
Inconsistent Response of Abundant and Rare Bacterial Communities to the Developmental Chronosequence of Pinus massoniana
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Region
2.2. Soil Sampling and Physicochemical Analysis
2.3. DNA Extraction, Amplification, and Sequencing
2.4. Data Analyses
3. Results
3.1. Variations in Soil Variables
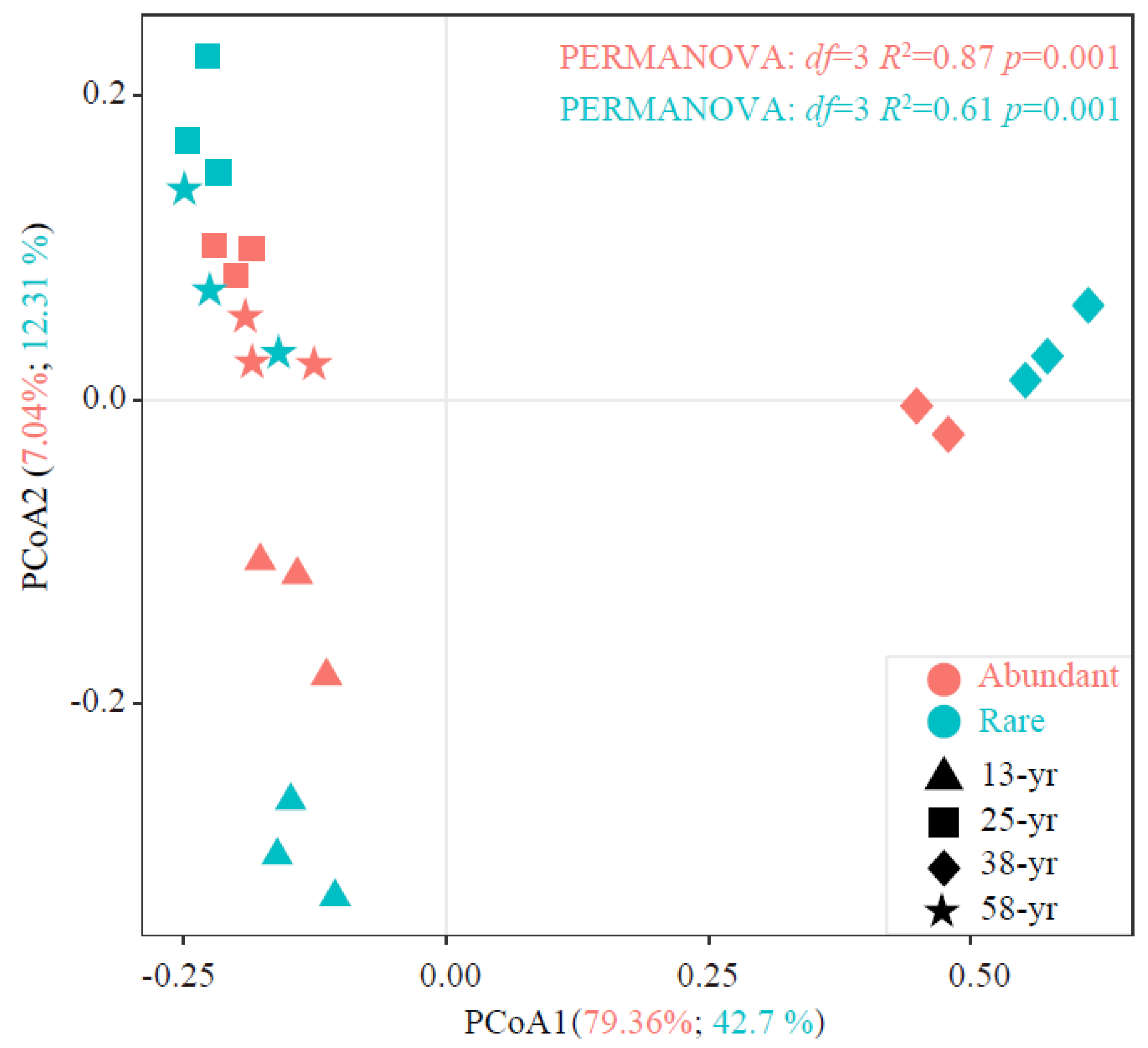
3.2. Characteristics of Abundant and Rare Bacterial Taxa
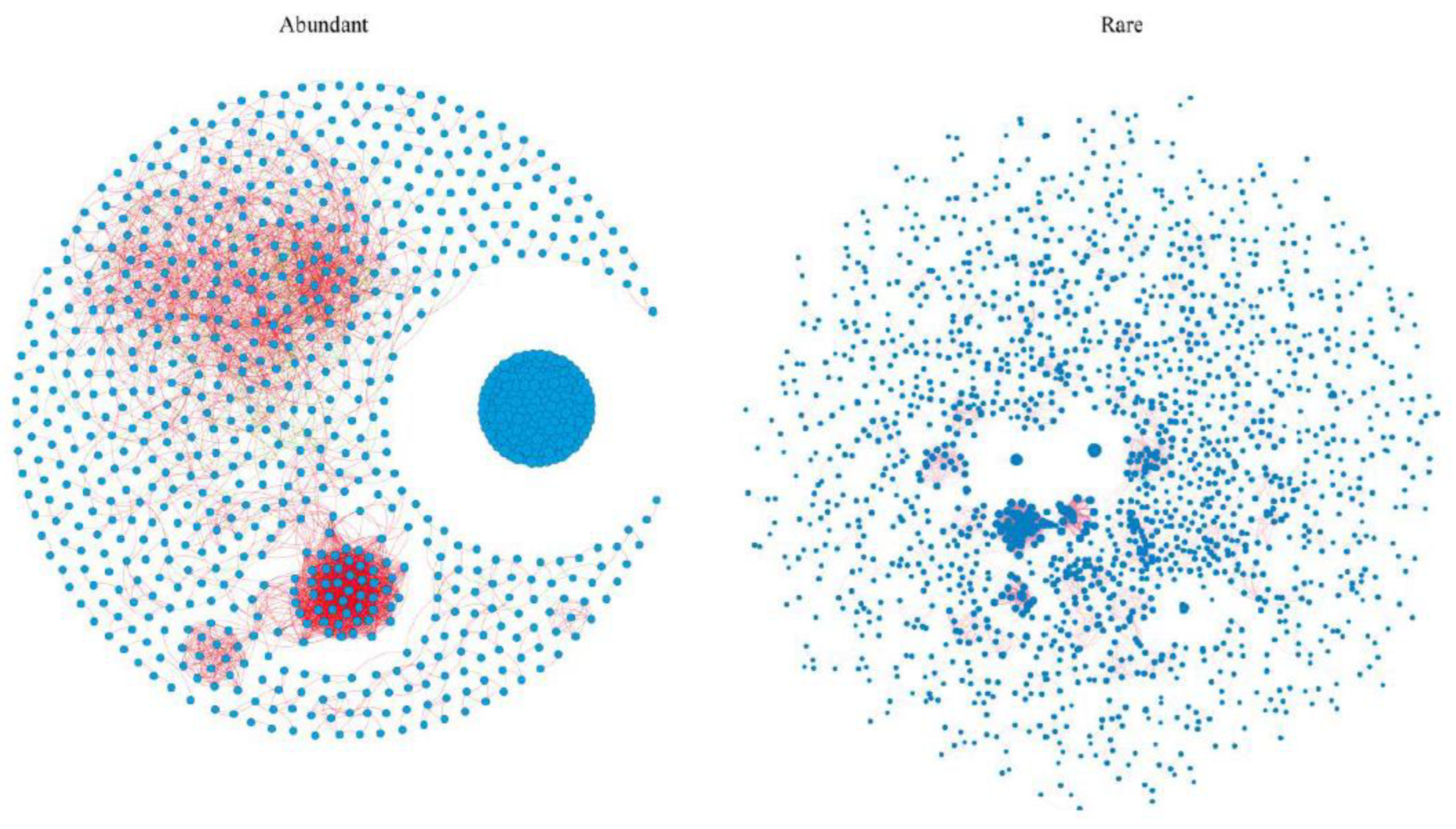
3.3. Co-Occurrence Patterns in Abundant and Rare Bacterial Taxa
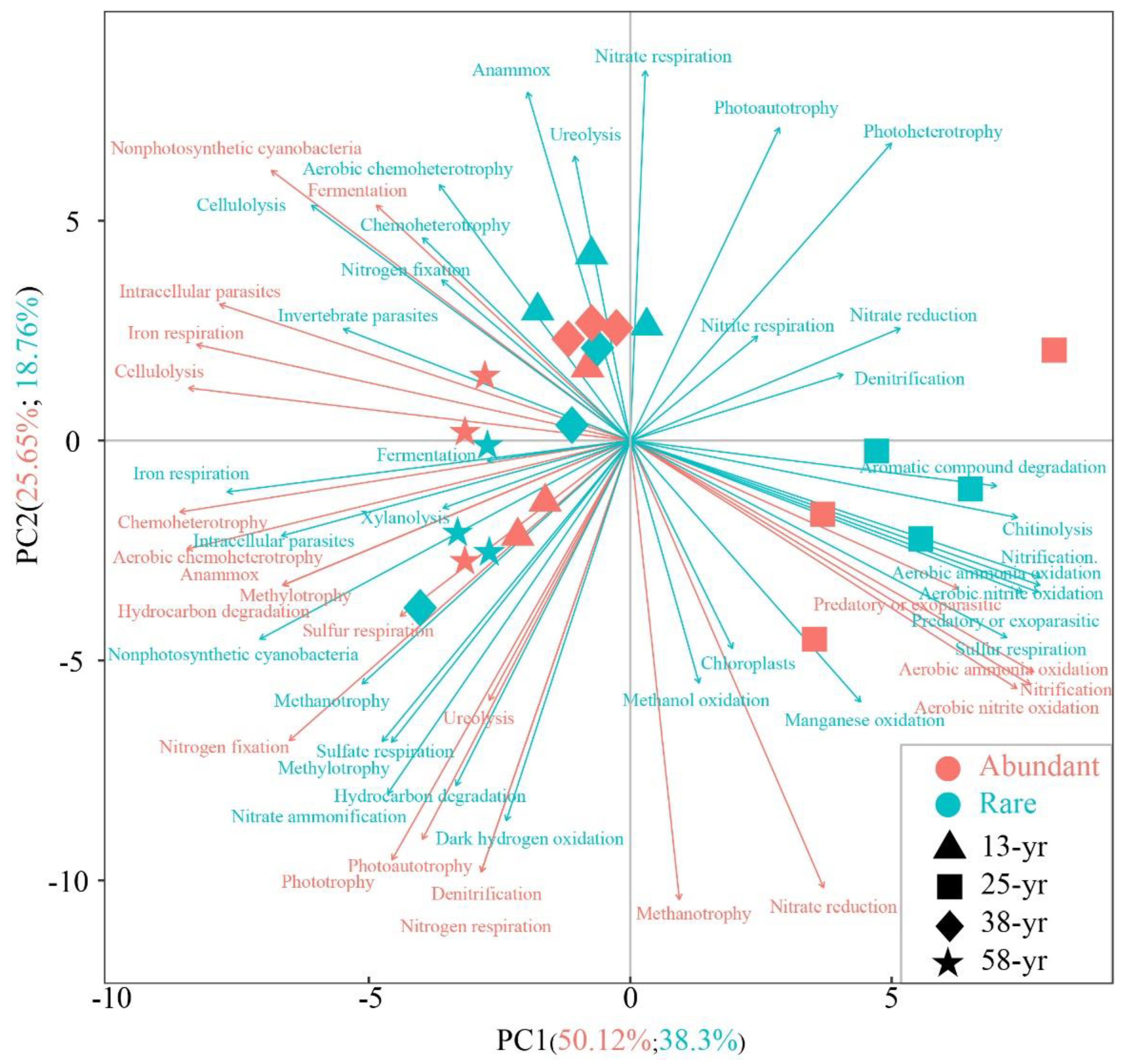
3.4. Variations in the Potential Functions of Abundant and Rare Bacterial Taxa
3.5. Associations between Soil Physicochemical Properties and Bacterial Functions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wallander, H.; Johansson, U.; Sterkenburg, E.; Durling, M.B.; Lindahl, B.D. Production of ectomycorrhizal mycelium peaks during canopy closure in Norway spruce forests. New Phytol. 2010, 187, 1124–1134. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, C.; Luo, Y. Meta-analysis of the impacts of global change factors on soil microbial diversity and functionality. Nat. Commun. 2020, 11, 3072. [Google Scholar] [CrossRef] [PubMed]
- Bukoski, J.J.; Cook-Patton, S.C.; Melikov, C.; Ban, H.Y.; Chen, J.L.; Goldman, E.D.; Harris, N.L.; Potts, M.D. Rates and drivers of aboveground carbon accumulation in global monoculture plantation forests. Nat. Commun. 2022, 13, 4206. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Zhao, S.; Yang, B.; Zhang, K.F.; Fan, Y.X.; Zhang, L.L.; Yang, X.D. The carbon and nitrogen stoichiometry in litter-soil-microbe continuum rather than plant diversity primarily shapes the changes in bacterial communities along a tropical forest restoration chronosequence. Catena 2022, 213, 106202. [Google Scholar] [CrossRef]
- Curtis, P.S.; Gough, C.M. Forest aging, disturbance and the carbon cycle. New Phytol. 2018, 219, 1188–1193. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Chen, W.M.; Wang, J.L.; Du, L.L.; Li, Q.P.; Wei, G.H. Soil microbiomes with distinct assemblies through vertical soil profiles drive the cycling of multiple nutrients in reforested ecosystems. Microbiome 2018, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.L.; Liu, B.; Ma, Y.; Sun, H. Differences in bacterial community structure and potential functions among Eucalyptus plantations with different ages and species of trees. Appl. Soil Ecol. 2020, 149, 103515. [Google Scholar] [CrossRef]
- Sun, S.; Badgley, B.D. Changes in microbial functional genes within the soil metagenome during forest ecosystem restoration. Soil Biol. Biochem. 2019, 135, 163–172. [Google Scholar] [CrossRef]
- Ding, X.X.; Liu, G.L.; Fu, S.L.; Chen, H.Y.H. Tree species composition and nutrient availability affect soil microbial diversity and composition across forest types in subtropical China. Catena 2021, 201, 105224. [Google Scholar] [CrossRef]
- Kyaschenko, J.; Clemmensen, K.; Hagenbo, A.; Karltun, E.; Lindahl, B.D. Shift in fungal communities and associated enzyme activities along an age gradient of managed Pinus sylvestris stands. ISME J. 2017, 11, 863–874. [Google Scholar] [CrossRef]
- Chen, X.L.; Chen, H.Y.H. Plant mixture balances terrestrial ecosystem C: N: P stoichiometry. Nat. Commu. 2021, 12, 4562. [Google Scholar] [CrossRef] [PubMed]
- Ling, N.; Wang, T.; Kuzyakov, Y. Rhizosphere bacteriome structure and functions. Nat. Communs. 2022, 13, 836. [Google Scholar] [CrossRef] [PubMed]
- Pedrós-Alió, C. The rare bacterial biosphere. Ann. Rev. Mar. Sci. 2012, 4, 449–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jousset, A.; Bienhold, C.; Chaztzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Küsel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F.; et al. Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 2017, 11, 853–862. [Google Scholar] [CrossRef]
- Jones, S.E.; Lennon, J.T. Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 5881–5886. [Google Scholar] [CrossRef] [Green Version]
- Sauret, C.; Séverin, T.; Vétion, G.; Guigue, C.; Goutx, M.; Pujo-Pay, M.; Conan, P.; Fagervold, S.K.; Ghiglione, J.F. ‘Rare biosphere’ bacteria as key phenanthrene degraders in coastal seawaters. Environ. Pollut. 2014, 194, 246–253. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Paszkiewicz, K.; Field, D.; Knight, R.; Gilbert, J.A. The Western English Channel contains a persistent microbial seed bank. ISME J. 2012, 6, 1089–1093. [Google Scholar] [CrossRef] [Green Version]
- Shade, A.; Jones, S.E.; Caporaso, J.G.; Handelsman, J.; Knight, R.; Fierer, N.; Gilbert, J.A. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. MBio 2014, 5, e01371-14. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Wang, J.M.; Wei, G.H.; Chen, W.M.; Lu, Y.H. Dominant role of abundant rather than rare bacterial taxa in maintaining agro-soil microbiomes under environmental disturbances. Chemosphere 2019, 235, 248–259. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef]
- Lynch, M.D.J.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 2015, 13, 217–229. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.M.; Wei, G.H. Linking phylogenetic niche conservatism to soil archaeal biogeography, community assembly and species coexistence. Glob. Ecol. Biogeogr. 2021, 30, 1488–1501. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Maestre, F.T.; Reich, P.B.; Jeffries, T.C.; Gaitan, J.J.; Encinar, D.; Berdugo, M.; Campbell, C.D.; Singh, B.K. Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat. Communs. 2016, 7, 10541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Forestry and Grassland Administration P.R. China (NFGA). China Forest Resources Report (2014–2018); China Forestry Publishing House: Beijing, China, 2019. [Google Scholar]
- Bai, Y.X.; Zhou, Y.C.; He, H.Z. Effects of rehabilitation through afforestation on soil aggregate stability and aggregate-associated carbon after forest fires in subtropical China. Geoderma 2020, 376, 114548. [Google Scholar] [CrossRef]
- Dong, H.Y.; Ge, J.F.; Sun, K.; Wang, B.Z.; Xue, J.M.; Wakelin, S.A.; Wu, J.S.; Sheng, W.X.; Liang, C.F.; Xu, Q.F.; et al. Change in root-associated fungal communities affects soil enzymatic activities during Pinus massoniana forest development in subtropical China. Forest Ecol Manag. 2021, 482, 118817. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, Y.C.; Ren, Q.F. Evolution of the Soil Bacterial Community Structure during the Development of Pinus massoniana Plantations in Subtropical China. Int. J. Agric. Biol. 2019, 22, 73–82. [Google Scholar]
- Bao, S.D. Soil Agricultural Chemistry Analysis, 3rd ed.; Agriculture Press: Beijing, China, 2000; pp. 22–110. (In Chinese) [Google Scholar]
- Beck, T.; Joergensen, R.G.; Kandeler, E.; Makeschin, F.; Nuss, E.; Oberholzer, H.R.; Scheu, S. An inter-laboratory comparison of ten different ways of measuring soil microbial biomass C. Soil Biol. Biochem. 1997, 29, 1023–1032. [Google Scholar] [CrossRef]
- Mori, H.; Maruyama, F.; Kato, H.; Toyoda, A.; Dozono, A.; Ohtsubo, Y.; Nagata, Y.; Fujiyama, A.; Tsuda, M.; Kurokawa, K. Design and experimental application of a novel non-degenerate universal primer set that amplifies prokaryotic 16S rRNA genes with a low possibility to amplify eukaryotic rRNA genes. DNA Res. 2014, 21, 217–227. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Mo, Y.; Zhang, W.; Yang, J.; Lin, Y.; Yu, Z.; Lin, S. Biogeographic patterns of abundant and rare bacterioplankton in three subtropical bays resulting from selective and neutral processes. ISME J. 2018, 12, 2198–2210. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Liu, Y.X.; Huang, L.Q. ImageGP: An easy-to-use data visualization web server for scientific researchers. iMeta 2022, 1, e5. [Google Scholar] [CrossRef]
- Bardgett, R.D.; van der Putten, W.H. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505–511. [Google Scholar] [CrossRef]
- Liu, L.; Yang, J.; Yu, Z.; Wilkinson, D.M. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 2015, 9, 2068–2077. [Google Scholar] [CrossRef] [Green Version]
- Logares, R.; Audic, S.; Bass, D.; Bittner, L.; Boutte, C.; Christen, R.; Claverie, J.M.; Decelle, J.; Dolan, J.R.; Dunthorn, M. Patterns of rare and abundant marine microbial eukaryotes. Curr. Biol. 2014, 24, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Uroz, S.; Buée, M.; Deveau, A.; Mieszkin, S.; Martin, F. Ecology of the forest microbiome: Highlights of temperate and boreal ecosystems. Soil Biol. Biochem. 2016, 103, 471–488. [Google Scholar] [CrossRef]
- Perring, M.P.; Bernhardt-Römermann, M.; Baeten, L.; Midolo, G.; Blondeel, H.; Depauw, L.; Landuyt, D.; Maes, S.L.; Lombaerde, E.D.; Carón, M.M.; et al. Global environmental change effects on plant community composition trajectories depend upon management legacies. Global Chang. Biol. 2018, 24, 1722–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeDuc, S.D.; Rothstein, D.E. Plant-available organic and mineral nitrogen shift in dominance with forest stand age. Ecology 2010, 91, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, B.; Liu, Z. Impacts of plant secondary metabolites from conifer litter on the decomposition of Populus purdomii litter. J. For. Res. 2019, 30, 2237–2245. [Google Scholar] [CrossRef]
- Xu, H.D.; Yu, M.K.; Cheng, X.R. Abundant fungal and rare bacterial taxa jointly reveal soil nutrient cycling and multifunctionality in uneven-aged mixed plantations. Ecol. Indic. 2021, 129, 107923. [Google Scholar] [CrossRef]
- Chomel, M.; Guittonny-Larchevêque, F.C.; Gallet, C.; DesRochers, A.; Paré, D.; Jackson, B.G.; Baldy, V. Plant secondary metabolites: A key driver of litter decomposition and soil nutrient cycling. J. Ecol. 2016, 104, 1527–1541. [Google Scholar] [CrossRef] [Green Version]
- Lucas-Borja, M.E.; Hedoa, J.; Cerdáb, A.; Candel-Pérez, D.; Viñegla, B. Unravelling the importance of forest age stand and forest structure driving microbiological soil properties, enzymatic activities and soil nutrients content in Mediterranean Spanish black pine (Pinus nigra Ar. ssp. salzmannii). Forest. Sci. Total Environ. 2016, 562, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Hagenbo, A.; Kyaschenko, J.; Clemmensen, K.E.; Lindahl, B.D.; Fransson, P. Fungal community shifts underpin declining mycelial production and turnover across a Pinus sylvestris chronosequence. J. Ecol. 2018, 106, 490–501. [Google Scholar] [CrossRef]
- Pan, C.C.; Feng, Q.; Li, Y.L.; Li, Y.Q.; Liu, L.D.; Yu, X.Y.; Ren, S.L. Rare soil bacteria are more responsive in desertification restoration than abundant bacteria. Environ. Sci. Pollut R. 2022, 29, 33323–33334. [Google Scholar] [CrossRef]
- Xu, L.; Cao, H.L.; Li, C.N.; Wang, C.H.; He, N.P.; Hu, S.Y.; Yao, M.J.; Wang, C.T.; Wang, J.M.; Zhou, S.G.; et al. The importance of rare versus abundant phoD- harboring subcommunities in driving soil alkaline phosphatase activity and available P content in Chinese steppe ecosystems. Soil Biol. Biochem. 2022, 164, 108491. [Google Scholar] [CrossRef]
- He, Z.B.; Liu, D.; Shi, Y.; Wu, X.J.; Dai, Y.X.; Shang, Y.W.; Peng, J.J.; Cui, Z.L. Broader environmental adaptation of rare rather than abundant bacteria in reforestation succession soil. Sci. Total Environ. 2022, 828, 154364. [Google Scholar] [CrossRef]
- Bell, C.W.; Asao, S.; Calderon, F.; Wolk, B.; Wallenstein, M.D. Plant nitrogen uptake drives rhizosphere bacterial community assembly during plant growth. Soil Biol. Biochem. 2015, 85, 170–182. [Google Scholar] [CrossRef]
- Zhou, X.L.; Wang, A.; Hobbie, E.A.; Zhu, F.F.; Qu, Y.Y.; Dai, L.M.; Li, D.J.; Liu, X.Y.; Zhu, W.X.; Koba, K.; et al. Mature conifers assimilate nitrate as efficiently as ammonium from soils in four forest plantations. New Phytol. 2021, 229, 3184–3194. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.G.; Binkley, D.; Fownes, J.H. Age-related decline in forest productivity: Pattern and process. In Advances in Ecological Research; Begon, M., Fitter, A.H., Eds.; Academic Press Ltd.: London, UK; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1997; Volume 27, pp. 213–262. [Google Scholar]
- Škerlep, M.; Nehzati, S.; Johansson, U.; Kleja, D.B.; Persson, P.; Kritzberg, E.S. Spruce Forest afforestation leading to increased Fe mobilization from soils. Biogeochemistry 2022, 57, 273–290. [Google Scholar] [CrossRef]
- Gahan, J.; Schmalenberger, A. The role of bacteria and mycorrhiza in plant sulfur supply. Front. Plant Sci. 2014, 5, 723. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.Y.; Yang, T.; Ma, Y.Y.; Zhang, K.P.; Soltis, P.S.; Soltis, D.E.; Gilbert, J.A.; Zhao, Y.P.; Fu, C.X.; Chu, H.Y. Soil pH determines bacterial distribution and assembly processes in natural mountain forests of eastern China. Global Ecol. Biogeogr. 2021, 30, 2164–2177. [Google Scholar] [CrossRef]
- Scarlett, K.; Denman, S.; Clark, D.R.; Forster, J.; Vanguelova, E.; Brown, N.; Whitby, C. Relationships between nitrogen cycling microbial community abundance and composition reveal the indirect effect of soil pH on oak decline. ISME J. 2021, 15, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.M.; Liu, D.Y.; Yin, H.J. Roots regulate microbial N processes to achieve an efficient NH4+ supply in the rhizosphere of alpine coniferous forests. Biogeochemistry 2021, 155, 39–57. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 13-yr | 25-yr | 38-yr | 58-yr | F Value | |
|---|---|---|---|---|---|
| MBC (mg kg−1) | 334.17 ± 12.97 a | 326 ± 11.61 a | 207.94 ± 1.65 b | 253.05 ± 10.82 b | 21.58 ** |
| MBN (mg kg−1) | 73.22 ± 4.5 b | 116.81 ± 5.7 a | 35.54 ± 2.42 c | 33.13 ± 2.98 c | 100 ** |
| Nalkali (mg kg−1) | 135.09 ± 10.92 b | 235.12 ± 21.27 a | 102.79 ± 6.31 b | 136.88 ± 16.93 b | 7.89 ** |
| TP (g kg−1) | 0.42 ± 0.03 a | 0.3 ± 0.01 b | 0.13 ± 0.01 c | 0.12 ± 0.01 c | 60.87 ** |
| AP (mg kg−1) | 2.33 ± 0.2 a | 1.83 ± 0.14 b | 1.79 ± 0.1 b | 2.24 ± 0.05 ab | 4.07 |
| TK (g kg−1) | 2.55 ± 0.12 a | 2.09 ± 0.22 a | 1.72 ± 0.42 a | 1.38 ± 0.32 a | 1.23 |
| AK (mg kg−1) | 54.05 ± 4.48 ab | 68.79 ± 8.25 a | 43.09 ± 5.77 b | 28.32 ± 4.58 b | 5.67 * |
| Nitrite reductase (μg g−1) | 4.99 ± 0.58 b | 11.18 ± 1.04 a | 7.76 ± 2.48 a | 3.98 ± 0.02 b | 26.26 ** |
| Urease (mg g−1) | 0.65 ± 0.09 b | 0.99 ± 0.06 a | 0.13 ± 0.02 c | 0.19 ± 0.04 c | 57.88 ** |
| Invertase (mg g−1) | 28.2 ± 2.2 a | 18.25 ± 1.85 b | 19.64 ± 0.38 b | 23.4 ± 1.08 b | 7.53 * |
| Modularity | Clustering Coefficient | Average Path Length | Network Diameter | Graph Density | Average Degree | |
|---|---|---|---|---|---|---|
| Abundant | 0.41 | 0.81 | 3.17 | 12.09 | 0.05 | 63.81 |
| Rare | 0.58 | 0.61 | 5.85 | 16 | 0.03 | 186.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Q.; Zhou, Y.; Zhao, H.; Bai, Y. Inconsistent Response of Abundant and Rare Bacterial Communities to the Developmental Chronosequence of Pinus massoniana. Forests 2022, 13, 1904. https://doi.org/10.3390/f13111904
Cao Q, Zhou Y, Zhao H, Bai Y. Inconsistent Response of Abundant and Rare Bacterial Communities to the Developmental Chronosequence of Pinus massoniana. Forests. 2022; 13(11):1904. https://doi.org/10.3390/f13111904
Chicago/Turabian StyleCao, Qianbin, Yunchao Zhou, Hui Zhao, and Yunxing Bai. 2022. "Inconsistent Response of Abundant and Rare Bacterial Communities to the Developmental Chronosequence of Pinus massoniana" Forests 13, no. 11: 1904. https://doi.org/10.3390/f13111904