
Genome-Wide Comprehensive Analysis of PtLACs: Prediction and Verification of the Functional Divergence of Tandem-Duplicated Genes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of the Laccase Gene Family from P. trichocarpa
2.2. In Silico Characterisation of Identified Laccases
2.3. Multiple Sequence Alignment and Phylogenetic Analysis
2.4. Detection of Positive Selection
2.5. Synonymous Substitutions per Synonymous Site Analysis
2.6. Gene Genomic Distribution and Segmental Duplication Analyses
2.7. Expression Patterns of PtLAC Using RNA-Seq Data
2.8. Plant Growth and RNA Isolation
2.9. Subcellular Localisation Analysis of PtLAC
2.10. Homozygous Line Acquisition and Treatment of Transgenic A. thaliana
3. Results
3.1. Identification of the LAC Gene Family in the Populus Genome
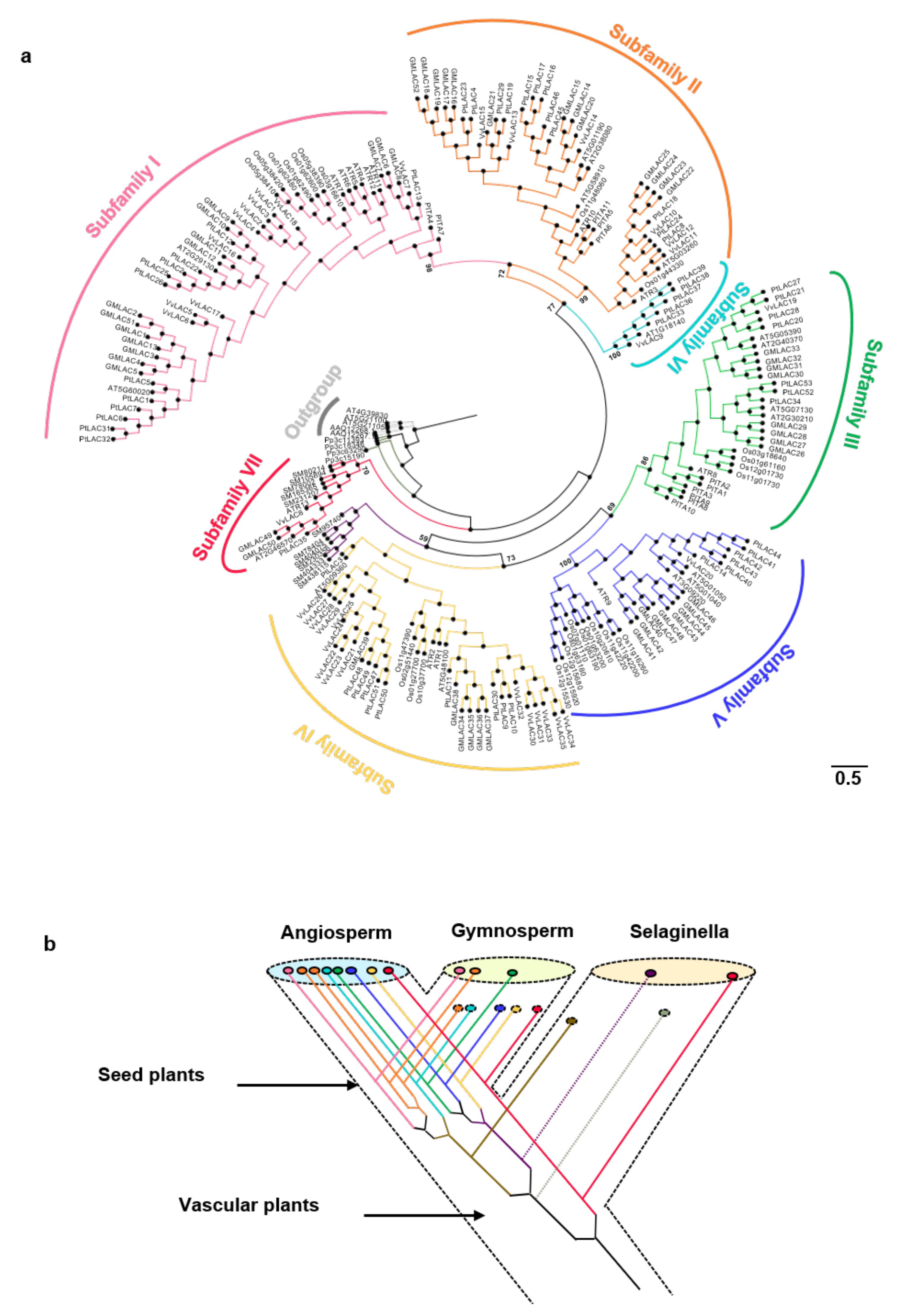
3.2. Evolutionary Analysis of the Laccase Gene Family in Terrestrial Plants
3.3. Positive Selection Analysis of Plant Laccases
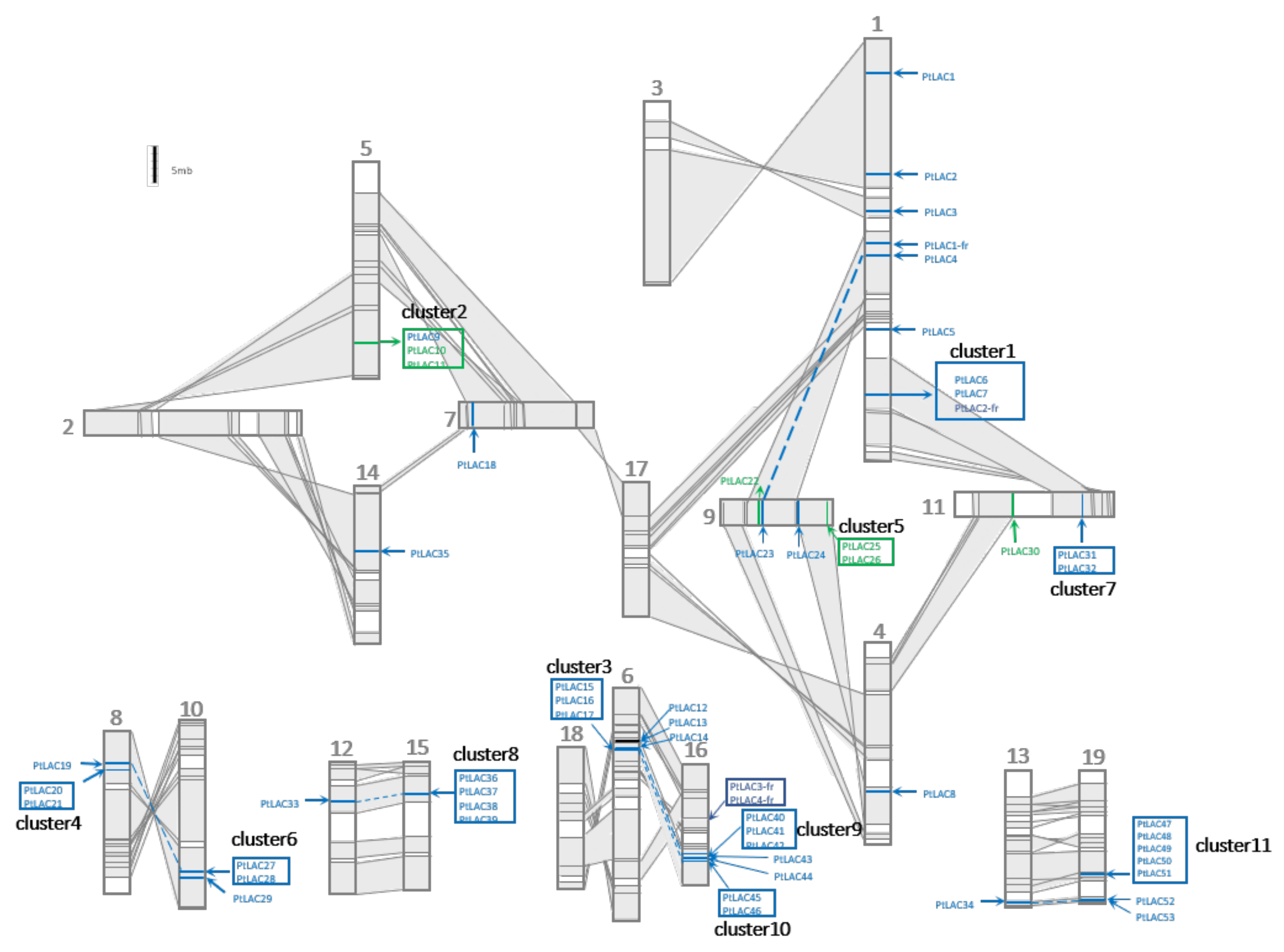
3.4. Formation Mechanism Analysis of the Laccase Gene Family in P. trichocarpa
3.5. Gene Structure Analysis and Motif Detection of PtLACs
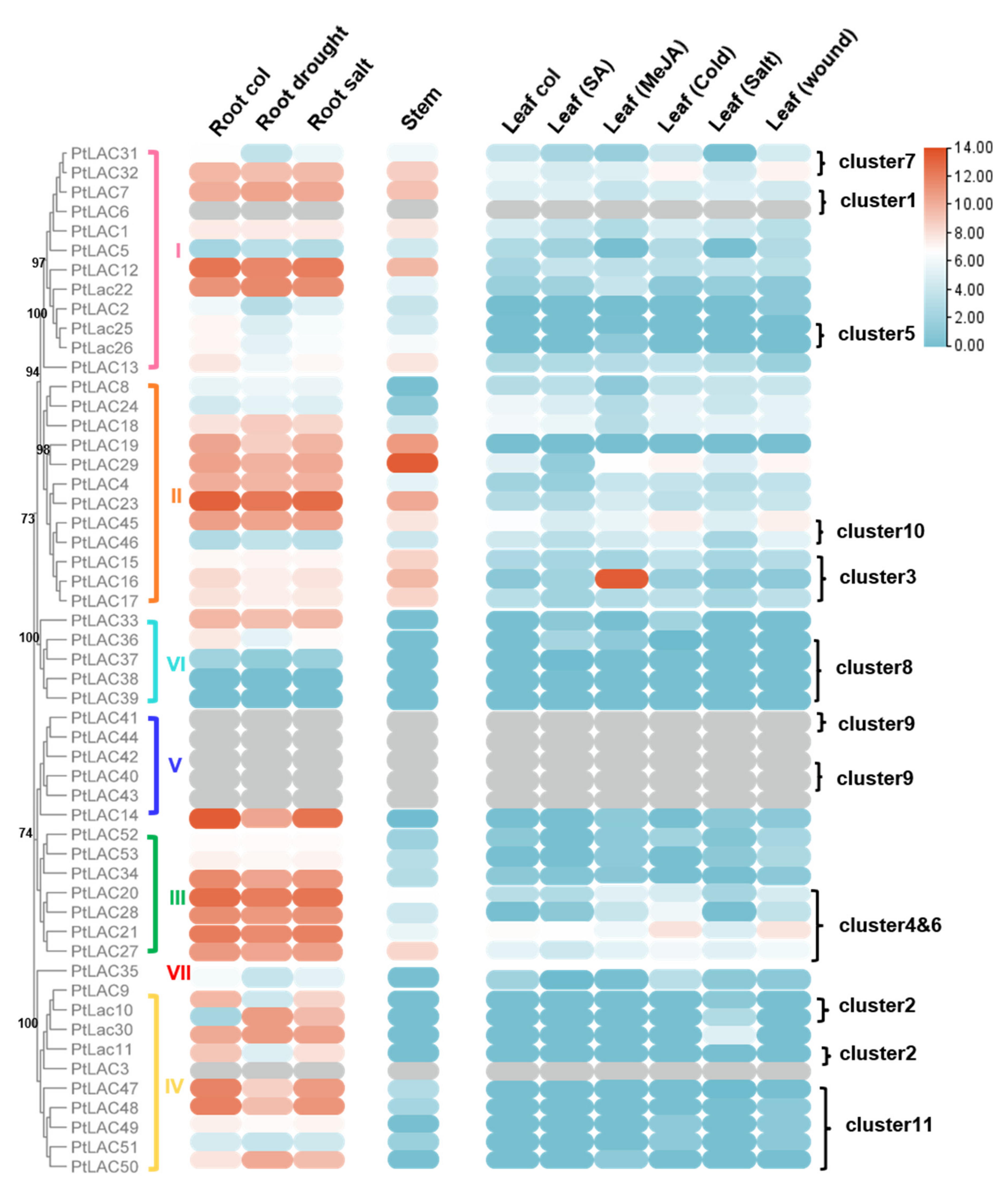
3.6. Expression Patterns of the PtLAC Genes Revealed by Transcriptome Analysis
3.7. Molecular Evolution Analysis of the Laccase Gene Cluster in P. trichocarpa
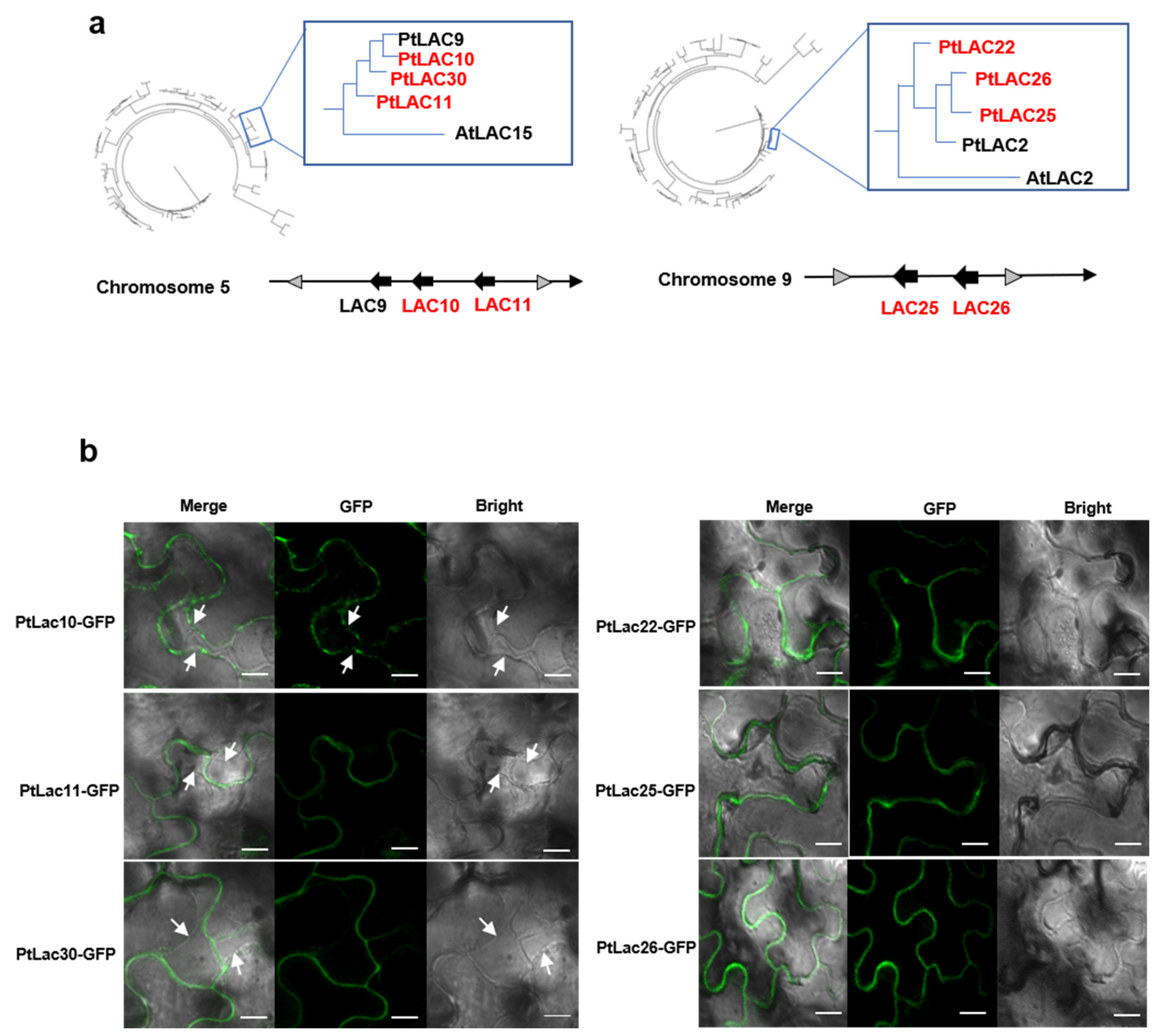
3.8. Subcellular Localization of P. trichocarpa LACs
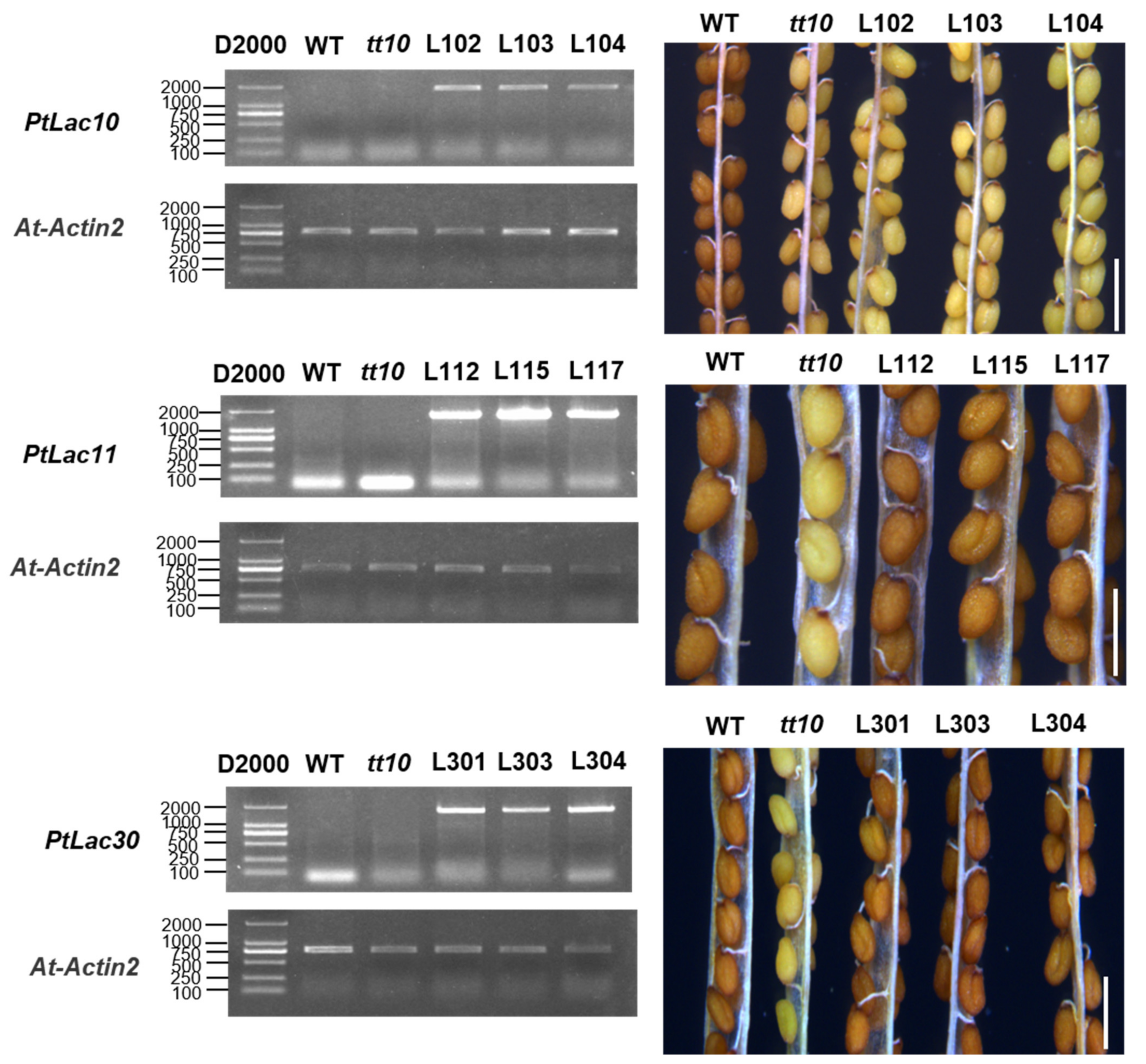
3.9. Identification of the Functional Divergence of Genes in Cluster 2
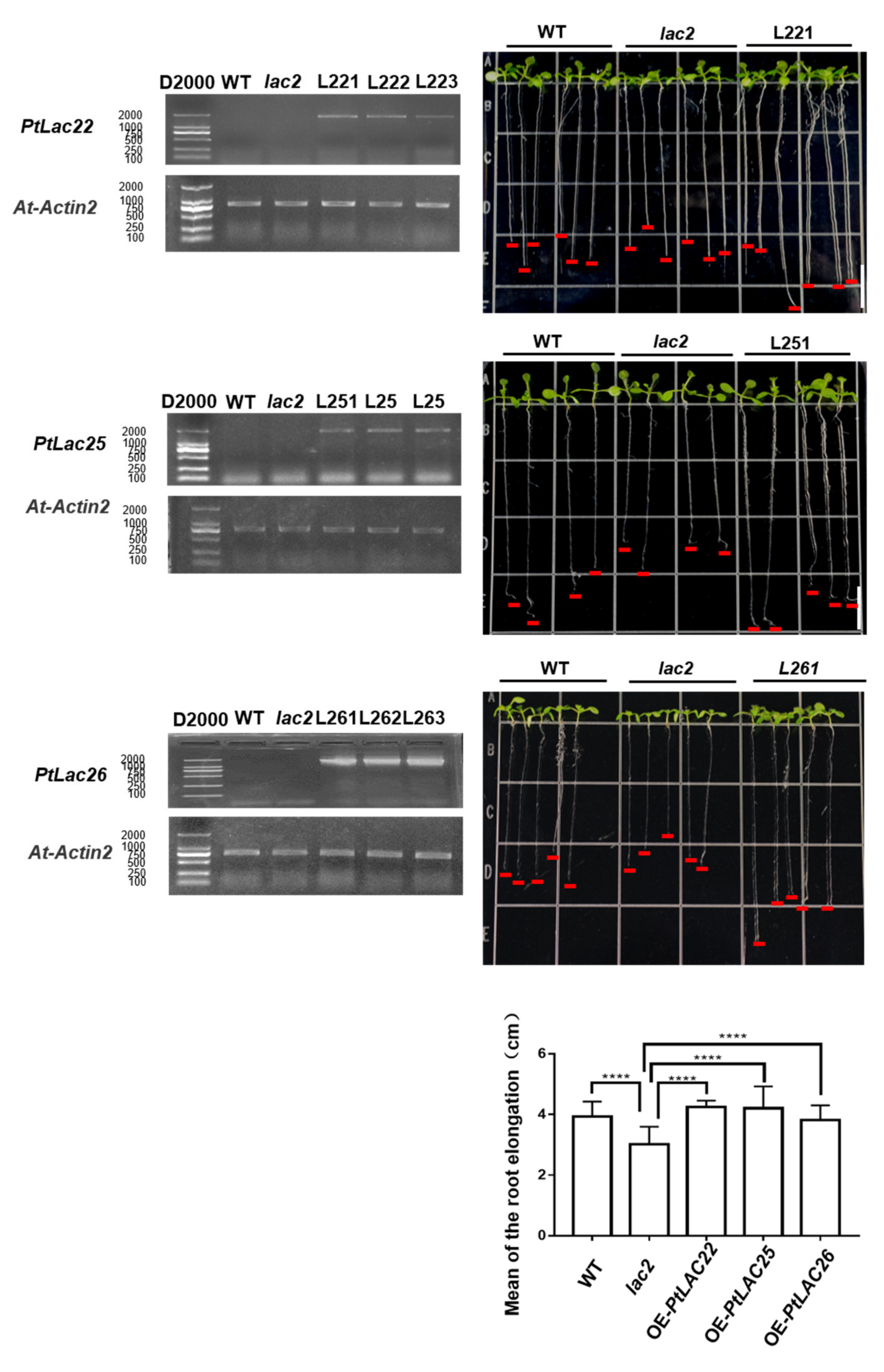
3.10. Identification of the Functional Divergence of Genes in Cluster 5
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Q.; Luo, L.; Wang, X.; Shen, Z.; Zheng, L. Comprehensive Analysis of Rice Laccase Gene (OsLAC) Family and Ectopic Expression of OsLAC10 Enhances Tolerance to Copper Stress in Arabidopsis. Int. J. Mol. Sci. 2017, 18, 209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bouchabke-Coussa, O.; Lebris, P.; Antelme, S.; Soulhat, C.; Gineau, E.; Dalmais, M.; Bendahmane, A.; Morin, H.; Mouille, G.; et al. LACCASE5 Is Required for Lignification of the Brachypodium distachyon Culm. Plant Physiol. 2015, 168, 192–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turlapati, P.V.; Kim, K.-W.; Davin, L.B.; Lewis, N.G. The laccase multigene family in Arabidopsis thaliana: Towards addressing the mystery of their gene function(s). Planta 2010, 233, 439–470. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Nakashima, J.; Chen, F.; Yin, Y.; Fu, C.; Yun, J.; Shao, H.; Wang, X.; Wang, Z.-Y.; Dixon, R.A. LACCASE Is Necessary and Nonredundant with PEROXIDASE for Lignin Polymerization during Vascular Development in Arabidopsis. Plant Cell 2013, 25, 3976–3987. [Google Scholar] [CrossRef] [Green Version]
- Pourcel, L.; Routaboul, J.M.; Kerhoas, L.; Caboche, M.; Lepiniec, L.; Debeaujon, I. TRANSPARENT TESTA10 encodes a laccase-like enzyme involved in oxidative polymerization of flavonoids in Arabidopsis seed coat. Plant Cell 2005, 17, 2966–2980. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.T.; Lu, Y.C.; Zhang, S.; Luo, F.; Yang, H. Rice (Oryza sativa) Laccases Involved in Modification and Detoxification of Herbicides Atrazine and Isoproturon Residues in Plants. J. Agric. Food Chem. 2016, 64, 6397–6406. [Google Scholar] [CrossRef]
- Liang, M.; Haroldsen, V.; Cai, X.; Wu, Y. Expression of a putative laccase gene, ZmLAC1, in maize primary roots under stress*. Plant Cell Environ. 2006, 29, 746–753. [Google Scholar] [CrossRef]
- Wang, G.-D.; Li, Q.-J.; Luo, B.; Chen, X.-Y. Ex planta phytoremediation of trichlorophenol and phenolic allelochemicals via an engineered secretory laccase. Nat. Biotechnol. 2004, 22, 893–897. [Google Scholar] [CrossRef]
- Rensing, S.A.; Lang, D.; Zimmer, A.D.; Terry, A. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 2008, 319, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Velasco, R.; Zharkikh, A.; Troggio, M. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2007, 2, e1326. [Google Scholar] [CrossRef] [Green Version]
- Ming, R.; Hou, S.; Feng, Y. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008, 452, 991–996. [Google Scholar] [CrossRef] [Green Version]
- Flagel, L.E.; Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar] [CrossRef]
- Hanada, K.; Zou, C.; Lehti-Shiu, M.D.; Shinozaki, K.; Shiu, S.H. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 2008, 148, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Rodgers-Melnick, E.; Mane, S.P.; Dharmawardhana, P.; Slavov, G.T.; Crasta, O.R.; Strauss, S.H.; Brunner, A.M.; Difazio, S.P. Contrasting patterns of evolution following whole genome versus tandem duplication events in Populus. Genome Res. 2012, 22, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Freeling, M. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar]
- Ranocha, P.; Chabannes, M.; Chamayou, S.; Danoun, S.; Jauneau, A.; Boudet, A.M.; Goffner, D. Laccase down-regulation causes alterations in phenolic metabolism and cell wall structure in poplar. Plant Physiol. 2002, 129, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Bryan, A.C.; Jawdy, S.; Gunter, L.; Gjersing, E.; Sykes, R.; Hinchee, M.A.; Winkeler, K.A.; Collins, C.M.; Engle, N.; Tschaplinski, T.J.; et al. Knockdown of a laccase in Populus deltoides confers altered cell wall chemistry and increased sugar release. Plant Biotechnol. J. 2016, 14, 2010–2020. [Google Scholar] [CrossRef]
- Alzohairy, A.M. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Bailey, T.; Elkan, C. Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Lang, D.; Ullrich, K.K.; Murat, F.; Fuchs, J.; Jenkins, J. The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J. 2018, 93, 515–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; dePamphilis, C.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A.; et al. The Selaginella Genome Identifies Genetic Changes Associated with the Evolution of Vascular Plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amborella Genome, P. The Amborella genome and the evolution of flowering plants. Science 2013, 342, 1241089. [Google Scholar] [CrossRef]
- Ouyang, S.; Zhu, W.; Hamilton, J.; Lin, H.; Campbell, M.; Childs, K.; Thibaud-Nissen, F.; Malek, R.L.; Lee, Y.; Zheng, L.; et al. The TIGR Rice Genome Annotation Resource: Improvements and new features. Nucleic Acids Res. 2007, 35, D883–D887. [Google Scholar] [CrossRef] [Green Version]
- Jaillon, O.; Aury, J.M. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; McLnerney, J.O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Anisimova, M.; Bielawski, J.P.; Yang, Z. Accuracy and Power of the Likelihood Ratio Test in Detecting Adaptive. Mol. Biol. Evol. 2001, 18, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.M.K. Codon-Substitution Models for Heterogeneous Selection Pressure at Amino Acid Sites. Genetics 2000, 155, 431–449. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Rivers, C.S.; Wardlaw, J.M.; Armitage, P.A.; Bastin, M.E.; Carpenter, T.K.; Cvoro, V.; Hand, P.J.; Dennis, M.S. Persistent Infarct Hyperintensity on Diffusion-Weighted Imaging Late after Stroke Indicates Heterogeneous, Delayed, Infarct Evolution. Stroke 2006, 37, 1418–1423. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Roychoudhury, A. Epigenetic regulation during salinity and drought stress in plants: Histone modifications and DNA methylation. Plant Gene 2017, 11, 199–204. [Google Scholar] [CrossRef]
- Xing, H.; Fu, X.; Yang, C.; Tang, X.; Guo, L.; Li, C.; Xu, C.; Luo, K. Genome-wide investigation of pentatricopeptide repeat gene family in poplar and their expression analysis in response to biotic and abiotic stresses. Sci. Rep. 2018, 8, 2817. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalitha, S. Primer Premier 5. Biotech Softw. Internet Rep. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Cai, X.; Davis, E.J.; Ballif, J.; Liang, M.; Bushman, E.; Haroldsen, V.; Torabinejad, J.; Wu, Y. Mutant identification and characterization of the laccase gene family in Arabidopsis. J. Exp. Bot. 2006, 57, 2563–2569. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.J.; Ding, L.; Zhu, J.K. SOS1, a Genetic Locus Essential for Salt Tolerance and Potassium Acquisition. Plant Cell 1996, 8, 617–627. [Google Scholar] [CrossRef] [Green Version]
- McCaig, B.C.; Meagher, R.B.; Dean, J.F. Gene structure and molecular analysis of the laccase-like multicopper oxidase (LMCO) gene family in Arabidopsis thaliana. Planta 2005, 221, 619–636. [Google Scholar] [CrossRef]
- Hakulinen, N.; Rouvinen, J. Three-dimensional structures of laccases. Cell. Mol. Life Sci. 2015, 72, 857–868. [Google Scholar] [CrossRef]
- Xie, T.; Liu, Z.; Wang, G. Structural basis for monolignol oxidation by a maize laccase. Nat. Plants 2020, 6, 231–237. [Google Scholar] [CrossRef]
- Boruc, J.; Mylle, E.; Duda, M.; De Clercq, R.; Rombauts, S.; Geelen, D.; Hilson, P.; Inze, D.; Van Damme, D.; Russinova, E. Systematic localization of the Arabidopsis core cell cycle proteins reveals novel cell division complexes. Plant Physiol. 2010, 152, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Marques, A.C.; Vinckenbosch, N.; Brawand, D.; Kaessmann, H. Functional diversification of duplicate genes through subcellular adaptation of encoded proteins. Genome Biol. 2008, 9, R54. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Zhang, J. Protein subcellular relocalization in the evolution of yeast singleton and duplicate genes. Genome Biol. Evol. 2009, 1, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Shirley, B.W.; Hanley, S.; Goodman, H.M. Effects of Ionizing Radiation on a Plant Genome: Analysis of Two Arabidopsis transparent testa Mutations. Plant Cell 1992, 4, 333–347. [Google Scholar]
- Shirley, B.W.; Kubasek, W.L.; Storz, G.; Bruggemann, E.; Koornneef, M.; Ausubel, F.M.; Goodman, H.M. Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. Plant J. 1995, 8, 659–671. [Google Scholar] [CrossRef]
- Simoes, M.S.; Carvalho, G.G.; Ferreira, S.S.; Hernandes-Lopes, J.; de Setta, N.; Cesarino, I. Genome-wide characterization of the laccase gene family in Setaria viridis reveals members potentially involved in lignification. Planta 2020, 251, 46. [Google Scholar] [CrossRef]
- Xu, X.; Zhou, Y.; Wang, B.; Ding, L.; Wang, Y.; Luo, L.; Zhang, Y.; Kong, W. Genome-wide identification and characterization of laccase gene family in Citrus sinensis. Gene 2019, 689, 114–123. [Google Scholar] [CrossRef]
- Liu, M.; Dong, H.; Wang, M.; Liu, Q. Evolutionary divergence of function and expression of laccase genes in plants. J. Genet. 2020, 99, 23. [Google Scholar] [CrossRef]
- Wang, Q.; Li, G.; Zheng, K.; Zhu, X.; Ma, J.; Wang, D.; Tang, K.; Feng, X.; Leng, J.; Yu, H.; et al. The Soybean Laccase Gene Family: Evolution and Possible Roles in Plant Defense and Stem Strength Selection. Genes 2019, 10, 701. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.C.; Purugganan, M.D. The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 15682–15687. [Google Scholar] [CrossRef] [Green Version]
- Kong, H.; Landherr, L.L.; Frohlich, M.W.; Leebens-Mack, J.; Ma, H.; dePamphilis, C.W. Patterns of gene duplication in the plant SKP1 gene family in angiosperms: Evidence for multiple mechanisms of rapid gene birth. Plant J. 2007, 50, 873–885. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Davis, E.; Gardner, D.; Cai, X.; Wu, Y. Involvement of AtLAC15 in lignin synthesis in seeds and in root elongation of Arabidopsis. Planta 2006, 224, 1185–1196. [Google Scholar] [CrossRef]
- Khandal, H.; Singh, A.P.; Chattopadhyay, D. The MicroRNA397b-LACCASE2 Module Regulates Root Lignification under Water and Phosphate Deficiency. Plant Physiol. 2020, 182, 1387–1403. [Google Scholar] [CrossRef]
- Liu, L.; Dean, J.F.D.; Friedman, W.E.; Eriksson, K.-E.L. A laccase-like phenoloxidase is correlated with lignin biosynthesis in Zinnia elegans stem tissues. Plant J. 1994, 6, 213–224. [Google Scholar] [CrossRef]
- Qin, S.; Fan, C.; Li, X.; Li, Y.; Hu, J.; Li, C.; Luo, K. LACCASE14 is required for the deposition of guaiacyl lignin and affects cell wall digestibility in poplar. Biotechnol. Biofuels 2020, 13, 197. [Google Scholar] [CrossRef]
- Fan, L.; Linker, R.; Gepstein, S.; Tanimoto, E.; Yamamoto, R.; Neumann, P.M. Progressive inhibition by water deficit of cell wall extensibility and growth along the elongation zone of maize roots is related to increased lignin metabolism and progressive stelar accumulation of wall phenolics. Plant Physiol. 2006, 140, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Dinneny, J.R. Developmental Responses to Water and Salinity in Root Systems. Annu. Rev. Cell Dev. Biol. 2019, 35, 239–257. [Google Scholar] [CrossRef]
- Rellán-Álvarez, R.; Lobet, G.; Dinneny, J.R. Environmental Control of Root System Biology. Annu. Rev. Plant Biol. 2016, 67, 619–642. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 3. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Model | Amino Acid | Isoelectronic Point (pI) | Molecular Weight | Gene | Gene Model | Amino Acid | Isoelectronic Point (pI) | Molecular Weight |
|---|---|---|---|---|---|---|---|---|---|
| PtLAC1 | Potri.001G054600 | 581 | 9.29 | 64.262KD | PtLAC30 | Potri.011G071100 | 555 | 8.72 | 60.919KD |
| PtLAC2 | Potri.001G184300 | 581 | 9.3 | 64.015KD | PtLAC31 | Potri.011G120200 | 562 | 6.67 | 63.079KD |
| PtLAC3 | Potri.001G206200 | 564 | 9.19 | 63.345KD | PtLAC32 | Potri.011G120300 | 581 | 9.03 | 63.935KD |
| PtLAC4 | Potri.001G248700 | 557 | 9.05 | 61.145KD | PtLAC33 | Potri.012G048900 | 581 | 9.18 | 64.027KD |
| PtLAC5 | Potri.001G341600 | 580 | 8.91 | 64.163KD | PtLAC34 | Potri.013G152700 | 579 | 9.35 | 64.456KD |
| PtLAC6 | Potri.001G401100 | 581 | 9.19 | 64.264KD | PtLAC35 | Potri.014G100600 | 576 | 9.29 | 64.491KD |
| PtLAC7 | Potri.001G401300 | 581 | 9.2 | 64.197KD | PtLAC36 | Potri.015G040400 | 572 | 7.26 | 63.456KD |
| PtLAC8 | Potri.004G156400 | 573 | 6.46 | 64.341KD | PtLAC37 | Potri.015G040600 | 579 | 8.52 | 64.38KD |
| PtLAC9 | Potri.005G200500 | 564 | 8.94 | 62.557KD | PtLAC38 | Potri.015G040700 | 579 | 8.96 | 64.499KD |
| PtLAC10 | Potri.005G200600 | 566 | 7.04 | 63.558KD | PtLAC39 | Potri.015G040800 | 559 | 9.91 | 62.010KD |
| PtLAC11 | Potri.005G200700 | 566 | 7.62 | 63.558KD | PtLAC40 | Potri.016G106000 | 568 | 6.82 | 61.924KD |
| PtLAC12 | Potri.006G087100 | 565 | 6.5 | 63.026KD | PtLAC41 | Potri.016G106100 | 569 | 8.49 | 62.01KD |
| PtLAC13 | Potri.006G087500 | 580 | 9.19 | 63.79KD | PtLAC42 | Potri.016G106300 | 562 | 8.31 | 61.538KD |
| PtLAC14 | Potri.006G094100 | 579 | 9.84 | 64.094KD | PtLAC43 | Potri.016G107500 | 568 | 6.82 | 61.958KD |
| PtLAC15 | Potri.006G096900 | 562 | 8.3 | 61.508KD | PtLAC44 | Potri.016G107900 | 569 | 8.05 | 62.029KD |
| PtLAC16 | Potri.006G097000 | 558 | 9.52 | 61.311KD | PtLAC45 | Potri.016G112000 | 557 | 9.3 | 60.916KD |
| PtLAC17 | Potri.006G097100 | 559 | 9.56 | 61.285KD | PtLAC46 | Potri.016G112100 | 564 | 9.53 | 62.027KD |
| PtLAC18 | Potri.007G023300 | 559 | 9.56 | 61.426KD | PtLAC47 | Potri.019G088500 | 567 | 7.01 | 62.76KD |
| PtLAC19 | Potri.008G064000 | 562 | 7.26 | 62.356KD | PtLAC48 | Potri.019G088600 | 567 | 7.29 | 62.781KD |
| PtLAC20 | Potri.008G073700 | 556 | 7.27 | 60.948KD | PtLAC49 | Potri.019G088700 | 567 | 7.68 | 62.77KD |
| PtLAC21 | Potri.008G073800 | 574 | 9.23 | 63.889KD | PtLAC50 | Potri.019G088800 | 566 | 6.49 | 62.735KD |
| PtLAC22 | Potri.009G034500 | 582 | 6.62 | 63.906KD | PtLAC51 | Potri.019G088900 | 567 | 6.78 | 62.919KD |
| PtLAC23 | Potri.009G042500 | 576 | 9.35 | 63.523KD | PtLAC52 | Potri.019G121700 | 576 | 8.23 | 63.9KD |
| PtLAC24 | Potri.009G102700 | 556 | 9.03 | 60.74KD | PtLAC53 | Potri.019G124300 | 576 | 8.23 | 63.888KD |
| PtLAC25 | Potri.009G156600 | 561 | 8.91 | 62.735KD | * PtLAC1-fr | Potri.001G243200 | 259 | ||
| PtLAC26 | Potri.009G156800 | 576 | 9.2 | 63.512KD | * PtLAC2-fr | Potri.001G401000 | 215 | ||
| PtLAC27 | Potri.010G183500 | 576 | 9.31 | 63.481KD | * PtLAC3-fr | Potri.016G083300 | 147 | ||
| PtLAC28 | Potri.010G183600 | 582 | 7.63 | 64.164KD | * PtLAC4-fr | Potri.016G083400 | 189 | ||
| PtLAC29 | Potri.010G193100 | 575 | 8.75 | 63.672KD | * PtLAC5-fr | Potri.T096500 | 183 |
| Foreground Branches | Model | np | LNL | 2Δ (LNL) | Estimates of Parameters | Model Compared | LRT p-Value | Positive Sites | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subfamily I | Model A | 420 | −105,416.831390 | 6.62418 | Site class | 0 | 1 | 2a | 2b | Model A vs.Model A null | p < 0.05 | 163 L 334 F (p > 0.95) |
| f | 0.83718 | 0.09491 | 0.06099 | 0.00691 | ||||||||
| ω0 | 0.12738 | 1.00000 | 0.12738 | 1.00000 | ||||||||
| ω1 | 0.12738 | 1.00000 | 18.62163 | 18.62163 | ||||||||
| Model A null | 419 | −105,420.143482 | 1 | Not Allowed | ||||||||
| Subfamily II | Model A | 412 | −103,339.374083 | 5.42867 | Site class | 0 | 1 | 2a | 2b | Model A vs.Model A null | p < 0.05 | 26 E (p > 0.95) |
| f | 0.85422 | 0.09827 | 0.04261 | 0.00490 | ||||||||
| ω0 | 0.12861 | 1.00000 | 0.12861 | 1.00000 | ||||||||
| ω1 | 0.12861 | 1.00000 | 49.21745 | 49.21745 | ||||||||
| Model A null | 411 | −103,342.088418 | 1 | Not Allowed | ||||||||
| Subfamily III | Model A | 420 | −105,420.468381 | 5.27829 | Site class | 0 | 1 | 2a | 2b | Model A vs.Model A null | p < 0.05 | 26 E (p > 0.95) |
| f | 0.85571 | 0.09694 | 0.04254 | 0.00482 | ||||||||
| ω0 | 0.12741 | 1.00000 | 0.12741 | 1.00000 | ||||||||
| ω1 | 0.12741 | 1.00000 | 43.09093 | 43.09093 | ||||||||
| Model A null | 419 | −105,423.107524 | 1 | Not Allowed | ||||||||
| Subfamily IV | Model A | 420 | −105,405.397615 | 13.5655 | Site class | 0 | 1 | 2a | 2b | Model A vs.Model A null | p < 0.01 | 55 C 93 T 95 L 170 M 227 G 236 S 242 A 324 F (p > 0.95) |
| f | 0.80518 | 0.09121 | 0.09307 | 0.01054 | ||||||||
| ω0 | 0.1271 | 1.00000 | 0.12713 | 1.00000 | ||||||||
| ω1 | 0.12713 | 1.00000 | 7.23646 | 7.23646 | ||||||||
| Model A null | 419 | −105,412.180374 | 1 | Not Allowed | ||||||||
| Subfamily V | Model A | 420 | −105,411.671727 | 6.82632 | Site class | 0 | 1 | 2a | 2b | Model A vs.Model A null | p < 0.01 | 2 V 33 H 50 R 179 I 252 R (p > 0.95) |
| f | 0.73746 | 0.08362 | 0.16070 | 0.01822 | ||||||||
| ω0 | 0.12711 | 1.00000 | 0.12711 | 1.00000 | ||||||||
| ω1 | 0.12711 | 1.00000 | 9.12388 | 9.12388 | ||||||||
| Model A null | 419 | −105,415.084887 | 1 | Not Allowed | ||||||||
| Subfamily VI | Model A | 420 | −105,420.856612 | 3.31621 | Site class | 0 | 1 | 2a | 2b | Model A vs.Model A null | p > 0.05 | Not Found |
| f | 0.85708 | 0.09723 | 0.04103 | 0.00466 | ||||||||
| ω0 | 0.12743 | 1.00000 | 0.12743 | 1.00000 | ||||||||
| ω1 | 0.12743 | 1.00000 | 4.04102 | 4.04102 | ||||||||
| Model A null | 419 | −105,422.514717 | 1 | Not Allowed | ||||||||
| Subfamily VII | Model A | 420 | −105,423.378874 | 0.05589 | Site class | 0 | 1 | 2a | 2b | Model A vs.Model A null | p > 0.05 | Not Found |
| f | 0.00193 | 0.00022 | 0.89619 | 0.10167 | ||||||||
| ω0 | 0.12745 | 1.00000 | 0.12745 | 1.00000 | ||||||||
| ω1 | 0.12745 | 1.00000 | 5.99537 | 5.99537 | ||||||||
| Model A null | 419 | −105,423.406821 | 1 | Not Allowed | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, N.; Han, X.-M.; Xue, Y.; Zhuge, X.-L.; Guan, C.-N.; Yang, H.-L. Genome-Wide Comprehensive Analysis of PtLACs: Prediction and Verification of the Functional Divergence of Tandem-Duplicated Genes. Forests 2022, 13, 157. https://doi.org/10.3390/f13020157
Xu N, Han X-M, Xue Y, Zhuge X-L, Guan C-N, Yang H-L. Genome-Wide Comprehensive Analysis of PtLACs: Prediction and Verification of the Functional Divergence of Tandem-Duplicated Genes. Forests. 2022; 13(2):157. https://doi.org/10.3390/f13020157
Chicago/Turabian StyleXu, Nan, Xue-Min Han, Yuan Xue, Xiang-Lin Zhuge, Chao-Nan Guan, and Hai-Ling Yang. 2022. "Genome-Wide Comprehensive Analysis of PtLACs: Prediction and Verification of the Functional Divergence of Tandem-Duplicated Genes" Forests 13, no. 2: 157. https://doi.org/10.3390/f13020157