

Comparison of Endophytic and Epiphytic Microbial Communities in Surviving and Dead Korean Fir (Abies koreana) Using Metagenomic Sequencing
 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Tree Sampling
2.2. DNA Extraction and Next-Generation Sequencing of 16S rRNA Genes and ITS Sequences
2.3. Diversity Analysis and Functional Prediction of 16S rRNA Genes and ITS Sequences
2.4. Statistical Analysis
3. Results
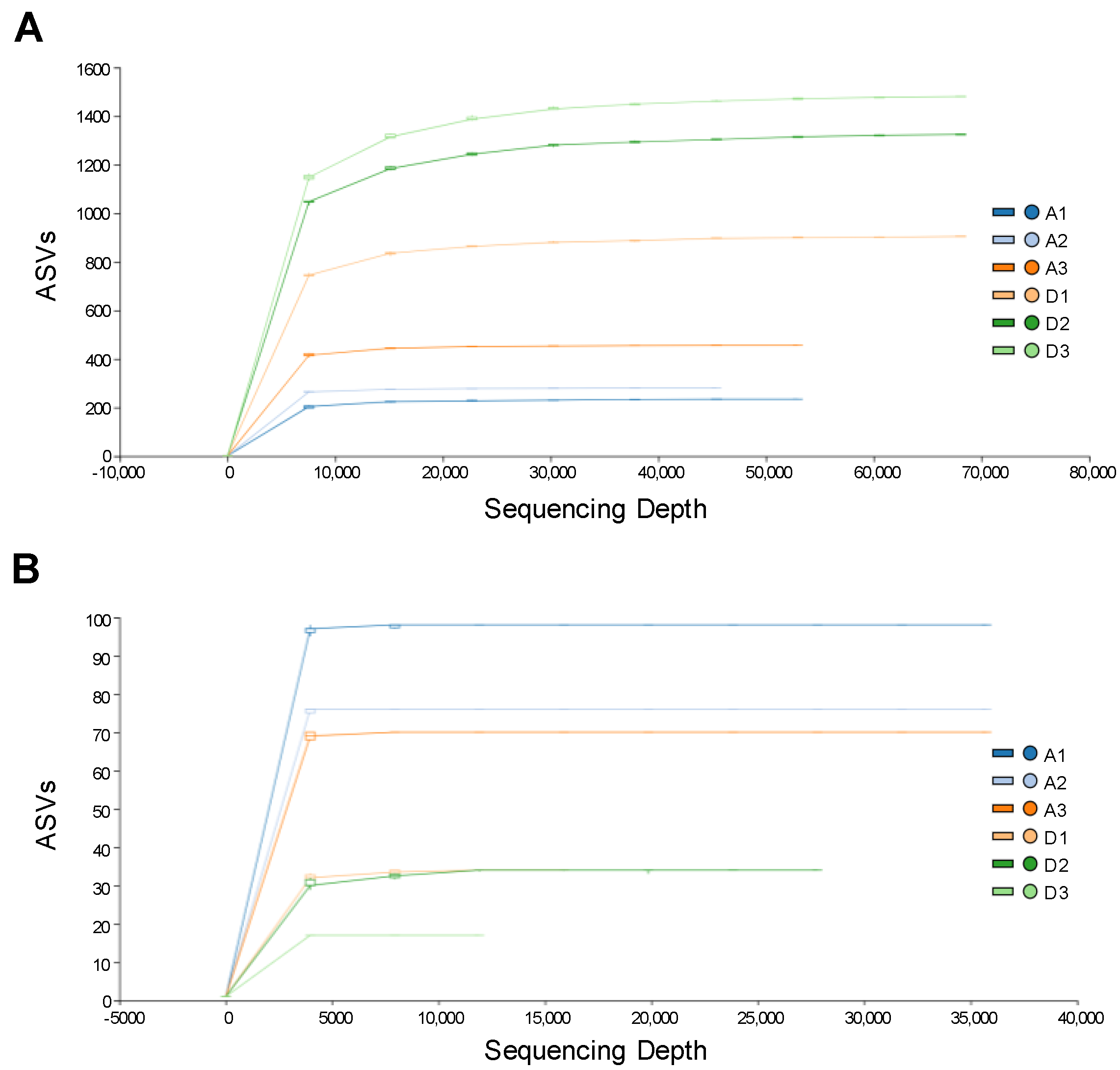
3.1. 16S rRNA and ITS Metagenomic Sequencing Analyses
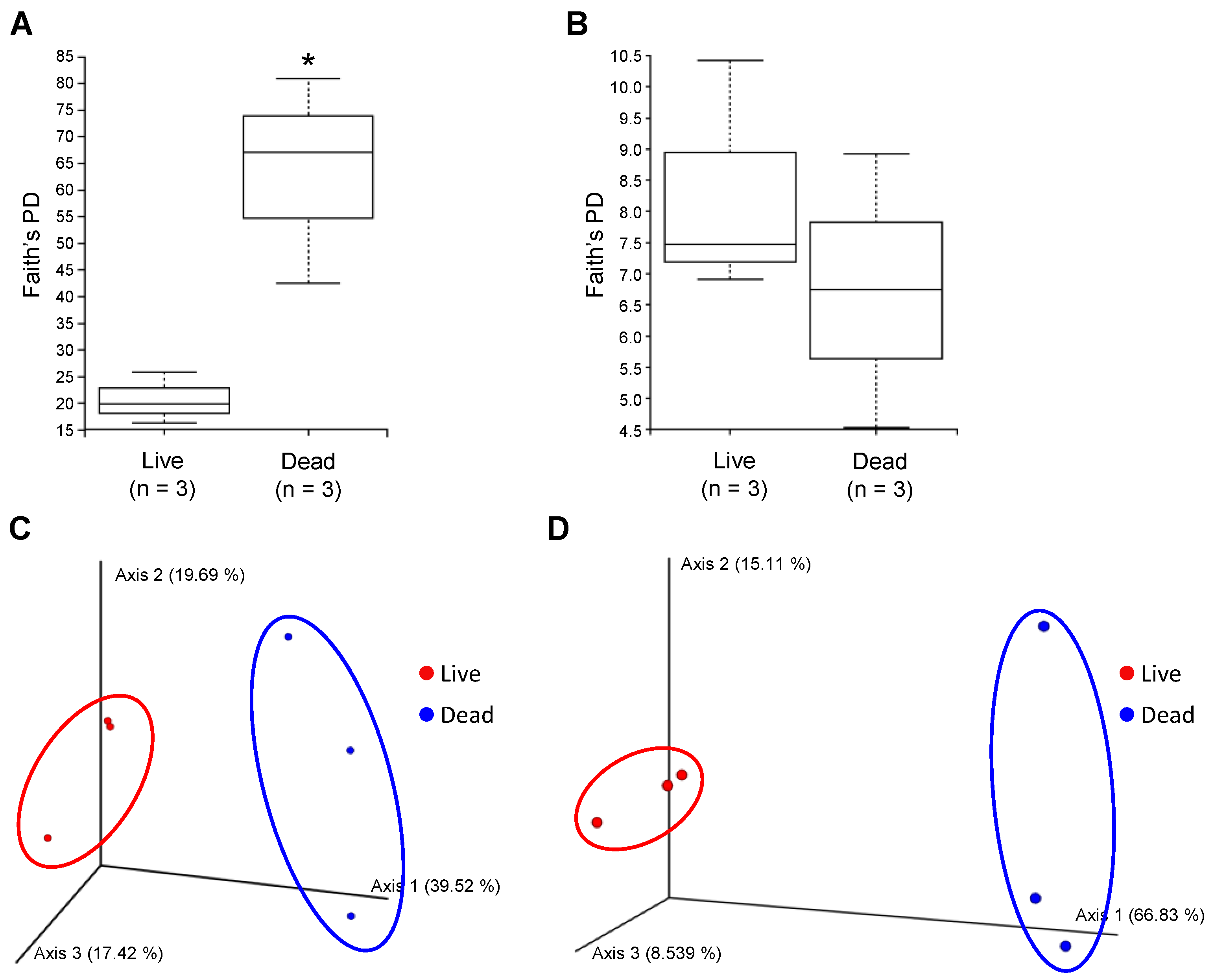
3.2. Diversity of Endophytic and Epiphytic Communities in Live and Dead Korean Fir Trees
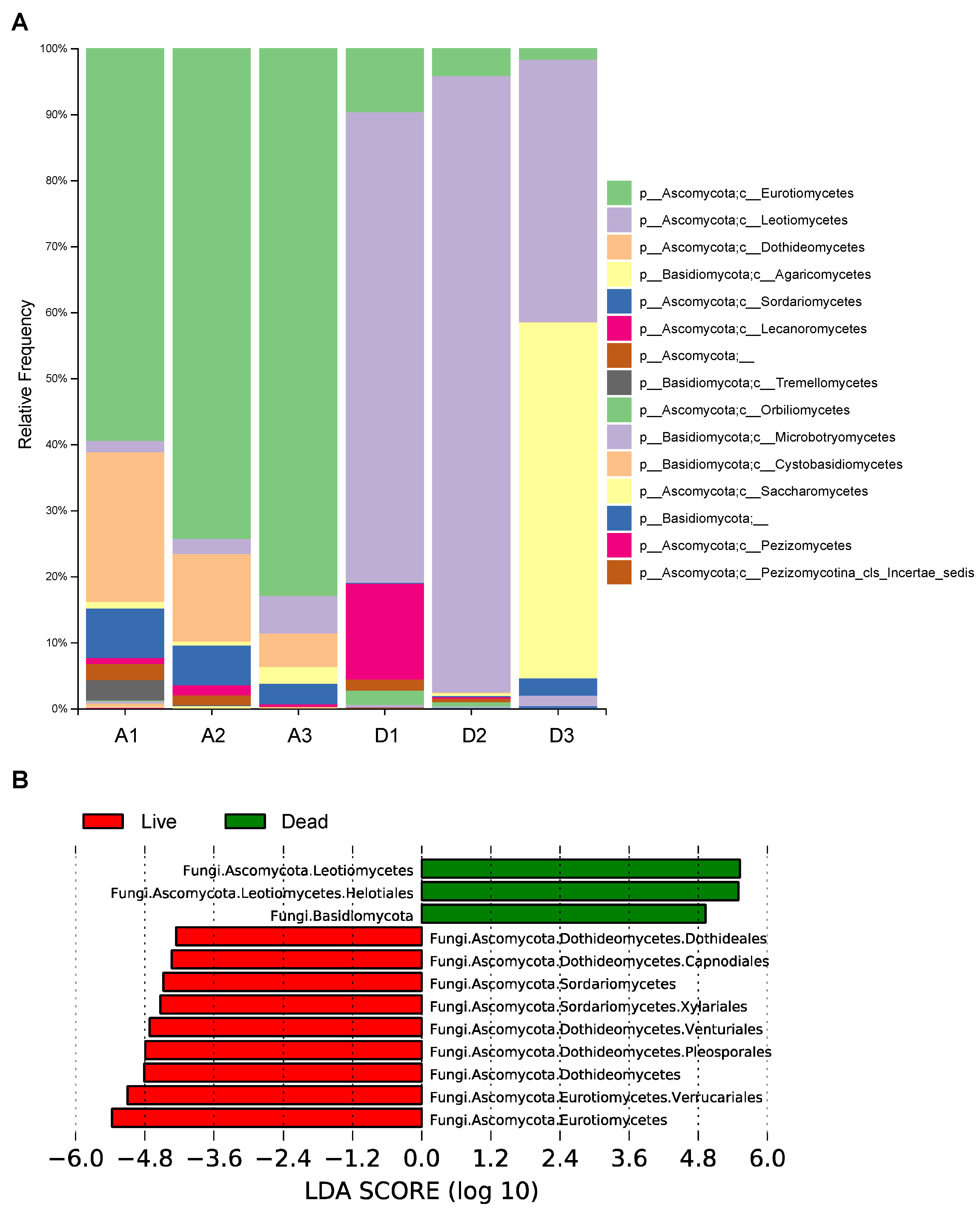
3.3. Taxonomic Profiling and Comparative Analysis of the Bacterial Communities between Live and Dead Korean Fir Trees
3.4. The Functional Inference of Bacterial Communities between Live and Dead Korean Fir Trees
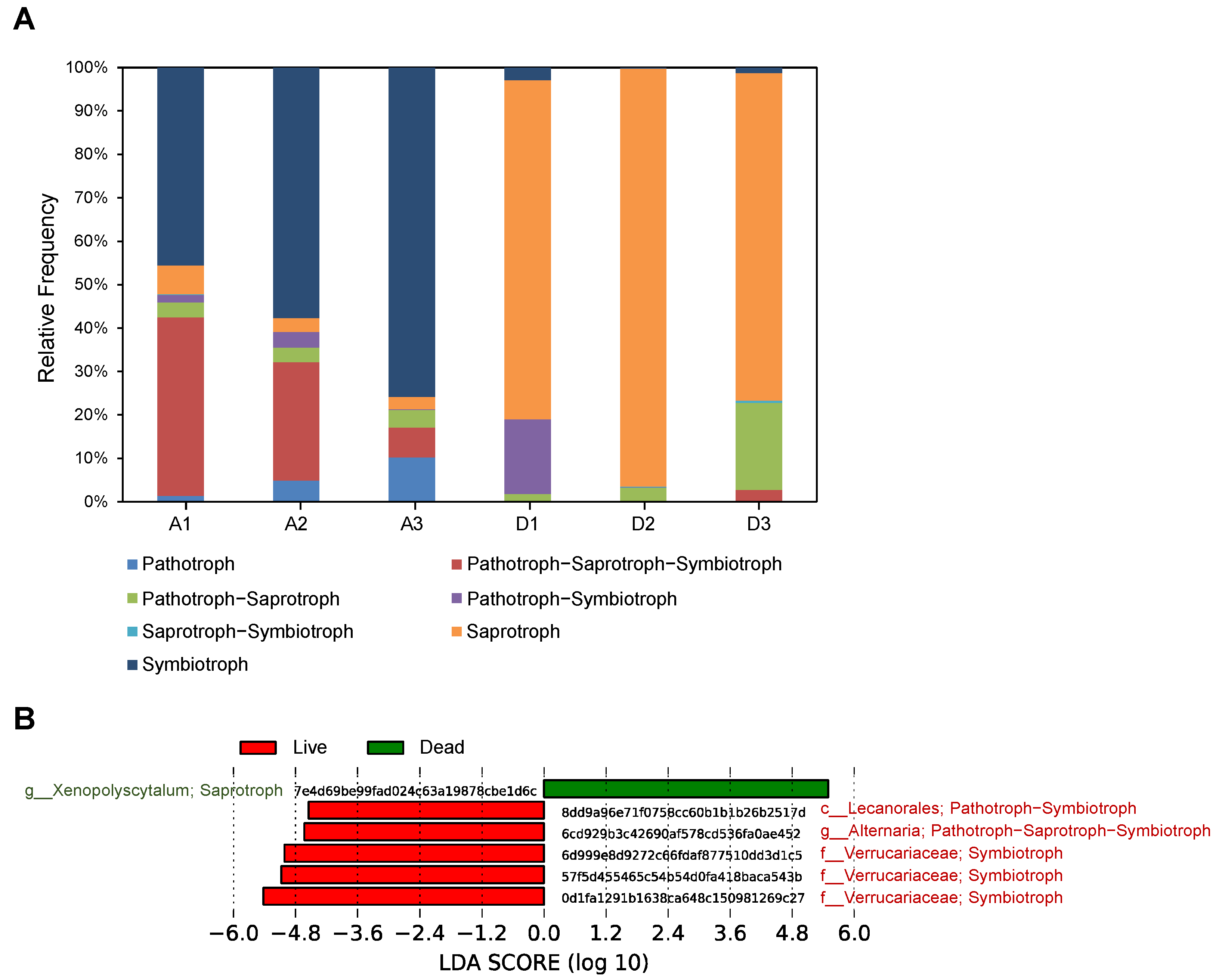
3.5. Taxonomic Profiling and Comparative Analysis of the Fungal Communities between Live and Dead Korean Fir Trees
3.6. The Functional Prediction of Fungal Communities between Live and Dead Korean Fir Trees
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kwak, M.; Hong, J.-K.; Park, J.H.; Lee, B.Y.; Suh, M.H.; Kim, C.S. Genetic assessment of Abies koreana (Pinaceae), the endangered Korean fir, and conservation implications. Conserv. Genet. 2017, 18, 1165–1176. [Google Scholar] [CrossRef]
- Nzokou, P.; Leefers, L.A. Costs and Returns in Michigan Christmas Tree Production, 2006; Michigan State University Extension: East Lansing, MI, USA, 2007. [Google Scholar]
- Kim, H.J.; Choi, E.H.; Lee, I.-S. Two lanostane triterpenoids from Abies koreana. Phytochemistry 2004, 65, 2545–2549. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Bu, Y.; Jeong, S.; Lim, J.; Kwon, Y.; Cha, D.S.; Kim, J.; Jeon, S.; Eun, J.; Jeon, H. Memory-Enhancing Effect of a Supercritical Carbon Dioxide Fluid Extract of the Needles of Abies koreana on Scopolamine-Induced Amnesia in Mice. Biosci. Biotechnol. Biochem. 2006, 70, 1821–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Le, Q.K.; Lee, M.H.; Kim, T.S.; Lee, H.-K.; Kim, Y.H.; Bae, K.; Lee, I.-S. A cytotoxic secocycloartenoid fromAbies koreana. Arch. Pharmacal Res. 2001, 24, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chang, C.; Kim, C.; Gardner, M. Abies Koreana. The IUCN Red List of Threatened Species 2011:E. T31244A9618913; IUCN: Gland, Switzerland, 2011. [Google Scholar]
- Koo, K.A.; Kim, D.-B. Review Forty-year Studies of Korean fir(Abies koreana Wilson). Korean J. Environ. Ecol. 2020, 34, 358–371. [Google Scholar] [CrossRef]
- Kim, N.-S.; Lee, H.-C. A study on changes and distributions of Korean fir in sub-alpine zone. J. Korean Soc. Environ. Restor. Technol. 2013, 16, 49–57. [Google Scholar] [CrossRef]
- Woo, S.Y.; Lim, J.-H.; Lee, D.K. Effects of Temperature on Photosynthetic Rates in Korean Fir (Abies koreana) between Healthy and Dieback Population. J. Integr. Plant Biol. 2008, 50, 190–193. [Google Scholar] [CrossRef]
- Kim, D.W.; Jeong, D.Y.; Park, H.C. Identification of Molecular Markers for Population Diagnosis of Korean Fir (Abies koreana) Vulnerable to Climate Change. Proc. Natl. Inst. Ecol. Repub. Korea 2020, 1, 68–73. [Google Scholar]
- Seo, J.-W.; Choi, E.-B.; Park, J.-H.; Kim, Y.-J.; Lim, H.-I. The Role of Aging and Wind in Inducing Death and/or Growth Reduction in Korean Fir (Abies Koreana Wilson) on Mt. Halla, Korea. Atmosphere 2021, 12, 1135. [Google Scholar] [CrossRef]
- Ahn, U.S.; Yun, Y.S. Causes of Decline in the Korean Fir Based on Spatial Distribution in the Mt. Halla Region in Korea: A Meta-Analysis. Forests 2020, 11, 391. [Google Scholar] [CrossRef] [Green Version]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Meaden, S.; Crowther, W.J.; Leimu, R.; Metcalf, C.J.E. A signature of tree health? Shifts in the microbiome and the ecological drivers of horse chestnut bleeding canker disease. New Phytol. 2017, 215, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganley, R.J.; Sniezko, R.A.; Newcombe, G. Endophyte-mediated resistance against white pine blister rust in Pinus monticola. For. Ecol. Manag. 2008, 255, 2751–2760. [Google Scholar] [CrossRef]
- Kim, C.S.; Jo, J.W.; Lee, H.; Kwag, Y.-N.; Cho, S.E.; Oh, S.H. Comparison of Soil Higher Fungal Communities between Dead and Living Abies koreana in Mt. Halla, the Republic of Korea. Mycobiology 2020, 48, 364–372. [Google Scholar] [CrossRef]
- Han, G.; Mannaa, M.; Jeon, H.; Jung, H.; Kim, J.-C.; Park, A.R.; Seo, Y.-S. Dysbiosis in the Rhizosphere Microbiome of Standing Dead Korean Fir (Abies koreana). Plants 2022, 11, 990. [Google Scholar] [CrossRef]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, e1. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guide Methods Appl. 1990, 18, 315–322. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2018, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Barbera, P.; Kozlov, A.M.; Czech, L.; Morel, B.; Darriba, D.; Flouri, T.; Stamatakis, A. EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences. Syst. Biol. 2018, 68, 365–369. [Google Scholar] [CrossRef]
- Louca, S.; Doebeli, M. Efficient comparative phylogenetics on large trees. Bioinformatics 2017, 34, 1053–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Doak, T.G. A Parsimony Approach to Biological Pathway Reconstruction/Inference for Genomes and Metagenomes. PLoS Comput. Biol. 2009, 5, e1000465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2013, 42, D459–D471. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Pii, Y.; Borruso, L.; Brusetti, L.; Crecchio, C.; Cesco, S.; Mimmo, T. The interaction between iron nutrition, plant species and soil type shapes the rhizosphere microbiome. Plant Physiol. Biochem. 2016, 99, 39–48. [Google Scholar] [CrossRef]
- Compant, S.; Saikkonen, K.; Mitter, B.; Campisano, A.; Mercado-Blanco, J. Editorial special issue: Soil, plants and endophytes. Plant Soil 2016, 405, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yang, H.; Zhang, T.; Sun, J.; Lou, K. Illumina-based analysis of endophytic bacterial diversity and space-time dynamics in sugar beet on the north slope of Tianshan mountain. Appl. Microbiol. Biotechnol. 2014, 98, 6375–6385. [Google Scholar] [CrossRef]
- Peñuelas, J.; Rico, L.; Ogaya, R.; Jump, A.S.; Terradas, J. Summer season and long-term drought increase the richness of bacteria and fungi in the foliar phyllosphere of Quercus ilex in a mixed Mediterranean forest. Plant Biol. 2012, 14, 565–575. [Google Scholar] [CrossRef]
- Schmidt, C.S.; Mrnka, L.; Lovecká, P.; Frantík, T.; Fenclová, M.; Demnerová, K.; Vosátka, M. Bacterial and fungal endophyte communities in healthy and diseased oilseed rape and their potential for biocontrol of Sclerotinia and Phoma disease. Sci. Rep. 2021, 11, 3810. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wei, Z.; Friman, V.-P.; Gu, S.-H.; Wang, X.-F.; Eisenhauer, N.; Yang, T.-J.; Ma, J.; Shen, Q.-R.; Xu, Y.-C.; et al. Probiotic Diversity Enhances Rhizosphere Microbiome Function and Plant Disease Suppression. mBio 2016, 7, e01790-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miliute, I.; Buzaite, O.; Baniulis, D.; Stanys, V. Bacterial endophytes in agricultural crops and their role in stress tolerance: A review. Zemdirb. Agric. 2015, 102, 465–478. [Google Scholar] [CrossRef]
- Santoyo, G.; Moreno-Hagelsieb, G.; Orozco-Mosqueda, M.D.C.; Glick, B.R. Plant growth-promoting bacterial endophytes. Microbiol. Res. 2016, 183, 92–99. [Google Scholar] [CrossRef]
- Tláskal, V.; Zrůstová, P.; Vrška, T.; Baldrian, P. Bacteria associated with decomposing dead wood in a natural temperate forest. FEMS Microbiol. Ecol. 2017, 93, fix157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, J.; Kellner, H.; Leonhardt, S.; Stengel, E.; Dahl, A.; Bässler, C.; Buscot, F.; Hofrichter, M.; Hoppe, B. Bacteria inhabiting deadwood of 13 tree species are heterogeneously distributed between sapwood and heartwood. Environ. Microbiol. 2018, 20, 3744–3756. [Google Scholar] [CrossRef] [Green Version]
- Garcia, R.; Müller, R. The Family Polyangiaceae. In The Prokaryotes: Deltaproteobacteria and Epsilonproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 247–279. [Google Scholar]
- Lee, K.C.Y.; Dunfield, P.F.; Stott, M.B. The Phylum Armatimonadetes. In The Prokaryotes: Other Major Lineages of Bacteria and The Archaea; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 447–458. [Google Scholar]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef]
- Lim, J.-H.; Woo, S.-Y.; Kwon, M.J.; Kim, Y.K. Antioxidant enzyme activities and soil properties of healthy and declining Abies koreana (Wils.) in Mt. Halla. J. Korean Soc. For. Sci. 2007, 96, 14–20. [Google Scholar]
- Palviainen, M.; Laiho, R.; Mäkinen, H.; Finér, L. Do decomposing Scots pine, Norway spruce, and silver birch stems retain nitrogen? Can. J. For. Res. 2008, 38, 3047–3055. [Google Scholar] [CrossRef]
- Rinne, K.T.; Rajala, T.; Peltoniemi, K.; Chen, J.; Smolander, A.; Mäkipää, R. Accumulation rates and sources of external nitrogen in decaying wood in a Norway spruce dominated forest. Funct. Ecol. 2017, 31, 530–541. [Google Scholar] [CrossRef]
- Terhonen, E.; Blumenstein, K.; Kovalchuk, A.; Asiegbu, F.O. Forest Tree Microbiomes and Associated Fungal Endophytes: Functional Roles and Impact on Forest Health. Forests 2019, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Kellogg, J.J.; Raja, H.A. Endolichenic fungi: A new source of rich bioactive secondary metabolites on the horizon. Phytochem. Rev. 2017, 16, 271–293. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, N.B.; Matthiesen, H.; Blanchette, R.A.; Alfredsen, G.; Held, B.W.; Westergaard-Nielsen, A.; Hollesen, J. Fungal attack on archaeological wooden artefacts in the Arctic—Implications in a changing climate. Sci. Rep. 2020, 10, 14577. [Google Scholar] [CrossRef] [PubMed]
- Kwaśna, H.; Szewczyk, W.; Baranowska, M.; Gallas, E.; Wiśniewska, M.; Behnke-Borowczyk, J. Mycobiota Associated with the Vascular Wilt of Poplar. Plants 2021, 10, 892. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, B.Y.; Lee, S.; Kim, J.; Park, H.; Kim, J.-H.; Kim, M.; Park, S.-J.; Kim, K.-T.; Ryu, H.; Shim, D. Comparison of Endophytic and Epiphytic Microbial Communities in Surviving and Dead Korean Fir (Abies koreana) Using Metagenomic Sequencing. Forests 2022, 13, 1932. https://doi.org/10.3390/f13111932
Choi BY, Lee S, Kim J, Park H, Kim J-H, Kim M, Park S-J, Kim K-T, Ryu H, Shim D. Comparison of Endophytic and Epiphytic Microbial Communities in Surviving and Dead Korean Fir (Abies koreana) Using Metagenomic Sequencing. Forests. 2022; 13(11):1932. https://doi.org/10.3390/f13111932
Chicago/Turabian StyleChoi, Bae Young, Suhyeon Lee, Jaewook Kim, Hyeonseon Park, Joon-Hyeok Kim, Minji Kim, Soo-Je Park, Ki-Tae Kim, Hojin Ryu, and Donghwan Shim. 2022. "Comparison of Endophytic and Epiphytic Microbial Communities in Surviving and Dead Korean Fir (Abies koreana) Using Metagenomic Sequencing" Forests 13, no. 11: 1932. https://doi.org/10.3390/f13111932