. [Google Scholar] [CrossRef] [Green Version]
- Leininger, S.; Urich, T.; Schloter, M.; Schwark, L.; Qi, J.; Nicol, G.W.; Prosser, J.I.; Schuster, S.C.; Schleper, C. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 2006, 442, 806–809. [Google Scholar] [CrossRef]
- Borneman, J.; Hartin, R.J. PCR Primers That Amplify Fungal rRNA Genes from Environmental Samples. Appl. Environ. Microbiol. 2000, 66, 4356–4360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muyzer, G.; De Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.J.; Bruns, S.L.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gefland, D., Sninsky, J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.; Sánchez-García, M.; Ebersberger, I.; de Sousa, F.; et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- R Core Development Team R. A Lanaguage and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Agerer, R.; Rambold, G. DEEMY—An Information System for Characterization and Determination of Ectomycorrhizae. Available online: http://www.deemy.de (accessed on 28 October 2020).
- Buzzini, P.; Lachance, M.-A.; Yurkov, A. Yeasts in Natural Ecosystems: Ecology; Springer International Publishing AG: Manhattan, NY, USA, 2017; ISBN 9783319615745. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [Green Version]
- Lenth, R.; Singmann, H.; Love, J.; Buerkner, P.; Herve, M. Estimated Marginal Means, aka Least-Squares Means; CRAN R 2019; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package; R package version 2.4-0 2019; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Hervé, M. RVAideMemoire: Testing and Plotting Procedures for Biostatistics. R package version 0.9-78. 2020. Available online: https://CRAN.R-project.org/package=RVAideMemoire (accessed on 4 September 2020).
- Lladó, S.; Žifčáková, L.; Větrovský, T.; Eichlerová, I.; Baldrian, P. Functional screening of abundant bacteria from acidic forest soil indicates the metabolic potential of Acidobacteria subdivision 1 for polysaccharide decomposition. Biol. Fertil. Soils 2016, 52, 251–260. [Google Scholar] [CrossRef]
- Větrovský, T.; Steffen, K.T.; Baldrian, P. Potential of Cometabolic Transformation of Polysaccharides and Lignin in Lignocellulose by Soil Actinobacteria. PLoS ONE 2014, 9, e89108. [Google Scholar] [CrossRef]
- Cardman, Z.; Arnosti, C.; Durbin, A.; Ziervogel, K.; Cox, C.; Steen, A.D.; Teske, A. Verrucomicrobia are candidates for polysaccharide-degrading bacterioplankton in an Arctic fjord of Svalbard. Appl. Environ. Microbiol. 2014, 80, 3749–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, C.E.; Grayston, S.J. Tree species influence on microbial communities in litter and soil: Current knowledge and research needs. For. Ecol. Manag. 2013, 309, 19–27. [Google Scholar] [CrossRef]
- Kubartová, A.; Ranger, J.; Berthelin, J.; Beguiristain, T. Diversity and decomposing ability of saprophytic fungi from temperate forest litter. Microb. Ecol. 2009, 58, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Žifčáková, L.; Dobiášová, P.; Kolářová, Z.; Koukol, O.; Baldrian, P. Enzyme activities of fungi associated with Picea abies needles. Fungal Ecol. 2011, 4, 427–436. [Google Scholar] [CrossRef]
- Hansson, K.; Olsson, B.A.; Olsson, M.; Johansson, U.; Kleja, D.B. Differences in soil properties in adjacent stands of Scots pine, Norway spruce and silver birch in SW Sweden. For. Ecol. Manag. 2011, 262, 522–530. [Google Scholar] [CrossRef]
- Barabote, R.D.; Xie, G.; Leu, D.H.; Normand, P.; Necsulea, A.; Daubin, V.; Médigue, C.; Adney, W.S.; Xin, C.X.; Lapidus, A.; et al. Complete genome of the cellulolytic thermophile Acidothermus cellulolyticus IIB provides insights into its ecophysiological and evolutionary adaptations. Genome Res. 2009, 19, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Cha, G.; Gao, B. A phylogenomic and molecular markers based analysis of the class Acidimicrobiia. Front. Microbiol. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, H.; He, X.; Thomas, B.W.; Lupwayi, N.Z.; Hao, X.; Thomas, M.C.; Shi, X. Fertilization shapes bacterial community structure by alteration of soil pH. Front. Microbiol. 2017, 8, 1325. [Google Scholar] [CrossRef] [Green Version]
- Erbilgin, O.; McDonald, K.L.; Kerfeld, C.A. Characterization of a planctomycetal organelle: A novel bacterial microcompartment for the aerobic degradation of plant saccharides. Appl. Environ. Microbiol. 2014, 80, 2193–2205. [Google Scholar] [CrossRef] [Green Version]
- Kaňa, J.; Tahovská, K.; Kopáček, J.; Šantručková, H. Excess of organic carbon in mountain spruce forest soils after bark beetle outbreak altered microbial N transformations and mitigated N-saturation. PLoS ONE 2015, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Ambus, P.; Zechmeister-Boltenstern, S. Denitrification and N-Cycling in Forest Ecosystems; Elsevier B.V.: Amsterdam, The Netherlands, 2007; ISBN 9780444528575. [Google Scholar]
- Pandey, C.B.; Kumar, U.; Kaviraj, M.; Minick, K.J.; Mishra, A.K.; Singh, J.S. DNRA: A short-circuit in biological N-cycling to conserve nitrogen in terrestrial ecosystems. Sci. Total Environ. 2020, 738, 139710. [Google Scholar] [CrossRef]
- Sponseller, R.A.; Gundale, M.J.; Futter, M.; Ring, E.; Nordin, A.; Näsholm, T.; Laudon, H. Nitrogen dynamics in managed boreal forests: Recent advances and future research directions. Ambio 2016, 45, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Wallander, H.; Johansson, U.; Sterkenburg, E.; Brandström Durling, M.; Lindahl, B.D. Production of ectomycorrhizal mycelium peaks during canopy closure in Norway spruce forests. New Phytol. 2010, 187, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.F.S.; Alexander, I.J. The ectomycorrhizal symbiosis: Life in the real world. Mycologist 2005, 19, 102–112. [Google Scholar] [CrossRef]
- Courty, P.-E.; Buée, M.; Diedhiou, A.G.; Frey-Klett, P.; Le Tacon, F.; Rineau, F.; Turpault, M.-P.; Uroz, S.; Garbaye, J. The role of ectomycorrhizal communities in forest ecosystem processes: New perspectives and emerging concepts. Soil Biol. Biochem. 2010, 42, 679–698. [Google Scholar] [CrossRef]
- Jones, M.D.; Durall, D.M.; Cairney, J.W.G. Ectomycorrhizal fungal communities in young forest stands regenerating after clearcut logging. New Phytol. 2003, 157, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Sterkenburg, E.; Clemmensen, K.E.; Lindahl, B.D.; Dahlberg, A. The significance of retention trees for survival of ectomycorrhizal fungi in clear-cut Scots pine forests. J. Appl. Ecol. 2019, 56, 1367–1378. [Google Scholar] [CrossRef]
- Varenius, K.; Lindahl, B.D.; Dahlberg, A. Retention of seed trees fails to lifeboat ectomycorrhizal fungal diversity in harvested Scots pine forests. FEMS Microbiol. Ecol. 2017, 93, 1–11. [Google Scholar] [CrossRef]
- Chambers, S.M.; Burke, R.M.; Brooks, P.R.; Cairney, J.W.G. Molecular and biochemical evidence for manganese-dependent peroxidase activity in Tylospora fibrillosa. Mycol. Res. 1999, 103, 1098–1102. [Google Scholar] [CrossRef]
- Fukasawa, Y.; Ando, Y.; Song, Z. Comparison of fungal communities associated with spruce seedling roots and bryophyte carpets on logs in an old-growth subalpine coniferous forest in Japan. Fungal Ecol. 2017, 30, 122–131. [Google Scholar] [CrossRef]
- Lindahl, B.D.; Tunlid, A. Ectomycorrhizal fungi—Potential organic matter decomposers, yet not saprotrophs. New Phytol. 2015, 205, 1443–1447. [Google Scholar] [CrossRef] [PubMed]
- Zak, D.R.; Pellitier, P.T.; Argiroff, W.A.; Castillo, B.; James, T.Y.; Nave, L.E.; Averill, C.; Beidler, K.V.; Bhatnagar, J.; Blesh, J.; et al. Exploring the role of ectomycorrhizal fungi in soil carbon dynamics. New Phytol. 2019, 223, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.R.; Schaetzl, R.J. An examination of podzolization near Lake Michigan using chronofunctions. Can. J. Soil Sci. 1992, 72, 527–541. [Google Scholar] [CrossRef] [Green Version]
- Barrett, L.R.; Schaetzl, R.J. Soils—Genesis and Geomorphology, 2nd ed.; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 16 Years | 36 Years | 100 Years | 110 Years | 130 Years | 160 Years | |
|---|---|---|---|---|---|---|
| Time since the last stand-replacing disturbance a (years) | 16 | 36 | 100 | 110 | 130 | 160 |
| Canopy closure (%) | 0 | 20 | 95 | 94 | 89 | 83 |
| Soil unit | Entic Podzol | Entic Podzol | Entic Skeletic Andic Podzol | Entic Podzol | Albic Skeletic Podzol | Albic Podzol |
| Upper organic (O) horizons | T | T | L + F | L + F | L + F | L + F |
| H | H | H | H | H | ||
| Upper mineral (A) horizons | A | A | A | A | A | A |
| AB | AB | AeB | Ep | Ep | ||
| Spodic (B) horizons | AB | (A)Bs | Bs | Bs | Bhs | Bhs |
| Bs | (A)Bs | Bs | Bs | Bs | Bs |
| Horizons | 16 Years | 36 Years | 100 Years | 110 Years | 130 Years | 160 Years | ||
|---|---|---|---|---|---|---|---|---|
| pH | O | 3.0 | 3.2 | 3.0 | 3.0 | 2.8 | 2.6 | |
| A | 3.1 | 3.4 | 3.0 | 3.0 | 2.6 | 2.5 | ||
| B | 3.8 ab | 4.1 a | 4.0 a | 3.7 ab | 4.1 a | 3.4 b | ||
| Ctot | mg g−1 | O | 34.9 b | 39.0 ab | 44.2 a | 44.7 a | 43.8 a | 39.7 ab |
| A | 8.5 | 10.7 | 23.8 | 14.1 | 11.5 | 11.6 | ||
| B | 7.0 bc | 6.9 bc | 17.8 a | 8.9 b | 6.3 bc | 4.0 c | ||
| Ntot | mg g−1 | O | 1.86 | 2.02 | 1.63 | 1.64 | 1.73 | 1.63 |
| A | 0.52 | 0.59 | 1.24 | 0.69 | 0.65 | 0.62 | ||
| B | 0.33 b | 0.27 b | 0.71 a | 0.33 b | 0.38 b | 0.21 b | ||
| Ctot:Ntot | mol:mol | O | 21.8 c | 22.8 bc | 31.6 a | 32.1 a | 29.6 a | 28.5 ab |
| A | 19.5 | 21.8 | 22.4 | 23.9 | 20.4 | 25.3 | ||
| B | 25.6 b | 30.3 ab | 32.0 a | 30.8 ab | 28.2 ab | 27.3 b | ||
| DOC | µg g−1 | O | 2757 | 3277 | 3365 | 3737 | 4262 | 3259 |
| A | 603 | 543 | 1454 | 1011 | 1110 | 1147 | ||
| B | 319 | 214 | 294 | 282 | 301 | 328 | ||
| DN | µg g−1 | O | 372 ab | 454 a | 198 bc | 196 bc | 205 abc | 159 c |
| A | 42.2 a | 89.2 a | 99.4 a | 48.1 a | 51.6 a | 51.4 a | ||
| B | 18.5 | 19.3 | 19.7 | 16.6 | 12.6 | 11.8 | ||
| DOC:DN | mol:mol | O | 7.8 c | 9.4 bc | 17.4 ab | 20.4 a | 21.5 a | 21.2 a |
| A | 16.8 a | 6.7 b | 16.3 a | 21.1 a | 21.8 a | 23.4 a | ||
| B | 17.4 cd | 12.5 d | 15.6 cd | 17.5 c | 24.1 b | 28.2 a | ||
| CEC | µmol cheq g−1 | O | 312 | 216 | 303 | 308 | 260 | 298 |
| A | 324 ab | 259 c | 363 a | 297 bc | 204 d | 129 e | ||
| B | 212 | 140 | 217 | 184 | 135 | 166 |
| ANOVA Site | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Horizon | 16 Years | 36 Years | 100 Years | 110 Years | 130 Years | 160 Years | F | p | |
| Fungi OTU richness | O | 127 (41) | 143 (11) | 84 (37) | 131 (28) | 91 (31) | 127 (12) | n.s. | |
| OTUs | A | 95 (18) | 96 (30) | 90 (6) | 86 (18) | 77 (17) | 64 (22) | n.s. | |
| B | 78 (19) | 72 (19) | 75 (17) | 102 (12) | 81 (28) | 91 (24) | n.s. | ||
| Fungi Shannon | O | 2.86 (0.45) | 3.22 (0.10) | 2.32 (0.86) | 3.15 (0.34) | 2.36 (1.04) | 3.00 (0.21) | n.s. | |
| A | 2.16 (0.11) | 1.90 (0.81) | 2.48 (0.26) | 2.42 (0.34) | 2.53 (0.41) | 2.44 (0.76) | n.s. | ||
| B | 2.20 (0.38) | 2.45 (0.29) | 2.45 (0.51) | 2.58 (0.35) | 2.38 (1.25) | 2.57 (0.56) | n.s. | ||
| Bacteria OTU richness | O | 726 (84) | 756 (57) | 663 (171) | 623 (73) | 680 (87) | 727 (117) | n.s. | |
| OTUs | A | 695 (50) a | 655 (70) ab | 596 (89) abc | 548 (89) bc | 565 (50) bc | 533 (58) c | 4.49 | 0.004 |
| B | 612 (72) a | 620 (66) a | 607 (33) a | 637 (20) a | 565 (56) ab | 512 (39) b | 6.88 | <0.001 | |
| Bacteria Shannon | O | 5.55 (0.24) | 5.60 (0.18) | 5.34 (0.40) | 5.33 (0.19) | 5.41 (0.25) | 5.57 (0.23) | n.s. | |
| A | 5.38 (0.15) a | 5.23 (0.16) ab | 5.15 (0.22) ab | 4.91 (0.34) b | 4.91 (0.14) b | 4.99 (0.20) b | 4.83 | 0.003 | |
| B | 5.08 (0.25) a | 5.05 (0.19) ab | 5.13 (0.09) a | 5.14 (0.09) a | 4.92 (0.19) ab | 4.79 (0.15) b | 5.2 | 0.001 | |
| Fungal 18S rRNA gene | O | 129 (110) | 58 (85) | 147 (89) | 52 (51) | 13 (7) | 60 (78) | n.s. | |
| 107 copies g−1 | A | 13.1 (0.6) a | 7.4 (0.5) ab | 41.9 (5.5) a | 9.1 (0.6) a | 11.2 (1) a | 1.3 (0.1) b | 5.4 | 0.001 |
| B | 3.76 (0.35) | 0.28 (0.02) | 1.74 (0.15) | 1.17 (0.15) | 5.2 (0.53) | 2.43 (0.21) | n.s. | ||
| Bacterial 16S rRNA gene | O | 36.7 (1.9) a | 27.8 (5) ab | 22.6 (2.5) ab | 6.2 (1.6) c | 9 (1.4) bc | 8.6 (1.4) bc | 9.51 | <0.001 |
| 109 copies g−1 | A | 16.6 (0.6) ab | 29.2 (2.2) a | 20.1 (2.2) ab | 7.8 (1.2) bc | 4.2 (0.4) c | 4.5 (1.4) c | 11.7 | <0.001 |
| B | 9.65 (1.68) a | 7.83 (2.26) ab | 1.84 (0.49) c | 1.05 (0.16) bc | 3.98 (0.75) abc | 4.82 (0.84) abc | 3.99 | 0.007 | |
| Fungi-to-bacteria (F:B) | O | 0.035 (0.027) | 0.028 (0.046) | 0.069 (0.034) | 0.098 (0.118) | 0.017 (0.008) | 0.055 (0.059) | n.s. | |
| copy:copy | A | 0.008 (0.003) ab | 0.003 (0.002) a | 0.016 (0.017) ab | 0.011 (0.004) ab | 0.025 (0.021) b | 0.007 (0.01) ab | 3.56 | 0.014 |
| B | 0.003 (0.002) ab | 0.001 (0.001) a | 0.008 (0.007) b | 0.018 (0.031) b | 0.011 (0.009) b | 0.006 (0.006) b | 3.26 | 0.018 | |
| ANOVA Sites | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Horizon | 16 Years | 36 Years | 100 Years | 110 Years | 130 Years | 160 Years | F | p | |
| Amanita | O | 2.3 (4.1) | 0.8 (1.1) | <0.1 | 2.7 (6) | <0.1 | 3.4 (7.6) | n.s. | |
| A | 10.1 (22.1) | 0.5 (1.1) | <0.1 | 6.9 (12.2) | 7.4 (8.3) | 11.7 (23.1) | n.s. | ||
| B | 15.4 (26.5) | <0.1 | <0.1 | 1.2 (2.3) | <0.1 | <0.1 | n.s. | ||
| Boletus | O | <0.1 | <0.1 | 0.3 (0.5) | 0.4 (0.5) | <0.1 | <0.1 | n.s. | |
| A | <0.1 c | <0.1 c | 6.6 (3.8) a | 2.1 (2.7) b | <0.1 c | <0.1 c | 11.7 | <0.001 | |
| B | <0.1 b | <0.1 b | 16.8 (16) a | 2.8 (4.2) ab | <0.1 b | <0.1 b | 5.8 | 0.001 | |
| Cantharellales | O | 3.6 (7.1) | 0.3 (0.1) | 0.4 (1) | 2.5 (4.5) | 0.3 (0.5) | <0.1 | n.s. | |
| A | 4.2 (9.1) ab | <0.1 b | <0.1 b | 24.5 (24.2) a | 2 (4.5) b | <0.1 b | 4.6 | 0.006 | |
| B | 0.4 (0.8) a | <0.1 a | <0.1 a | 17.8 (24.1) a | 28.2 (48.5) a | <0.1 a | 3.0 | 0.029 | |
| Hygrophorus | O | 11.3 (8.7) | <0.1 | 4.8 (5.6) | 1.2 (1.9) | <0.1 | 4.1 (7.2) | n.s. | |
| A | 10.8 (20.2) | 0.2 (0.4) | 0.8 (1.1) | <0.1 | 0.2 (0.4) | 0.3 (0.6) | n.s. | ||
| B | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | n.s. | ||
| Meliniomyces | O | 0.2 (0.1) c | 2.8 (0.4) ab | 0.7 (0.7) bc | 4 (1.7) a | 1.1 (0.8) bc | 2.4 (1) ab | 12.6 | <0.001 |
| A | 0.1 (0.3) | <0.1 | 0.4 (0.5) | 0.4 (0.4) | 0.7 (0.8) | 1.6 (2.7) | n.s. | ||
| B | <0.1 | <0.1 | <0.1 | <0.1 | 6.1 (10.6) | 0.2 (0.1) | n.s. | ||
| Piloderma | O | 4.6 (8.5) | <0.1 | 14.1 (17.1) | 11.1 (9.6) | 2 (4) | 10.1 (12.7) | n.s. | |
| A | <0.1 | <0.1 | 8.3 (11.5) | 10.2 (16.6) | 13.1 (17.3) | 2.8 (3.9) | n.s. | ||
| B | <0.1 a | <0.1 a | 0.4 (0.5) a | <0.1 a | 0.2 (0.1) a | <0.1 a | 2.7 | 0.045 | |
| Russula | O | <0.1 | 9.6 (13.5) | 12.7 (27.7) | 9.1 (17.8) | 21.4 (24.3) | 10.7 (23.8) | n.s. | |
| A | <0.1 | 3.6 (7.8) | 30.6 (43.1) | 4.1 (9.2) | 9.6 (7.6) | 14.5 (21.5) | n.s. | ||
| B | <0.1 | <0.1 | 0.5 (0.5) | <0.1 | <0.1 | 5.6 (9.7) | n.s. | ||
| Thelephorales | O | 1.3 (1.4) | 0.4 (0.2) | 1.1 (2) | 0.3 (0.3) | 1.1 (2.1) | 0.6 (0.5) | n.s. | |
| A | 0.3 (0.7) | 0.2 (0.2) | <0.1 | <0.1 | 0.7 (1) | 4.2 (7.8) | n.s. | ||
| B | <0.1 | 2.1 (3) | 2.1 (3.6) | <0.1 | <0.1 | 0.1 (0.1) | n.s. | ||
| Tylospora | O | 32.6 (18.2) | 48.4 (13.9) | 16.3 (15.1) | 20.3 (20.3) | 22.1 (27.5) | 17.2 (22.6) | n.s. | |
| A | 48.6 (28.9) | 59.3 (33) | 25.9 (22.4) | 11.9 (16) | 25.9 (23.9) | 16.8 (11.7) | n.s. | ||
| B | 20.9 (23) | 14.8 (26.4) | 21.9 (24.2) | 20.7 (21.4) | 0.1 (0.1) | 24.2 (33.4) | n.s. | ||
| Wilcoxina | O | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | n.s. | |
| A | 5.7 (10.4) | 4.4 (8.7) | <0.1 | <0.1 | <0.1 | <0.1 | n.s. | ||
| B | 14.8 (20) | 4.6 (8.9) | 0.9 (2.1) | 2.2 (3.2) | <0.1 | <0.1 | n.s. | ||
| All ectomycorrhizal summed | O | 52.1 (26.8) | 59.3 (0.7) | 53.9 (32.3) | 45.2 (13.3) | 46.8 (23.7) | 48.4 (27.2) | n.s. | |
| A | 75.6 (12.6) | 68.4 (37.2) | 72.3 (6.5) | 42.2 (23.2) | 58.9 (16.7) | 46.8 (33.5) | n.s. | ||
| B | 51.1 (17.1) | 19.5 (24.6) | 41 (23.6) | 33.8 (28.8) | 28.9 (48.2) | 30.3 (30.9) | n.s. | ||
| ANOVA Sites | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Horizon | 16 Years | 36 Years | 100 Years | 110 Years | 130 Years | 160 Years | F | p | |
| Acidimicrobiia | O | 1.20 (0.35) c | 1.54 (0.72) bc | 2.08 (0.58) abc | 3.65 (1.25) a | 2.92 (1.11) ab | 3.10 (1.34) ab | 5.99 | <0.001 |
| A | 0.95 (0.41) ab | 0.62 (0.17) b | 1.54 (1.09) ab | 1.91 (1.06) a | 1.70 (0.41) a | 1.41 (0.69) ab | 3.57 | 0.013 | |
| B | 1.35 (0.45) ab | 0.96 (0.28) b | 1.46 (0.24) ab | 1.75 (0.29) a | 1.38 (0.23) ab | 1.84 (0.44) a | 5.64 | <0.001 | |
| Actinobacteria | O | 3.97 (1.03) | 4.14 (2.6) | 6.08 (2.22) | 8.43 (3.21) | 6.79 (2.59) | 6.65 (3.81) | n.s. | |
| A | 2.75 (1.28) bc | 1.85 (0.64) c | 6.50 (2.35) a | 4.73 (2.14) ab | 4.79 (1.18) ab | 4.17 (1.85) abc | 5.61 | 0.001 | |
| B | 1.54 (1.11) bc | 0.51 (0.31) c | 1.29 (0.72) bc | 1.76 (0.82) bc | 2.91 (2.18) b | 5.24 (0.92) a | 12.3 | <0.001 | |
| AD3 | O | 0.11 (0.04) | 0.20 (0.16) | 0.14 (0.13) | 0.12 (0.05) | 0.19 (0.14) | 0.22 (0.22) | n.s. | |
| A | 1.25 (0.78) a | 0.99 (0.77) a | 0.38 (0.33) a | 0.92 (1.19) a | 0.23 (0.17) a | 0.19 (0.16) a | 3.01 | 0.028 | |
| B | 4.36 (2.16) cd | 3.14 (0.98) bd | 6.82 (1.51) c | 3.09 (1.17) bd | 1.67 (1.0) ab | 1.19 (0.77) a | 13.6 | <0.001 | |
| Alphaproteobacteria | O | 15.6 (2.5) a | 13.8 (2.2) a | 13.7 (3.2) a | 17.3 (1.8) a | 17.9 (2.2) a | 17.6 (2.5) a | 2.87 | 0.030 |
| A | 11.4 (5.2) b | 9.5 (4.5) b | 17.5 (2.5) ab | 15.2 (2.8) ab | 18.7 (3.9) a | 20.9 (3.1) a | 6.83 | <0.001 | |
| B | 9.7 (3.4) bc | 7.6 (1.4) c | 9.8 (2.5) bc | 10.8 (3.2) bc | 13.3 (3.9) ab | 19.5 (5.6) a | 8.10 | <0.001 | |
| Bacteroidia | O | 7.51 (3.12) | 7.04 (1.21) | 7.93 (5.26) | 5.53 (3.03) | 5.96 (3.18) | 6.63 (3.40) | n.s. | |
| A | 2.22 (1.23) a | 2.40 (0.85) a | 3.00 (1.24) a | 1.35 (0.97) a | 1.22 (0.26) a | 1.41 (0.47) a | 3.14 | 0.024 | |
| B | 0.61 (0.21) | 0.82 (0.44) | 1.26 (0.86) | 0.79 (0.42) | 0.88 (0.50) | 0.66 (0.20) | n.s. | ||
| Chlamydiae | O | 0.65 (0.26) | 0.86 (0.59) | 0.88 (0.60) | 1.5 (1.43) | 0.86 (0.40) | 1.34 (1.06) | n.s. | |
| A | 1.33 (0.32) | 1.27 (0.26) | 0.85 (0.34) | 1.68 (0.79) | 0.9 (0.27) | 1.72 (0.93) | n.s. | ||
| B | 1.42 (0.23) b | 2.02 (0.71) ab | 1.36 (0.48) b | 2.75 (0.50) a | 1.7 (0.53) b | 1.29 (0.38) b | 6.72 | <0.001 | |
| Fimbriimonadia | O | 0.51 (0.17) b | 0.37 (0.02) ab | 0.27 (0.14) a | 0.22 (0.09) a | 0.25 (0.11) a | 0.33 (0.09) ab | 3.87 | 0.009 |
| A | 0.22 (0.15) b | 0.14 (0.06) ab | <0.1 ab | <0.1 a | <0.1 a | <0.1 a | 4.41 | 0.005 | |
| B | 0.1 (0.03) bc | 0.14 (0.04) c | 0.15 (0.04) c | <0.1 bc | <0.1 ab | <0.1 a | 8.68 | <0.001 | |
| Gammaproteobacteria | O | 13.12 (3.56) | 11.41 (5.46) | 9.66 (3.83) | 10.21 (5.62) | 9.12 (3.92) | 8.60 (3.10) | n.s. | |
| A | 5.37 (0.76) | 4.96 (1.05) | 6.77 (1.90) | 7.39 (5.64) | 3.67 (1.83) | 4.01 (1.03) | n.s. | ||
| B | 6.76 (1.37) b | 8.08 (2.03) b | 5.51 (0.82) b | 5.31 (1.24) b | 5.38 (1.96) b | 2.97 (1.00) a | 8.82 | <0.001 | |
| Gemmatimonadetes | O | 0.65 (0.26) b | 0.47 (0.14) ab | 0.32 (0.19) ab | 0.20 (0.11) a | 0.32 (0.15) ab | 0.36 (0.20) ab | 4.06 | 0.007 |
| A | 1.50 (0.28) c | 1.27 (0.53) bc | 0.75 (0.12) abc | 0.79 (0.71) ab | 0.74 (0.25) ab | 0.50 (0.24) a | 5.17 | 0.002 | |
| B | 1.80 (0.46) cd | 1.31 (0.33) bcd | 1.99 (0.57) d | 1.09 (0.43) bc | 0.97 (0.32) b | 3.01 (0.68) a | 14.0 | <0.001 | |
| Ktedonobacteria | O | 0.20 (0.27) | 0.47 (0.56) | 0.16 (0.23) | <0.1 | <0.1 | <0.1 | n.s. | |
| A | 5.20 (1.89) b | 6.15 (3.1) b | 2.27 (2.06) ab | 3.15 (4.58) a | 0.21 (0.30) a | <0.1 a | 6.81 | <0.001 | |
| B | 7.31 (1.62) | 11.02 (5.72) | 9.46 (3.31) | 10.62 (3.84) | 7.03 (4.35) | 4.88 (3.32) | n.s. | ||
| Myxococcia | O | 0.49 (0.11) | 0.23 (0.13) | 0.31 (0.18) | 0.31 (0.13) | 0.29 (0.07) | 0.45 (0.25) | n.s. | |
| A | 0.26 (0.06) | 0.28 (0.13) | 0.17 (0.06) | 0.20 (0.25) | 0.16 (0.05) | 0.15 (0.06) | n.s. | ||
| B | 0.55 (0.27) c | 0.46 (0.09) bc | 0.28 (0.13) ab | 0.21 (0.12) a | 0.22 (0.07) a | 0.14 (0.04) a | 9.53 | <0.001 | |
| Nitrososphaeria | O | 0.18 (0.12) | 0.26 (0.19) | 0.20 (0.25) | 0.16 (0.18) | 0.24 (0.36) | 0.23 (0.27) | n.s. | |
| A | 3.45 (1.58) bc | 4.07 (1.58) c | 1.16 (0.4) ab | 1.37 (1.27) a | 1.28 (0.39) a | 1.34 (0.79) a | 7.28 | <0.001 | |
| B | 5.61 (1.44) ab | 8.55 (1.38) b | 5.28 (1.67) ab | 5.12 (3.46) ab | 3.01 (1.49) a | 4.15 (1.52) a | 5.35 | 0.001 | |
| RCP2-54 | O | 1.70 (1.25) | 1.81 (1.80) | 1.21 (1.21) | 1.73 (1.66) | 1.54 (1.45) | 2.09 (1.77) | n.s. | |
| A | 4.54 (1.26) ab | 3.65 (1.25) b | 4.29 (1.59) ab | 5.30 (2.90) ab | 6.31 (1.22) ab | 7.69 (3.16) a | 2.87 | 0.034 | |
| B | 2.59 (0.78) bcd | 2.09 (0.71) cd | 1.69 (0.27) d | 3.17 (0.83) abc | 3.70 (1.40) ab | 4.57 (0.37) a | 10.2 | <0.001 | |
| Thermoleophilia | O | 1.05 (0.38) | 1.37 (1.06) | 1.60 (0.86) | 2.84 (1.04) | 2.24 (1.24) | 2.23 (1.48) | n.s. | |
| A | 0.60 (0.29) bc | 0.31 (0.14) c | 0.96 (0.41) ab | 1.29 (0.62) a | 0.90 (0.40) ab | 0.55 (0.13) bc | 6.18 | <0.001 | |
| B | 0.62 (0.3) bc | 0.50 (0.17) c | 1.16 (0.32) a | 0.93 (0.32) ab | 0.80 (0.12) abc | 0.79 (0.18) abc | 5.32 | 0.001 | |
| Verrucomicrobiae | O | 5.66 (1.19) | 5.88 (1.27) | 6.43 (1.93) | 5.00 (1.41) | 6.01 (1.61) | 6.57 (2.24) | n.s. | |
| A | 4.89 (0.77) b | 5.07 (1.57) b | 5.32 (0.28) b | 4.25 (0.87) ab | 3.16 (0.49) a | 4.39 (0.95) ab | 3.86 | 0.010 | |
| B | 3.03 (0.92) ab | 2.56 (0.86) ab | 3.94 (1.17) b | 2.85 (1.36) ab | 3.67 (0.63) ab | 2.19 (0.89) a | 2.75 | 0.037 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choma, M.; Šamonil, P.; Kaštovská, E.; Bárta, J.; Tahovská, K.; Valtera, M.; Šantrůčková, H. Soil Microbiome Composition along the Natural Norway Spruce Forest Life Cycle. Forests 2021, 12, 410. https://doi.org/10.3390/f12040410
Choma M, Šamonil P, Kaštovská E, Bárta J, Tahovská K, Valtera M, Šantrůčková H. Soil Microbiome Composition along the Natural Norway Spruce Forest Life Cycle. Forests. 2021; 12(4):410. https://doi.org/10.3390/f12040410
Chicago/Turabian StyleChoma, Michal, Pavel Šamonil, Eva Kaštovská, Jiří Bárta, Karolina Tahovská, Martin Valtera, and Hana Šantrůčková. 2021. "Soil Microbiome Composition along the Natural Norway Spruce Forest Life Cycle" Forests 12, no. 4: 410. https://doi.org/10.3390/f12040410
APA StyleChoma, M., Šamonil, P., Kaštovská, E., Bárta, J., Tahovská, K., Valtera, M., & Šantrůčková, H. (2021). Soil Microbiome Composition along the Natural Norway Spruce Forest Life Cycle. Forests, 12(4), 410. https://doi.org/10.3390/f12040410