
Epigenetic and Genetic Variability in Contrasting Latitudinal Fagus sylvatica L. Provenances
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Experimental Layout
2.2. Watering Treatment and Water Potential Measurements Layout
2.3. DNA Extraction
2.4. MSAP Analysis and Scoring
2.5. AFLP Analysis and Scoring
2.6. Statistical Analysis
3. Results
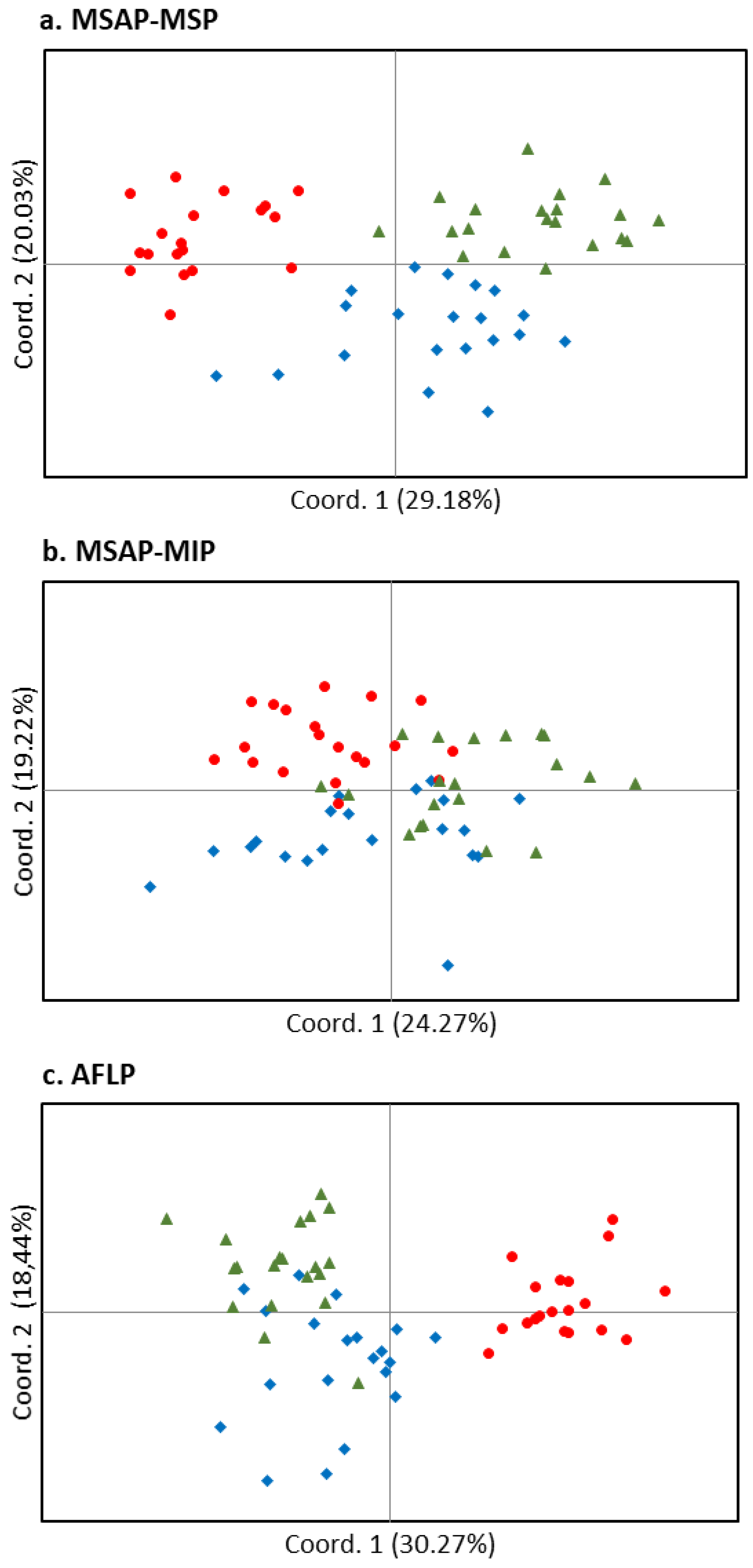
3.1. Epigenetic and Genetic Variability among Provenances
3.1.1. Epigenetic Variability
3.1.2. Genetic Variability
3.1.3. Mantel Test
3.2. Water Stress Response
3.2.1. Water Status of Seedlings
3.2.2. Epigenetic Analysis
3.2.3. Genetic Variation
3.2.4. Correlation between Genetic and Epigenetic Variability
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Douville, H.; Raghavan, K.; Renwick, J.; Allan, R.P.; Arias, P.A.; Barlow, M.; Cerezo-Mota, R.; Cherchi, A.; Gan, T.Y.; Gergis, J.; et al. 2021: Water Cycle Changes. In Climate Change 2021: The Physical Science Basis; Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S.L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M.I., et al., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2021; pp. 1055–1210. [Google Scholar] [CrossRef]
- IPCC. 2022: Climate Change 2022: Impacts, Adaptation and Vulnerability; Pörtner, H.O., Roberts, D.C., Tignor, M., Poloczanska, E.S., Mintenbeck, K., Alegría, A., Craig, M., Langsdorf, S., Löschke, S., Möller, V., et al., Eds.; Contribution of Working Group II to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2022; p. 3056. [Google Scholar] [CrossRef]
- Peñuelas, J.; Boada, M. A global change-induced biome shift in the Montseny mountains (NE Spain). Glob. Chang. Biol. 2003, 9, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Jump, A.S.; Hunt, J.M.; Peñuelas, J. Rapid climate change-related growth decline at the southern range edge of Fagus sylvatica. Glob. Chang. Biol. 2006, 12, 2163–2174. [Google Scholar] [CrossRef] [Green Version]
- Peñuelas, J.; Ogaya, R.; Boada, M.; Jump, A.S. Migration, invasion and decline: Changes in recruitment and forest structure in a warming-linked shift of European beech forest in Catalonia (NE Spain). Ecography 2007, 30, 829–838. [Google Scholar] [CrossRef]
- Rabasa, S.G.; Granda, E.; Benavides, R.; Kunstler, G.; Espelta, J.M.; Ogaya, R.; Peñuelas, J.; Scherer-Lorenzen, M.; Gil, W.; Grodzki, W.; et al. Disparity in elevational shifts of European trees in response to recent climate warming. Glob. Chang. Biol. 2013, 19, 2490–2499. [Google Scholar] [CrossRef]
- Shaw, R.G.; Etterson, J.R. Rapid climate change and the rate of adaptation: Insight from experimental quantitative genetics. New Phytol. 2012, 195, 752–765. [Google Scholar] [CrossRef]
- Alberto, F.J.; Aitken, S.N.; Alía, R.; González-Martínez, S.G.; Hänninenk, H.; Kremer, A.; Lefèvre, F.; Lenormand, T.; Yeaman, S.; Whetten, R.; et al. Potential for evolutionary responses to climate change—Evidence from tree populations. Glob. Chang. Biol. 2013, 19, 1645–1661. [Google Scholar] [CrossRef] [Green Version]
- Borthakur, D.; Busov, V.; Cao, H.; Du, Q.; Gailing, O.; Isik, F.; Ko, J.H.; Li, C.; Li, Q.; Niu, S.; et al. Current status and trends in forest genomics. For. Res. 2022, 2, 11. [Google Scholar] [CrossRef]
- Balao, F.; Paun, O.; Alonso, C. Uncovering the contribution of epigenetics to plant phenotypic variation in Mediterranean ecosystems. Plant Biol. 2018, 20, 38–49. [Google Scholar] [CrossRef]
- Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. A role for epigenetic regulation in the adaptation and stress responses of non-model plants. Front. Plant Sci. 2019, 10, 246. [Google Scholar] [CrossRef] [Green Version]
- Nicotra, A.B.; Atkin, O.K.; Bonser, S.P.; Davidson, A.M.; Finnegan, E.J.; Mathesius, U.; Poot, P.; Purugganan, M.D.; Richards, C.L.; Valladares, F.; et al. Plant phenotypic plasticity in a changing climate. Trends Plant Sci. 2010, 15, 684–692. [Google Scholar] [CrossRef]
- Bräutigam, K.; Vining, K.J.; Lafon-Placette, C.; Fossdal, C.G.; Mirouze, M.; Marcos, J.G.; Fluch, S.; Fernández Fraga, M.; Guevara, M.A.; Abarca, D.; et al. Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 2013, 3, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.; Ribeyre, Z.; Vigneaud, J.; Sow, M.D.; Fichot, R.; Messier, C.; Pinto, G.; Nolet, P.; Maury, S. Advances and promises of epigenetics for forest trees. Forests 2020, 11, 976. [Google Scholar] [CrossRef]
- Sahu, P.P.; Pandey, G.; Sharma, N.; Puranik, S.; Muthamilarasan, M.; Prasad, M. Epigenetic mechanisms of plant stress responses and adaptation. Plant Cell Rep. 2013, 32, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Arora, H.; Singh, R.K.; Sharma, S.; Sharma, N.; Panchal, A.; Das, T.; Prasad, A.; Prasad, M. DNA methylation dynamics in response to abiotic and pathogen stress in plants. Plant Cell Rep. 2022, 41, 1931–1944. [Google Scholar] [CrossRef]
- Wu, W.Q.; Yi, M.R.; Wang, X.F.; Ma, L.L.; Jiang, L.; Li, X.W.; Xiao, H.X.; Sun, M.Z.; Li, L.F.; Bao, L. Genetic and epigenetic differentiation between natural Betula ermanii (Betulaceae) populations inhabiting contrasting habitats. Tree Genet. Genomes 2013, 9, 1321–1328. [Google Scholar] [CrossRef]
- Klupczynska, E.A.; Ratajczak, E. Can forest trees cope with climate change?—Effects of DNA methylation on gene expression and adaptation to environmental change. Int. J. Mol. Sci. 2021, 22, 13524. [Google Scholar] [CrossRef]
- Bossdorf, O.; Richards, C.L.; Pigliucci, M. Epigenetics for ecologists. Ecol. Let. 2008, 1, 106–115. [Google Scholar] [CrossRef]
- Richards, E.J. Natural epigenetic variation in plant species: A view from the field. Curr. Opin. Plant Biol. 2011, 14, 204–209. [Google Scholar] [CrossRef]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Oberkofler, V.; Pratx, L.; Bäurle, I. Epigenetic regulation of abiotic stress memory: Maintaining the good things while they last. Curr. Opin. Plant Biol. 2021, 61, 102007. [Google Scholar] [CrossRef]
- Goodrich, J.; Puangsomlee, P.; Martin, M.; Long, D.; Meyerowitz, E.M.; Coupland, G. A Polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature 1997, 386, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Bouyer, D.; Roudier, F.; Heese, M.; Andersen, E.D.; Gey, D.; Nowack, M.K.; Goodrich, J.; Renou, J.-P.; Grini, P.E.; Colot, V.; et al. Polycomb repressive complex 2 controls the embryo-to-seedling phase transition. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, M.; Iwase, A.; Sugimoto, K. Control of plant cell differentiation by histone modification and DNA methylation. Curr. Opin. Plant Biol. 2015, 28, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.; Schrey, A.W.; Pigliucci, M. Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecol. Lett. 2012, 15, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.; Pigliucci, M. Epigenetic Inheritance. A Decade into the Extended Evolutionary Synthesis. Paradigmi 2020, 38, 463–494. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Fischer, M.; Colot, V.; Bossdorf, O. Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytol. 2013, 197, 314–322. [Google Scholar] [CrossRef]
- Herrera, C.M.; Bazaga, P. Epigenetic differentiation and relationship to adaptive genetic divergence in discrete populations of the violet Viola cazorlensis. New Phytol. 2010, 187, 867–876. [Google Scholar] [CrossRef]
- Herrera, C.M.; Bazaga, P. Epigenetic correlates of plant phenotypic plasticity: DNA methylation differs between prickly and non-prickly leaves in heterophyllous Ilex aquifolium (Aquifoliaceae) trees. J. Linn. Soc. Bot. 2013, 171, 441–452. [Google Scholar] [CrossRef]
- Herrera, C.M.; Medrano, M.; Bazaga, P. Comparative epigenetic and genetic spatial structure of the perennial herb Helleborus foetidus: Isolation by environment, isolation by distance, and functional trait divergence. Am. J. Bot. 2017, 104, 1195–1204. [Google Scholar] [CrossRef] [Green Version]
- Thumma, B.R.; Matheson, B.A.; Zhang, D.; Meeske, C.; Meder, R.; Downes, G.M.; Southerton, S.G. Identification of a Cis-acting regulatory polymorphism in a Eucalypt COBRA-like gene affecting cellulose content. Genetics 2009, 183, 1153–1164. [Google Scholar] [CrossRef] [Green Version]
- Bender, J. DNA methylation and epigenetics. Annu. Rev. Plant Biol. 2004, 55, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hrivnák, M.; Krajmerova, D.; Frýdl, J.; Gömöry, D. Variation of cytosine methylation patterns in European beech (Fagus sylvatica L.). Tree Genet. Genomes. 2017, 13, 117. [Google Scholar] [CrossRef]
- Inácio, V.; Barros, P.M.; Costa, A.; Roussado, C.; Goncalves, E.; Costa, R.; Graca, J.; Oliveira, M.M.; Morais-Cecílio, L. Differential DNA methylation patterns are related to phellogen origin and quality of Quercus suber cork. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.S.; Qin, Q.; Sun, F.; Wang, Y.X.; Xu, D.D.; Li, Z.K.; Fu, B.Y. Genome-wide differences in DNA methylation changes in two contrasting rice genotypes in response to drought conditions. Front. Plant Sci. 2016, 7, 1675. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xu, J.; Pu, X.M.; Lv, H.Z.; Liu, Y.J.; Ma, H.L.; Wu, F.K.; Wang, Q.J.; Feng, X.J.; Liu, T.H.; et al. Maize DNA methylation in response to drought stress is involved in target gene expression and alternative splicing. Int. J. Mol. Sci. 2021, 22, 8285. [Google Scholar] [CrossRef]
- Ackah, M.; Guo, L.; Li, S.; Jin, X.; Asakiya, C.; Aboagye, E.T.; Yuan, F.; Wu, M.; Essoh, L.G.; Adjibolosoo, D.; et al. DNA methylation changes and its associated genes in mulberry (Morus alba L.) Yu-711 response to drought stress using MethylRAD sequencing. Plants 2022, 11, 190. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef]
- Neves, D.M.; Almeida, L.A.D.H.; Santana-Vieira, D.D.S.; Freschi, L.; Ferreira, C.F.; Soares Filho, W.D.S.; Costa, M.G.C.; Micheli, F.; Coelho Filho, M.A.; Gesteira, A.D.S. Recurrent water deficit causes epigenetic and hormonal changes in citrus plants. Sci. Rep. 2017, 7, 13684. [Google Scholar] [CrossRef] [Green Version]
- Correia, B.; Valledor, L.; Hancock, R.D.; Jesus, C.; Amaral, J.; Meijon, M.; Pinto, G. Depicting how Eucalyptus globulus survives drought: Involvement of redox and DNA methylation events. Funct. Plant Biol. 2016, 43, 838–850. [Google Scholar] [CrossRef] [Green Version]
- Gourcilleau, D.; Bogeat-Triboulot, M.B.; Le Thiec, D.; Lafon-Placette, C.; Delaunay, A.; El-Soud, W.A. DNA methylation and histone acetylation: Genotypic variations in hybrid poplars, impact of water deficit and relationships with productivity. Ann. For. Sci. 2010, 67, 2009101. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Zhang, Z.; Wu, H.; Huang, C.; Shuai, P.; Ye, C.-Y.; Tang, S.; Wang, Y.; Yang, L.; Wang, J.; et al. Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014, 15, S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafon-Placette, C.; Le Gac, A.L.; Chauveau, D.; Segura, V.; Delaunay, A.; Lesage-Descauses, M.C.; Hummel, I.; Cohen, D.; Jesson, B.; Le Thiec, D.; et al. Changes in the epigenome and transcriptome of the poplar shoot apical meristem in response to water availability affect preferentially hormone pathways. J. Exp. Bot. 2018, 69, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Sow, M.D.; Le Gac, A.L.; Fichot, R.; Lanciano, S.; Delaunay, A.; Le Jan, I.; Lesage-Descauses, M.C.; Citerne, S.; Caius, J.; Brunaud, V.; et al. RNAi suppression of DNA methylation affects the drought stress response and genome integrity in transgenic poplar. New Phytol. 2021, 232, 80–97. [Google Scholar] [CrossRef]
- Raj, S.; Bräutigam, K.; Hamanishi, E.T.; Wilkins, O.; Thomas, B.R.; Schroeder, W.; Mansfield, S.D.; Plant, A.L.; Campbell, M.M. Clone history shapes Populus drought responses. Proc. Natl Acad. Sci. USA 2011, 108, 12521–12526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sow, M.D.; Segura, V.; Chamaillard, S.; Jorge, V.; Delaunay, A.; Lafon-Placette, C.; Fichot, R.; Faivre-Rampant, P.; Villar, M.; Brignolas, F.; et al. Narrow-sense heritability and pST estimates of DNA methylation in three Populus nigra L. populations under contrasting water availability. Tree Genet. Genomes 2018, 14, 78. [Google Scholar] [CrossRef]
- Backes, K.; Leuschner, C. Leaf water relations of competitive Fagus sylvatica and Quercus petraea trees during 4 years differing in soil drought. Can. J. For. Res. 2000, 30, 335–346. [Google Scholar] [CrossRef]
- Aranda, I.; Gil, L.; Pardos, J.A. Seasonal changes in apparent hydraulic conductance and their implications for water use of European beech (Fagus sylvatica L.) and sessile oak [Quercus petraea (Matt.) Liebl] in South Europe. Plant Ecol. 2005, 179, 155–167. [Google Scholar] [CrossRef]
- Cano, F.J.; Sánchez-Gómez, D.; Rodríguez-Calcerrada, J.; Warren, C.R.; Gil, L. Aranda, I. Effects of drought on mesophyll conductance and photosynthetic limitations at different tree canopy layers. Plant Cell Environ. 2013, 36, 1961–1980. [Google Scholar] [CrossRef]
- Leuzinger, S.; Zotz, G.; Asshoff, R.; Körner, C. Responses of deciduous forest trees to severe drought in Central Europe. Tree Physiol. 2005, 25, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Ciais, P.; Reichstein, M.; Viovy, N.; Granier, A.; Ogee, J.; Allard, V.; Aubinet, M.; Buchmann, N.; Bernhofer, C.; Carrara, A.; et al. Europe-wide reduction in primary productivity caused by the heat and drought in 2003. Nature 2005, 437, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Psomas, A.; Ginzler, C.; Thuiller, W.; Zappa, M.; Zimmermann, N.E. Large scale early-wilting response of central European forests to the 2018 extreme drought. Glob. Change Biol. 2020, 26, 7021–7035. [Google Scholar] [CrossRef] [PubMed]
- Magri, D.; Vendramin, G.G.; Comps, B.; Dupanloup, I.; Geburek, T.; Gömöry, D.; Latałowa, M.; Litt, T.; Paule, L.; Roure, J.M.; et al. A new scenario for the quaternary history of European beech populations: Palaeobotanical evidence and genetic consequences. New Phytol. 2006, 171, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Hampe, A.; Petit, R. Conserving biodiversity under climate change: The rear edge matters. Ecol. lett. 2005, 8, 461–467. [Google Scholar] [CrossRef] [Green Version]
- De Lafontaine, G.; Ducousso, A.; Lefevre, S.; Magnanou, E.; Petit, R.J. Stronger spatial genetic structure in recolonized areas than in refugia in the European beech. Mol. Ecol. 2013, 22, 4397–4412. [Google Scholar] [CrossRef]
- Postolache, D.; Oddou-Muratorio, S.; Vajana, E.; Bagnoli, F.; Guichoux, E.; Hampe, A.; Le Provost, G.; Lesur, I.; Popescu, F.; Scotti, I.; et al. Genetic signatures of divergent selection in European beech (Fagus sylvatica L.) are associated with the variation in temperature and precipitation across its distribution range. Mol. Ecol. 2021, 30, 5029–5047. [Google Scholar] [CrossRef]
- Sánchez-Gómez, D.; Robson, T.M.; Gasco, A.; Gil-Pelegrín, E.; Aranda, I. Differences in the leaf functional traits of six beech (Fagus sylvatica L.) populations are reflected in their response to water limitation. Environ. Exp. Bot. 2013, 87, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Reyna-López, G.; Simpson, J.; Ruiz-Herrera, J. Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol. Gen. Genet. 1997, 253, 703–710. [Google Scholar] [CrossRef]
- McClelland, M.; Nelson, M.; Raschke, E. Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res 1994, 22, 3640–3659. [Google Scholar] [CrossRef] [Green Version]
- Cervera, M.T.; Ruiz-García, L.; Martínez-Zapater, J.M. Analysis of DNA methylation in Arabidopsis thaliana based on methylation-sensitive AFLP markers. Mol. Genet. Genomics. 2002, 268, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Schulz, B.; Eckstein, R.L.; Durka, W. Scoring and analysis of methylation-sensitive amplification polymorphisms for epigenetic population studies. Mol. Ecol. Resour. 2013, 13, 642–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulneček, J.; Kovařík, A. How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genet. 2014, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara, M.Á.; de María, N.; Sáez-Laguna, E.; Vélez, M.D.; Cervera, M.T.; Cabezas, J.A. Analysis of DNA Cytosine Methylation Patterns Using Methylation-Sensitive Amplification Polymorphism (MSAP). In Plant Epigenetics. Methods in Molecular Biology; Kovalchuk, I., Ed.; Humana Press: Boston, MA, USA, 2017; Volume 1456, pp. 99–112. [Google Scholar] [CrossRef]
- Salmon, A.; Clotault, J.; Jenczewski, E.; Chable, V.; Manzanares-Dauleux, M.J. Brassica oleracea displays a high level of DNA methylation polymorphism. Plant Sci. 2008, 174, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Cervera, M.T.; Remington, D.; Frigerio, J.M.; Storme, V.; Ivens, B.; Boerjan, W.; Plomion, C. Improved AFLP analysis of tree species. Can. J. For. Res. 2000, 30, 1608–1616. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes. 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Medrano, M.; Alonso, C.; Bazaga, P.; López, E.; Herrera, C.M. Comparative genetic and epigenetic diversity in pairs of sympatric, closely related plants with contrasting distribution ranges in south-eastern Iberian mountains. AoB Plants 2020, 12, 2041–2851. [Google Scholar] [CrossRef]
- Herrera, C.M.; Medrano, M.; Bazaga, P. Comparative spatial genetics and epigenetics of plant populations: Heuristic value and a proof of concept. Mol. Ecol. 2016, 25, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Avramidou, E.V.; Ganopoulos, I.V.; Doulis, A.G.; Tsaftaris, A.S.; Aravanopoulos, F.A. Beyond population genetics: Natural epigenetic variation in wild cherry (Prunus avium). Tree Genet. Genomes. 2015, 11, 1–9. [Google Scholar] [CrossRef]
- Avramidou, E.V.; Doulis, A.G.; Aravanopoulos, F.A. Determination of epigenetic inheritance, genetic inheritance, and estimation of genome DNA methylation in a full-sib family of Cupressus sempervirens L. Gene 2015, 162, 180–187. [Google Scholar] [CrossRef]
- Sáez-Laguna, E.; Guevara, M.A.; Díaz, L.M.; Sánchez-Gómez, D.; Collada, C.; Aranda, I.; Cervera, M.T. Epigenetic variability in the genetically uniform forest tree species Pinus pinea L. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lira-Medeiros, C.F.; Parisod, C.; Fernandes, R.A.; Mata, C.S.; Cardoso, M.A.; Ferreira, P.C.G. Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Plomion, C.; Bastien, C.; Bogeat-Triboulot, M.B.; Bouffier, L.; Déjardin, A.; Sébastien Duplessis, S.; Bruno Fady, B.; Heuertz, M.; Le Gac, A.-L.; Le Provost, G.; et al. Forest tree genomics: 10 achievements from the past 10 years and future prospects. Ann. For. Sci. 2016, 73, 77–103. [Google Scholar] [CrossRef] [Green Version]
- García-García, I.; Méndez-Cea, B.; Martín-Gálvez, D.; Seco, J.I.; Gallego, F.J.; Linares, J.C. Challenges and Perspectives in the Epigenetics of Climate Change-Induced Forests Decline. Front. Plant Sci. 2022, 12, 797958. [Google Scholar] [CrossRef] [PubMed]
- Cadahía, E.; Fernández de Simón, B.; Aranda, I.; Sanz, M.; Sánchez-Gómez, D.; Pinto, E. Non-targeted metabolomic profile of Fagus Sylvatica L. leaves using liquid chromatography with mass spectrometry and gas chromatography with mass spectrometry. Phytochem. Anal. 2015, 26, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Aranda, I.; Bahamonde, H.A.; Sánchez-Gómez, D. Intra-population variability in the drought response of a beech (Fagus sylvatica L.) population in the southwest of Europe. Tree Physiol. 2017, 37, 938–949. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.F.; Song, Y.P.; Yang, X.H.; Zhang, Z.Y.; Zhang, D.Q. Variation in genomic methylation in natural populations of Chinese white poplar. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Ci, D.; Song, Y.; Du, Q.; Tian, M.; Han, S.; Zhang, D. Variation in genomic methylation in natural populations of Populus simonii is associated with leaf shape and photosynthetic traits. J. Exp. Bot. 2016, 67, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, F.S.; Zhou, S.; Zhan, Y.G.; Dong, J. Drought resistance and DNA methylation of interspecific hybrids between Fraxinus mandshurica and Fraxinus americana. Trees 2014, 28, 1679–1692. [Google Scholar] [CrossRef]
- Alakärppä, E.; Salo, H.M.; Valledor, L.; Cañal, M.J.; Häggman, H.; Vuosku, J. Natural variation of DNA methylation and gene expression may determine local adaptations of Scots pine populations. J. Exp. Bot. 2018, 69, 5293–5305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Song, W.Q.; Chen, C.B.; Zhou, Y.N.; Qi, L.W.; Wang, C.G. DNA methylation status is associated with the formation of heterosis in Larix kaempferi intraspecific hybrids. Mol. Breeding 2013, 31, 463–475. [Google Scholar] [CrossRef]
- Gugger, P.F.; Fitz-Gibbon, S.; Pellegrini, M.; Sork, V.L. Species-wide patterns of DNA methylation variation in Quercus lobate and their association with climate gradients. Mol. Ecol. 2016, 25, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Rico, L.; Ogaya, R.; Barbeta, A.; Peñuelas, J. Changes in DNA methylation fingerprint of Quercus ilex trees in response to experimental field drought simulating projected climate change. Plant Biol. 2014, 16, 419–427. [Google Scholar] [CrossRef]
- Cascales, J.; Acevedo, R.M.; Paiva, D.I.; Gottlieb, A.M. Differential DNA methylation and gene expression during development of reproductive and vegetative organs in Ilex species. J. Plant Res. 2021, 134, 559–575. [Google Scholar] [CrossRef]
- Carsjens, C.; Ngoc, Q.N.; Guzy, J.; Knutzen, F.; Meier, I.C.; Müller, M.; Finkeldey, R.; Leuschner, C.; Polle, A. Intra-specific variations in expression of stress-related genes in beech progenies are stronger than drought-induced responses. Tree Physiol. 2014, 34, 1348–1361. [Google Scholar] [CrossRef] [Green Version]
- Aranda, I.; Sánchez-Gómez, D.; Cadahía, E.; Fernández de Simón, M.B. Ecophysiological and metabolic response patterns to drought under controlled condition in open-pollinated maternal families from a Fagus sylvatica L. population. Environ. Exp. Bot. 2018, 150, 209–221. [Google Scholar] [CrossRef]
- Platt, A.; Gugger, P.F.; Pellegrini, M.; Sork, V.L. Genome-wide signature of local adaptation linked to variable CpG methylation in oak populations. Mol. Ecol. 2015, 24, 3823–3830. [Google Scholar] [CrossRef]
- Yakovlev, I.A.; Carneros, E.; Lee, Y.; Jorunn, E.; Olsen, J.E.; Fossdal, C.G. Transcriptional profiling of epigenetic regulators in somatic embryos during temperature induced formation of an epigenetic memory in Norway spruce. Planta 2016, 243, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Mounger, J.; Boquete, M.T.; Marc, W.; Schmid, M.W.; Granado, R.; Robertson, M.H.; Voors, S.A.; Langanke, K.L.; Alvarez, M.; Wagemaker, C.A.M.; et al. Inheritance of DNA methylation differences in the mangrove Rhizophora mangle. Evol. Dev. 2021, 23, 351–374. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z.; Zhang, G.; Song, Y.; Diao, S.; He, C.; Zhang, J. Transcriptome and DNA methylome provide insights into the molecular regulation of drought stress in sea buckthorn. Genomics 2022, 114, 110345. [Google Scholar] [CrossRef]
- Tang, X.M.; Tao, X.; Wang, Y.; Ma, D.W.; Li, D.; Yang, H.; Ma, X.R. Analysis of DNA methylation of perennial ryegrass under drought using the methylation-sensitive amplification polymorphism (MSAP) technique. Mol. Genet. Genomics 2014, 289, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Abid, G.; Mingeot, D.; Muhovski, Y.; Mergeai, G.; Aouida, M.; Abdelkarim, S.; Aroua, I.; Ayed, M.E.; M´hamdi, M.; Sassi, K.; et al. Analysis of DNA methylation patterns associated with drought stress response in faba bean (Vicia faba L.) using methylation-sensitive amplification polymorphism (MSAP). Environ. Exp. Bot. 2017, 142, 34–44. [Google Scholar] [CrossRef]
- Liu, N.; Staswick, P.E.; Avramova, Z. Memory responses of jasmonic acid-associated Arabidopsis genes to a repeated dehydration stress. Plant Cell. Environ. 2016, 39, 2515–2529. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Provenance Code | Assay | Country | Location | Latitude | Longitude | Altitude (m. a. s. l) | Rainfall (mm) | Average Temperature (°C) |
|---|---|---|---|---|---|---|---|---|
| DE | 1 | Germany | Kempten | 47° 44′ | 10° 23′ | 860–900 | 1316 | 6.9 |
| ES | 1, 2 | Spain | Montejo de la Sierra | 42° 01′ | 3° 05′ | 1250–1400 | 950–1100 | 8.1 |
| SE | 1 | Sweden | Blaviksliarna | 57° 90′ | 13° 14′ | 75 | 860 | 6.5 |
| SE | 2 | Sweden | Falkenberg | 56° 52′ | 12° 51′ | 150 | 900 | 7 |
| HpaII/MspI-ATC EcoRI-AAC | HpaII/MspI-ACT EcoRI-ACG | HpaII/MspI-AAT EcoRI-AAC | Total | |
|---|---|---|---|---|
| No. total markers | 64 | 72 | 69 | 205 |
| No. scorable markers | 49 | 47 | 48 | 144 |
| Methylation-insensitive markers (MI) | ||||
| No. polymorphic markers (MIP) | 13 | 10 | 11 | 34 |
| No. monomorphic markers (MIM) | 9 | 10 | 8 | 27 |
| Methylation-sensitive fragments (MS) | ||||
| No. polymorphic markers (MSP) | 27 | 25 | 29 | 81 |
| No. monomorphic markers (MSM) | 0 | 2 | 0 | 2 |
| DE | ES | SE | |
|---|---|---|---|
| Full or hemi-methylated internal C | 31.1% ± 2.9% | 30.7% ± 2.4% | 30.8% ± 2.5% |
| Hemi-methylated external C | 0.9% ± 1.0% | 1.5% ± 0.7% | 1.2% ± 1.1% |
| Total | 32.1% ± 2.7% | 32.3% ± 2.7% | 32.0% ± 2.7% |
| a) | |||||||
| Provenance | Sample size | No. loci | P% | Na | Ne | I | He |
| DE | 20 | 81 | 75.31 | 1.519 ± 0.095 | 1.324 ± 0.038 | 0.313 ± 0.028 | 0.200 ± 0.020 |
| ES | 19 | 81 | 67.90 | 1.432 ± 0.096 | 1.268 ± 0.035 | 0.273 ± 0.027 | 0.171 ± 0.019 |
| SE | 20 | 81 | 75.31 | 1.543 ± 0.091 | 1.337 ± 0.038 | 0.322 ±0.028 | 0.207 ± 0.020 |
| b) | |||||||
| Provenance | Sample size | No. loci | P% | Na | Ne | I | He |
| DE | 20 | 34 | 85.29 | 1.735 ± 0.114 | 1.355 ± 0.065 | 0.327 ± 0.045 | 0.210 ± 0.033 |
| ES | 20 | 34 | 61.76 | 1.235 ± 0.096 | 1.326 ± 0.064 | 0.289 ± 0.049 | 0.191 ± 0.035 |
| SE | 20 | 34 | 67.65 | 1.412 ± 0.153 | 1.319 ± 0.062 | 0.297 ±0.046 | 0.192 ± 0.033 |
| c) | |||||||
| Provenance | Sample size | No. loci | P% | Na | Ne | I | He |
| DE | 20 | 105 | 87.62 | 1.810 ± 0.053 | 1.428 ± 0.034 | 0.397 ± 0.023 | 0.258 ± 0.017 |
| ES | 19 | 105 | 65.71 | 1.419 ± 0.083 | 1.369 ± 0.036 | 0.326 ± 0.027 | 0.216 ± 0.019 |
| SE | 20 | 105 | 74.29 | 1.562 ± 0.076 | 1.374 ± 0.033 | 0.353 ± 0.025 | 0.229 ± 0.018 |
| EcoRI + ATC MseI + CAT | EcoRI + ATA MseI + CAT | Total | |
|---|---|---|---|
| No. total markers | 121 | 106 | 227 |
| No. scorable markers | 90 | 90 | 180 |
| No. polymorphic markers | 51 | 54 | 105 |
| No. monomorphic markers | 39 | 36 | 75 |
| HpaII/MspI-AAT EcoRI-AAC | HpaII/MspI-ACT EcoRI-ACG | HpaII/MspI-ATC EcoRI-AAC | HpaII/MspI-ACT EcoRI-AAC | Total | |
|---|---|---|---|---|---|
| No. total markers | 48 | 63 | 60 | 64 | 235 |
| No. scorable markers | 30 | 59 | 54 | 45 | 188 |
| Methylation-insensitive markers (MI) | |||||
| No. polymorphic markers (MIP) | 2 | 10 | 16 | 1 | 29 |
| No. monomorphic markers (MIM) | 7 | 16 | 12 | 15 | 50 |
| Methylation-sensitive fragments (MS) | |||||
| No. polymorphic markers (MSP) | 21 | 29 | 26 | 29 | 105 |
| No. monomorphic markers (MSM) | 0 | 4 | 0 | 0 | 4 |
| SE-WW | SE-WS | ES-WW | ES-WS | |
|---|---|---|---|---|
| Full or hemi-methylated internal C | 30.9% ± 2.7% | 31.0% ± 2.7% | 29.5% ± 2.1% | 30.5% ± 2.6% |
| Hemi-methylated external C | 1.6% ±1.4% | 1.5% ± 1.1% | 2.4% ± 1.7% | 1.2% ± 1.1% |
| Total | 32.5% ± 2.9% | 32.5% ± 2.7% | 32.0% ± 2.8% | 31.7% ± 2.8% |
| a) | ||||||||
| Provenance | Treatment | Sample size | No. loci | P% | Na | Ne | I | He |
| SE | WS | 13 | 104 | 74.04 | 1.519 ± 0.082 | 1.335 ± 0.033 | 0.321 ± 0.025 | 0.206 ± 0.018 |
| SE | WW | 13 | 104 | 65.38 | 1.356 ± 0.090 | 1.335 ± 0.036 | 0.304 ± 0.027 | 0.199 ± 0.019 |
| ES | WS | 14 | 104 | 66.35 | 1.346 ± 0.091 | 1.331 ± 0.033 | 0.311 ±0.026 | 0.202 ± 0.018 |
| ES | WW | 11 | 104 | 75.00 | 1.529 ± 0.082 | 1.353 ± 0.035 | 0.331 ± 0.025 | 0.214 ± 0.018 |
| b) | ||||||||
| Provenance | Treatment | Sample size | N loci | P% | Na | Ne | I | He |
| SE | WS | 14 | 29 | 58.62 | 1.207 ± 0.182 | 1.348 ± 0.066 | 0.313 ± 0.053 | 0.208 ± 0.037 |
| SE | WW | 14 | 29 | 79.31 | 1.586 ± 0.153 | 1.413 ± 0.070 | 0.370 ± 0.049 | 0.243 ± 0.036 |
| ES | WS | 15 | 29 | 65.52 | 1.379 ± 0.168 | 1.328 ± 0.069 | 0.297 ± 0.051 | 0.195 ± 0.037 |
| ES | WW | 12 | 29 | 65.52 | 1.379 ± 0.168 | 1.369 ± 0.072 | 0.322 ± 0.053 | 0.214 ± 0.038 |
| c) | ||||||||
| Provenance | Treatment | Sample size | N loci | P% | Na | Ne | I | He |
| SE | WS | 14 | 232 | 78.02 | 1.621 ± 0.049 | 1.394 ± 0.023 | 0.364 ± 0.017 | 0.237 ± 0.012 |
| SE | WW | 14 | 232 | 77.16 | 1.603 ± 0.050 | 1.359 ± 0.022 | 0.346 ± 0.016 | 0.222 ± 0.011 |
| ES | WS | 15 | 232 | 68.97 | 1.453 ± 0.056 | 1.390 ± 0.024 | 0.349 ± 0.018 | 0.231 ± 0.013 |
| ES | WW | 12 | 232 | 69.83 | 1.487 ± 0.054 | 1.373 ± 0.024 | 0.339 ± 0.018 | 0.223 ± 0.012 |
| EcoRI + ATC | EcoRI + ATA | EcoRI-ACC | EcoRI-AAT | EcoRI-ACA | Total | |
|---|---|---|---|---|---|---|
| MseI + CAT | MseI + CAT | Mse-CAT | Mse-CCA | Mse-CCA | ||
| No. total markers | 124 | 136 | 46 | 65 | 81 | 452 |
| No. scorable markers | 101 | 84 | 41 | 61 | 66 | 352 |
| No. polymorphic markers | 78 | 57 | 26 | 31 | 41 | 232 |
| No. monomorphic markers | 23 | 27 | 15 | 30 | 25 | 120 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guevara, M.Á.; Sánchez-Gómez, D.; Vélez, M.D.; de María, N.; Díaz, L.M.; Ramírez-Valiente, J.A.; Mancha, J.A.; Aranda, I.; Cervera, M.T. Epigenetic and Genetic Variability in Contrasting Latitudinal Fagus sylvatica L. Provenances. Forests 2022, 13, 1971. https://doi.org/10.3390/f13121971
Guevara MÁ, Sánchez-Gómez D, Vélez MD, de María N, Díaz LM, Ramírez-Valiente JA, Mancha JA, Aranda I, Cervera MT. Epigenetic and Genetic Variability in Contrasting Latitudinal Fagus sylvatica L. Provenances. Forests. 2022; 13(12):1971. https://doi.org/10.3390/f13121971
Chicago/Turabian StyleGuevara, María Ángeles, David Sánchez-Gómez, María Dolores Vélez, Nuria de María, Luis Miguel Díaz, José Alberto Ramírez-Valiente, José Antonio Mancha, Ismael Aranda, and María Teresa Cervera. 2022. "Epigenetic and Genetic Variability in Contrasting Latitudinal Fagus sylvatica L. Provenances" Forests 13, no. 12: 1971. https://doi.org/10.3390/f13121971