
Incompatibility Phylogenetic Signals between Double-Digest Restriction Site-Associated DNA Sequencing and Plastid Genomes in Chinese Curcuma (Zingiberaceae)—A Recent Qinghai–Tibetan Plateau Diversification Genera


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. Sequencing and Clustering
2.3. Phylogenetic Analyses
2.4. The Inference of Combination with Multi-Locus Species Trees
2.5. Consistency Analysis of Inferred Trees
3. Results
3.1. Sequence Characters
3.2. Phylogenetic Inference
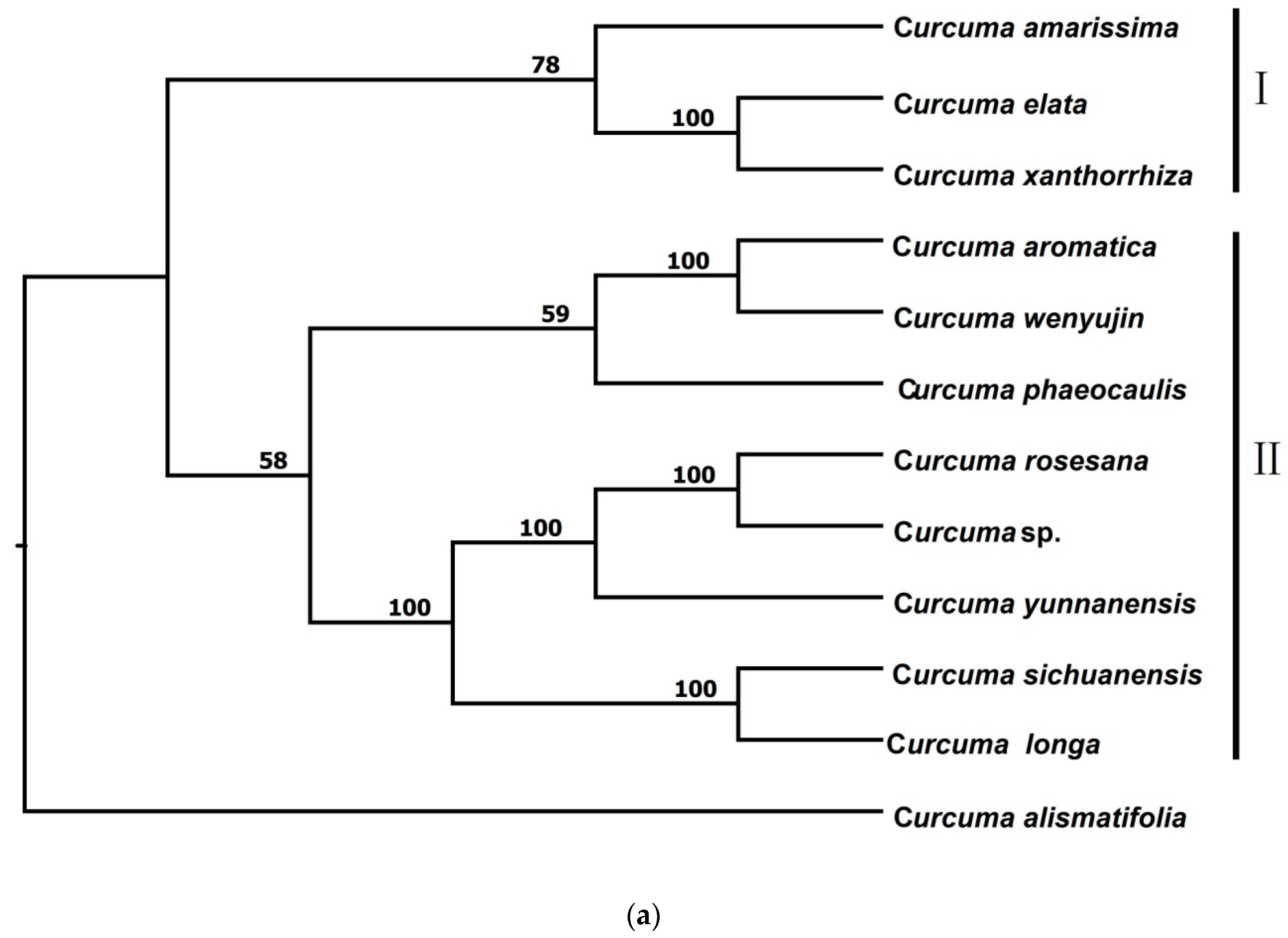
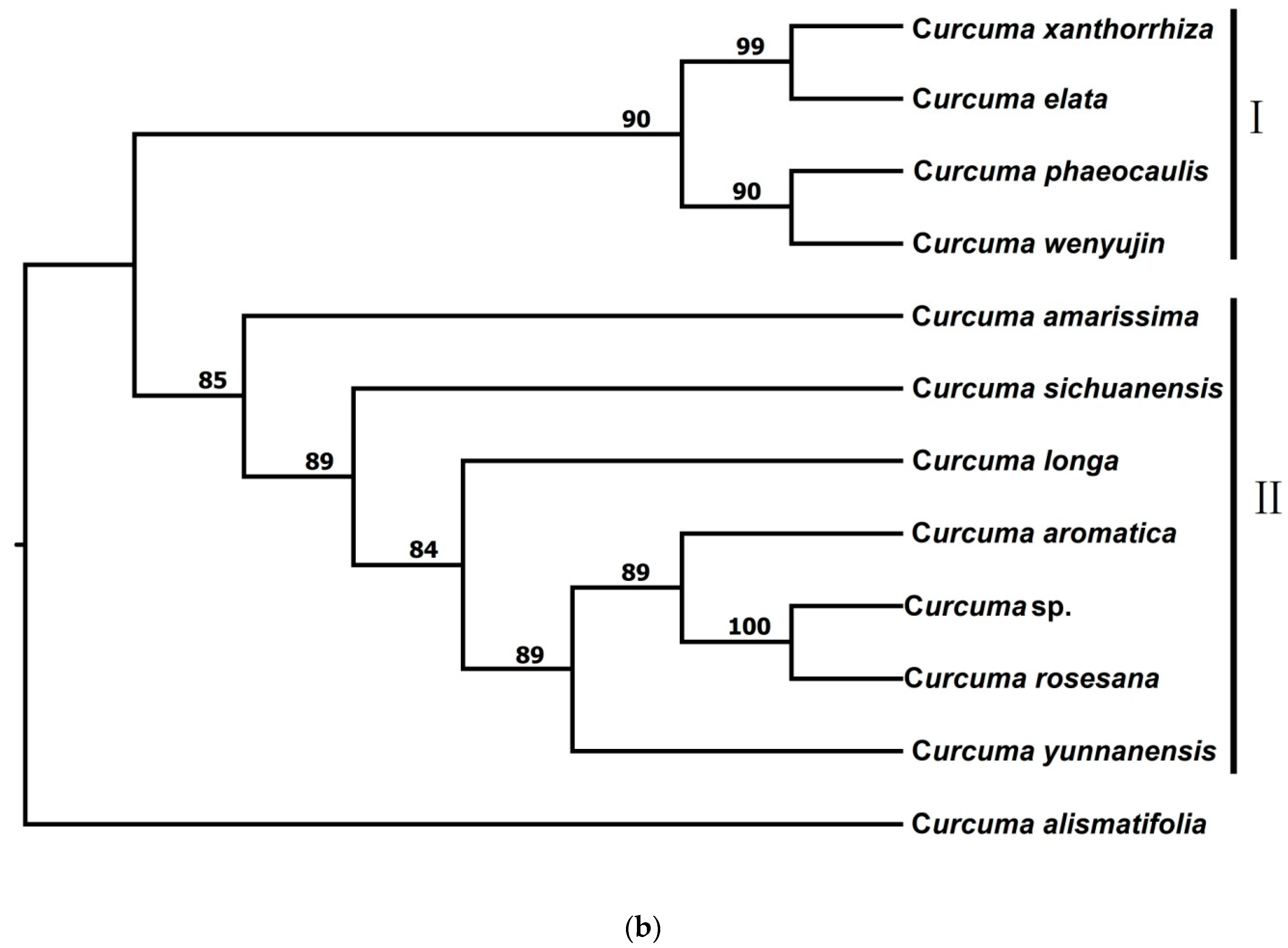
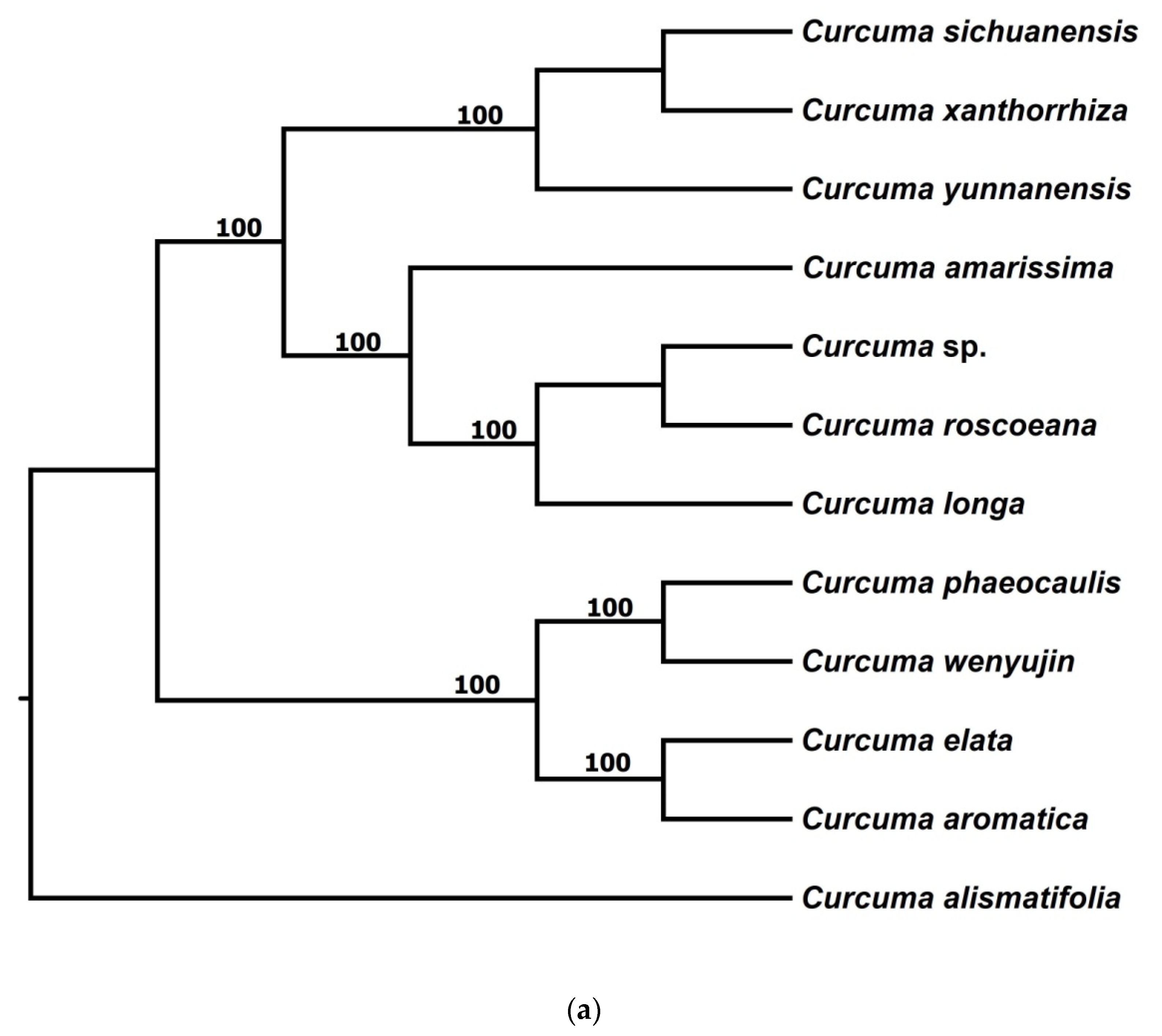
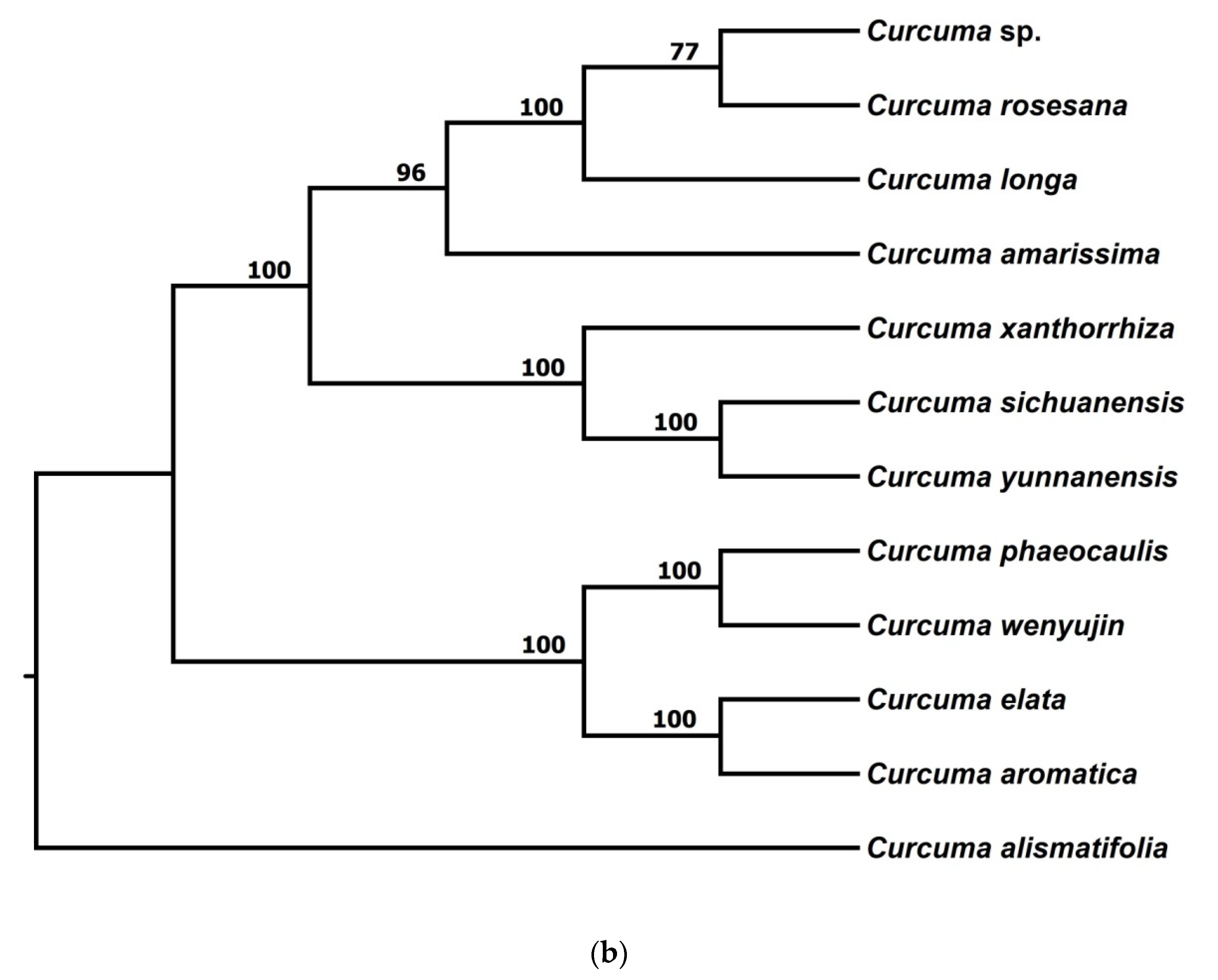
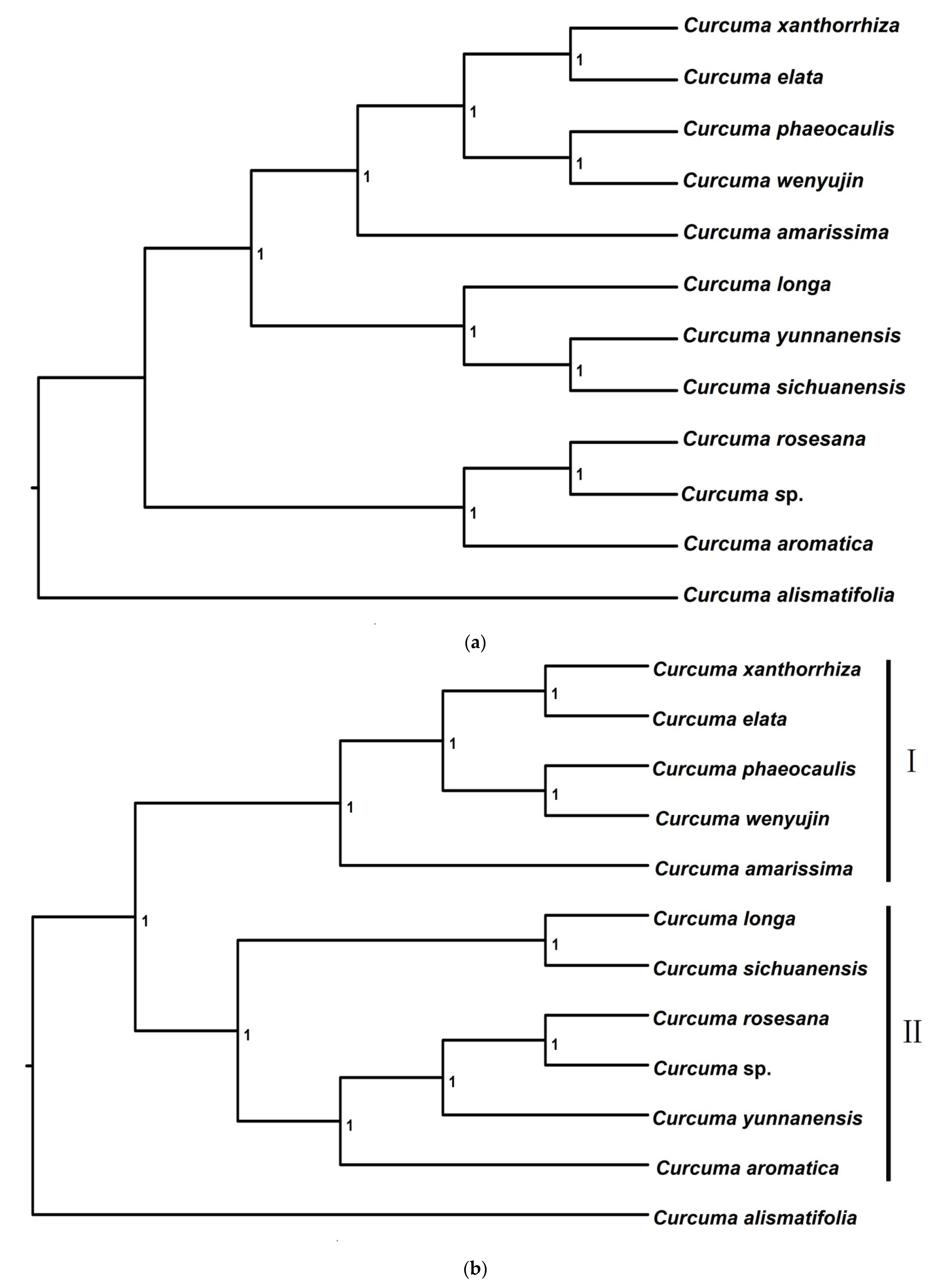
3.2.1. Phylogenetic Trees from pyRAD
3.2.2. Phylogenetic Trees from STACKS
3.2.3. Phylogenetic Trees from Chloroplast Genome Data
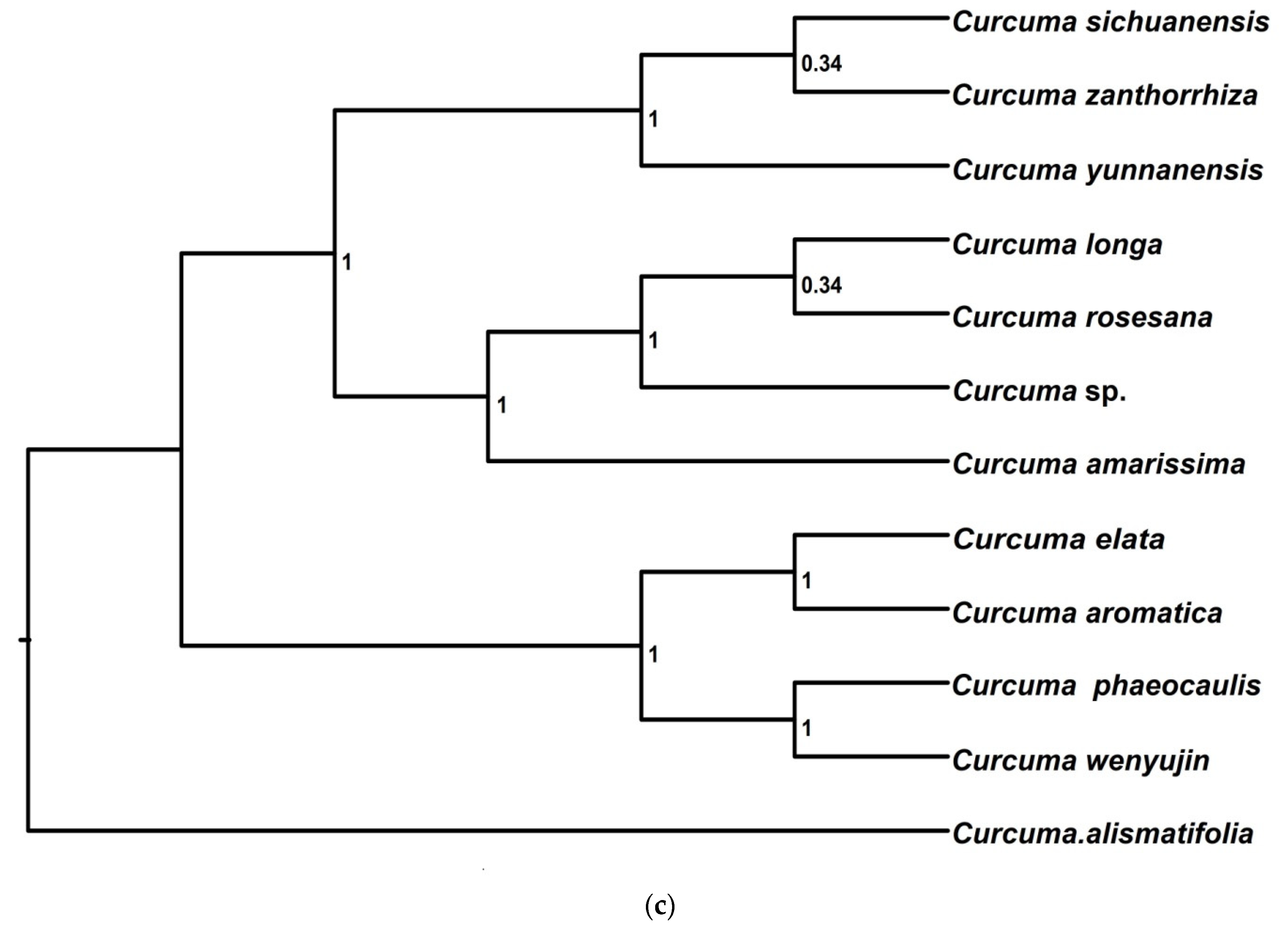
3.2.4. Species Tree Inference
3.2.5. Inconsistency of Inferred Trees
4. Discussion
4.1. Phylogenetic Incompatibility and Biological Implications
4.2. The Genomic Data for Inferring the Phylogeny of Curcuma
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Givnish, T. Adaptive radiation and molecular systematics: Aims and conceptual issues. In Molecular Evolution and Adaptive Radiation; Givnish, T.J.S.K., Ed.; Cambridge University Press: New York, NY, USA, 1997; pp. 1–54. [Google Scholar]
- Li, J. The Qinghai-Tibet Plateau Uplifting and Environmental Evolution in Asia: Article Collection of Academician; Science Press: Beijing, China, 2006. [Google Scholar]
- Liu, X.; Dong, B. Influence of the Tibetan Plateau uplift on the Asian monsoon-arid environment evolution. Chin. Sci. Bull. 2013, 58, 4277–4291. [Google Scholar] [CrossRef] [Green Version]
- Hoorn, C.; Mosbrugger, V.; Mulch, A.; Antonelli, A. Biodiversity from mountain building. Nat. Geosci. 2013, 6, 154. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, J.-Q.; Nie, Z.-L.; Zhong, Y.; Sun, H. Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Front. Genet. 2014, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Favre, A.; Päckert, M.; Pauls, S.U.; Jähnig, S.C.; Uhl, D.; Michalak, I.; Muellner-Riehl, A.N. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol. Rev. 2015, 90, 236–253. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.W.; Ree, R.H. Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proc. Natl. Acad. Sci. USA 2017, 114, E3444–E3451. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.M.; Yue, J.P.; Nie, Z.L.; Li, Z.M.; Comes, H.P.; Sun, H. Phylogeography of Sophora davidii (Leguminosae) across the ‘Tanaka-Kaiyong Line’, an important phytogeographic boundary in Southwest China. Mol. Ecol. 2013, 22, 4270–4288. [Google Scholar] [CrossRef]
- Liang, H.; Deng, J.; Gao, G.; Ding, C.; Zhang, L.; Xu, K.; Wang, H.; Yang, R. Inferring the Phylogeny and Divergence of Chinese Curcuma (Zingiberaceae) in the Hengduan Mountains of the Qinghai–Tibet Plateau by Reduced Representation Sequencing. Forests 2021, 12, 520. [Google Scholar] [CrossRef]
- Záveská, E.; Fér, T.; Šída, O.; Krak, K.; Marhold, K.; Leong-Škorničková, J. Phylogeny of Curcuma (Zingiberaceae) based on plastid and nuclear sequences: Proposal of the new subgenus Ecomata. TAXON 2012, 61, 747–763. [Google Scholar] [CrossRef]
- Kress, W.J.; Prince, L.M.; Williams, K.J. The phylogeny and a new classification of the gingers (Zingiberaceae): Evidence from molecular data. Am. J. Bot. 2002, 89, 1682–1696. [Google Scholar] [CrossRef]
- Nian, L.; Telin, W. Notes on Curcuma in China. J. Trop. Subtrop. Bot. 1999, 7, 146–150. [Google Scholar]
- Chen, J.; Zhao, J.; Erickson, D.L.; Xia, N.; Kress, W.J. Testing DNA barcodes in closely related species of Curcuma (Zingiberaceae) from Myanmar and China. Mol. Ecol. Resour. 2015, 15, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.B.; Ding, C.; Zhang, L.; Zhou, Y.; Yang, R. Relationships among six herbal species (Curcuma) assessed by four isozymes. Phyton 2011, 80, 181–188. [Google Scholar]
- Deng, J.B.; Ding, C.; Zhang, L.; Yang, R.; Yang, R. Authentication of three related herbal species (Curcuma) by DNA barcoding. J. Med. Plants Res. 2011, 5, 6401–6406. [Google Scholar]
- Deng, J.; Liang, H.; Zhang, L.; Zhang, W.; Zhang, G.; Luo, X.; Yang, R.; Ahmad, K.S. Evaluation on genetic relationships among China’s endemic Curcuma L. herbs by mtDNA. Phyton 2018, 87, 156–161. [Google Scholar]
- Chen, J.; Xia, N.-H. Pollen morphology of Chinese Curcuma L. and Boesenbergia Kuntz (Zingiberaceae): Taxonomic implications. Flora-Morphol. Distrib. Funct. Ecol. Plants 2011, 206, 458–467. [Google Scholar] [CrossRef]
- Xiao, X.-H.; Shu, G.-M.; Li, L.Y.; Fang, Q.M.; Xia, W.J.; Su, Z.W. Histological and morphological studies on the rhizomes of Curcuma. China J. Chin. Mater. Med. 2004, 29, 395–399. [Google Scholar]
- Xiao, X.-H.; Zhao, Y.-L.; Jin, C.; Shu, G.-M.; Fang, Q.-M.; Shu, Z.W. Histological and morphological studies on leaves of Curcuma in China. China J. Chin. Mater. Med. 2004, 29, 203–207. [Google Scholar]
- Záveská, E.; Fér, T.; Šída, O.; Marhold, K.; Leong-Škorničková, J. Hybridization among distantly related species: Examples from the polyploid genus Curcuma (Zingiberaceae). Mol. Phylogenetics Evol. 2016, 100, 303–321. [Google Scholar] [CrossRef]
- Cao, H.; Sasaki, Y.; Fushimi, H.; Komatsu, K. Molecular Analysis of Medicinally-Used Chinese and Japanese Curcuma Based on 18S rRNA Gene and trnK Gene Sequences. Biol. Pharm. Bull. 2001, 24, 1389–1394. [Google Scholar] [CrossRef] [Green Version]
- Steven, G.; Subramanyam, R. Testing plant barcoding in a sister species complex of pantropical Acacia (Mimosoideae, Fabaceae). Mol. Ecol. Resour. 2009, 9, 172–180. [Google Scholar] [CrossRef]
- Lemmon, E.M.; Lemmon, A.R. High-Throughput Genomic Data in Systematics and Phylogenetics. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 99–121. [Google Scholar] [CrossRef] [Green Version]
- Harvey, M.G.; Smith, B.T.; Glenn, T.C.; Faircloth, B.C.; Brumfield, R.T. Sequence capture versus restriction site associated DNA sequencing for shallow systematics. Syst. Biol. 2016, 65, 910–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godden, G.T.; Jordon-Thaden, I.E.; Chamala, S.; Crowl, A.A.; García, N.; Germain-Aubrey, C.C.; Heaney, J.M.; Latvis, M.; Qi, X.; Gitzendanner, M.A. Making next-generation sequencing work for you: Approaches and practical considerations for marker development and phylogenetics. Plant Ecol. Divers. 2012, 5, 427–450. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of High-Density Genetic Maps for Barley and Wheat Using a Novel Two-Enzyme Genotyping-by-Sequencing Approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- McCormack, J.E.; Hird, S.M.; Zellmer, A.J.; Carstens, B.C.; Brumfield, R.T. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogenetics Evol. 2013, 66, 526–538. [Google Scholar] [CrossRef]
- Eaton, D.A.; Spriggs, E.L.; Park, B.; Donoghue, M.J. Data from: Misconceptions on missing data in RAD-seq phylogenetics with a deep-scale example from flowering plants. Syst. Biol. 2016, 66, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Massatti, R.; Reznicek, A.A.; Knowles, L.L. Utilizing RADseq data for phylogenetic analysis of challenging taxonomic groups: A case study in Carex sect. Racemosae. Am. J. Bot. 2016, 103, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Hipp, A.L.; Manos, P.S.; González-Rodríguez, A.; Hahn, M.; Kaproth, M.; McVay, J.D.; Avalos, S.V.; Cavender-Bares, J. Sympatric parallel diversification of major oak clades in the Americas and the origins of Mexican species diversity. New Phytol. 2018, 217, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-X.; Zhou, M.-Y.; Yang, G.-Q.; Zhang, Y.-X.; Ma, P.-F.; Guo, C.; Vorontsova, M.S.; Li, D.-Z. ddRAD analyses reveal a credible phylogenetic relationship of the four main genera of Bambusa-Dendrocalamus-Gigantochloa complex (Poaceae: Bambusoideae). Mol. Phylogenetics Evol. 2020, 146, 106758. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Hao, Y.-J.; Li, J.-H.; Fu, C.-X.; Soltis, P.S.; Soltis, D.E.; Zhao, Y.-P. Phylogenomic conflict resulting from ancient introgression following species diversification in Stewartia s.l. (Theaceae). Mol. Phylogenetics Evol. 2019, 135, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, E.L.; Eaton, D.A.R.; Sweeney, P.W.; Schlutius, C.; Edwards, E.J.; Donoghue, M.J. Restriction-Site-Associated DNA Sequencing Reveals a Cryptic Viburnum Species on the North American Coastal Plain. Syst. Biol. 2019, 68, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.A.; Keuter, A.; McVay, J.D.; Hipp, A.L.; Manos, P.S. The evolution and diversification of the red oaks of the California Floristic Province (Quercus section Lobatae, series Agrifoliae). Am. J. Bot. 2017, 104, 1581–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, O.M.; Ortiz, E.M.; Simpson, B.B. Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high-Andean diversification (Asteraceae: Astereae: Diplostephium). New Phytol. 2017, 214, 1736–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.-M.; Zhu, G.-F.; Xu, Y.-C.; Ye, Y.-J.; Liu, J.-M. Complete Chloroplast Genomes of Three Medicinal Alpinia Species: Genome Organization, Comparative Analyses and Phylogenetic Relationships in Family Zingiberaceae. Plants 2020, 9, 286. [Google Scholar] [CrossRef] [Green Version]
- Gui, L.; Jiang, S.; Xie, D.; Yu, L.; Huang, Y.; Zhang, Z.; Liu, Y. Analysis of complete chloroplast genomes of Curcuma and the contribution to phylogeny and adaptive evolution. Gene 2020, 732, 144355. [Google Scholar] [CrossRef]
- Gonçalves, D.J.; Simpson, B.B.; Ortiz, E.M.; Shimizu, G.H.; Jansen, R.K. Incongruence between gene trees and species trees and phylogenetic signal variation in plastid genes. Mol. Phylogenetics Evol. 2019, 138, 219–232. [Google Scholar] [CrossRef]
- Saarela, J.M.; Burke, S.V.; Wysocki, W.P.; Barrett, M.D.; Clark, L.G.; Craine, J.M.; Peterson, P.M.; Soreng, R.J.; Vorontsova, M.S.; Duvall, M.R. A 250 plastome phylogeny of the grass family (Poaceae): Topological support under different data partitions. PeerJ 2018, 6, e4299. [Google Scholar] [CrossRef]
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef]
- Palmer, J.D.; Jansen, R.K.; Michaels, H.J.; Chase, M.W.; Manhart, J.R. Chloroplast DNA Variation and Plant Phylogeny. Ann. Mo. Bot. Gard. 1988, 75, 1180. [Google Scholar] [CrossRef]
- Gu, C.; Ma, L.; Wu, Z.; Chen, K.; Wang, Y. Comparative analyses of chloroplast genomes from 22 Lythraceae species: Inferences for phylogenetic relationships and genome evolution within Myrtales. BMC Plant Biol. 2019, 19, 281. [Google Scholar] [CrossRef] [PubMed]
- Rogalski, M.; Vieira, L.D.N.; Fraga, H.; Guerra, M.P. Plastid genomics in horticultural species: Importance and applications for plant population genetics, evolution, and biotechnology. Front. Plant Sci. 2015, 6, 586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Chen, J. Comparison and Phylogenetic Analyses of Nine Complete Chloroplast Genomes of Zingibereae. Forests 2021, 12, 710. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, Y.; Deng, J.; Gao, G.; Ding, C.; Zhang, L.; Yang, R. The Complete Chloroplast Genome Sequences of 14 Curcuma Species: Insights Into Genome Evolution and Phylogenetic Relationships Within Zingiberales. Front. Genet. 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ma, P.F.; Yang, G.Q.; Ye, X.Y.; Guo, Y.; Liu, J.X.; Liu, Y.L.; Eaton, D.A.; Guo, Z.H.; Li, D.Z. Parallel ddRAD and genome skimming analyses reveal a radiative and reticulate evolutionary history of the temperate bamboos. Syst. Biol. 2021, 70, 756–773. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Soltis, D.E.; Soltis, P.S.; Zhu, X.; Burleigh, J.G.; Chen, Z. Data from: Deep phylogenetic incongruence in the angiosperm clade Rosidae. Mol. Phylogenetics Evol. 2015, 83, 156–166. [Google Scholar] [CrossRef]
- Seehausen, O. Hybridization and adaptive radiation. Trends Ecol. Evol. 2004, 19, 198–207. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, T.; Kanwal, N.; Zhao, Y.; Bai, G.; Zhao, G. Completion of Eight Gynostemma BL. (Cucurbitaceae) Chloroplast Genomes: Characterization, Comparative Analysis, and Phylogenetic Relationships. Front. Plant Sci. 2017, 8, 1583. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.-Q.; Chen, Y.-M.; Wang, J.-P.; Guo, C.; Zhao, L.; Wang, X.-Y.; Guo, Y.; Li, L.; Li, D.-Z.; Guo, Z.-H. Development of a universal and simplified ddRAD library preparation approach for SNP discovery and genotyping in angiosperm plants. Plant Methods 2016, 12, 39. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, D.; Ree, R.H. Inferring Phylogeny and Introgression using RADseq Data: An Example from Flowering Plants (Pedicularis: Orobanchaceae). Syst. Biol. 2013, 62, 689–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.-C.; Yu, Y.; Liu, X.; Pais, A.; Ranney, T.G.; Whetten, R.W.; Xiang, J. Phylogenomics of polyploid Fothergilla (Hamamelidaceae) by RAD-tag based GBS-insights into species origin and effects of software pipelines. J. Syst. Evol. 2015, 53, 432–447. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Kuma, K.I.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Heled, J.; Drummond, A.J. Bayesian Inference of Species Trees from Multilocus Data. Mol. Biol. Evol. 2010, 27, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Nye, T.M.W.; Liò, P.; Gilks, W.R. A novel algorithm and web-based tool for comparing two alternative phylogenetic trees. Bioinformatics 2005, 22, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Biasatti, D.; Wang, Y.; Gao, F.; Xu, Y.; Flynn, L. Paleoecologies and paleoclimates of late cenozoic mammals from Southwest China: Evidence from stable carbon and oxygen isotopes. J. Southeast Asian Earth Sci. 2012, 44, 48–61. [Google Scholar] [CrossRef]
- Sun, K.; Liu, Q.-Y.; Wang, A.; Gao, Y.-W.; Zhao, L.-C.; Guan, W.-B. Comparative Analysis and Phylogenetic Implications of Plastomes of Five Genera in Subfamily Amyridoideae (Rutaceae). Forests 2021, 12, 277. [Google Scholar] [CrossRef]
- Leong-Škorničková, J.; Šída, O.; Jarolímová, V.; Sabu, M.; Fér, T.; Trávníček, P.; Suda, J. Chromosome Numbers and Genome Size Variation in Indian Species of Curcuma (Zingiberaceae). Ann. Bot. 2007, 100, 505–526. [Google Scholar] [CrossRef] [Green Version]
- Mario, F.M.; Greg, M.; Beatriz, V.; Llorenç, S.; Pablo, V.; Glover, B.J. Resolving Recent Plant Radiations: Power and Robustness of Genotyping-by-Sequencing. Syst. Biol. 2017, 2, 2. [Google Scholar]
- Lee, J.Y.; Joseph, L.; Edwards, S.V. A Species Tree for the Australo-Papuan Fairy-wrens and Allies (Aves: Maluridae). Syst. Biol. 2012, 61, 253–271. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.E.; Keller, I.; Wittwer, S.; Selz, O.M.; Mwaiko, S.; Greuter, L.; Sivasundar, A.; Seehausen, O. Data from: Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Mol. Ecol. 2022, 3, 787–798. [Google Scholar]
- Dupuis, J.R.; Brunet, B.M.T.; Bird, H.M.; Lumley, L.M.; Fagua, G.; Boyle, B.; Levesque, R.; Cusson, M.; Powell, J.A.; Sperling, F.A.H. Genome-wide SNPs resolve phylogenetic relationships in the North American spruce budworm (Choristoneura fumiferana) species complex. Mol. Phylogenet Evol. 2017, 111, 158–168. [Google Scholar] [CrossRef]
- Tonini, J.; Moore, A.; Stern, D.; Shcheglovitova, M.; Ortí, G. Concatenation and Species Tree Methods Exhibit Statistically Indistinguishable Accuracy under a Range of Simulated Conditions. PLoS Curr. 2015, 7, 7. [Google Scholar] [CrossRef]
- Bombonato, J.R.; do Amaral, D.T.; Silva, G.A.R.; Khan, G.; Moraes, E.M.; da Silva Andrade, S.C.; Eaton, D.A.R.; Alonso, D.P.; Ribolla, P.E.M.; Taylor, N.; et al. The potential of genome-wide RAD sequences for resolving rapid radiations: A case study in Cactaceae. Mol. Phylogenetics Evol. 2020, 151, 106896. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Voucher Number | Locality | Total Loci After pyRAD | Total Loci after STACKS | Ploidy Level |
|---|---|---|---|---|---|
| C. xanthorrhiza | 38, 2003, 0324 | Thailand | 47,240 | 34,562 | 3 |
| C. elata | 00, 2005, 0073 | Yunnan, China | 50,784 | 32,430 | 3 |
| C. yunnanensis | C29064 | Yunnan, China | 51,896 | 34,011 | 3 |
| C. alismatifolia | 38, 2003, 0354 | Thailand | 47,062 | 22,622 | 3 |
| C. amarissima | 00, 2009, 0882 | Yunnan, China | 49,623 | 33,974 | 3 |
| C. sichuanensis | 00, 2001, 2519 | Yunnan, China | 53,508 | 34,797 | 3 |
| C. aromatica | 00, 2007, 0944 | Yunnan, China | 46,837 | 37,155 | 3 |
| C. wenyujin | 00, 2010, 0862 | Guangxi, China | 59,172 | 37,942 | 3 |
| C. longa | 00, 2008, 0868 | Yunnan, China | 52,467 | 35,504 | 3 |
| C. rosesana | 00, 2001, 1412 | Guangdong, China | 53,489 | 31,633 | 3 |
| C. sp. | 00, 2004, 0191 | Guangdong, China | 59,824 | 33,012 | 3 |
| C. phaeocaulis | 00, 2000, 0695 | Yunnan, China | 56,548 | 38,111 | 4 |
| . | pyRAD | STACKS | Chloroplast Genomes | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Full concatenated super-matrix with ML (Figure 1a) | Concatenated loci with ML (Figure 1b) | MCC with BEAST (Figure 4a) | Full concatenated super-matrix with ML (Figure 2a) | Concatenated loci with ML (Figure 2b) | MCC with BEAST (Figure 4b) | Coding sequence with ML (Figure 3a) | Complete chloroplast genomes with ML (Figure 3b) | MCC with BEAST (Figure 4c) | ||
| pyRAD | Full concatenated super-matrix with ML (Figure 1a) | 52.00% | 55.30% | 76.10% | 56.10% | 69.00% | 50.50% | 50.50% | 44.80% | |
| Concatenated loci with ML (Figure 1b) | — | 70.30% | 53.50% | 69.90% | 66.30% | 72.40% | 76.70% | 65.40% | ||
| MCC with BEAST (Figure 4a) | — | — | 61.50% | 78.90% | 81.50% | 57.50% | 63.30% | 56.30% | ||
| STACKS | Full concatenated super-matrix with ML (Figure 2a) | — | — | — | 64.00% | 75.60% | 50.90% | 50.90% | 45.70% | |
| Concatenated loci with ML (Figure 2b) | — | — | — | — | 83.30% | 58.40% | 57.90% | 52.50% | ||
| MCC with BEAST (Figure 4b) | — | — | — | — | — | 55.10% | 55.10% | 54.10% | ||
| Chloroplast genomes | coding sequence with ML (Figure 3a) | — | — | — | — | — | — | 92.60% | 92.60% | |
| complete chloroplast genomes with ML (Figure 3b) | — | — | — | — | — | — | — | 85.20% | ||
| MCC with BEAST (Figure 4c) | — | — | — | — | — | — | — | — | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, H.; Deng, J.; Gao, G.; Ding, C.; Zhang, L.; Yang, R. Incompatibility Phylogenetic Signals between Double-Digest Restriction Site-Associated DNA Sequencing and Plastid Genomes in Chinese Curcuma (Zingiberaceae)—A Recent Qinghai–Tibetan Plateau Diversification Genera. Forests 2022, 13, 280. https://doi.org/10.3390/f13020280
Liang H, Deng J, Gao G, Ding C, Zhang L, Yang R. Incompatibility Phylogenetic Signals between Double-Digest Restriction Site-Associated DNA Sequencing and Plastid Genomes in Chinese Curcuma (Zingiberaceae)—A Recent Qinghai–Tibetan Plateau Diversification Genera. Forests. 2022; 13(2):280. https://doi.org/10.3390/f13020280
Chicago/Turabian StyleLiang, Heng, Jiabin Deng, Gang Gao, Chunbang Ding, Li Zhang, and Ruiwu Yang. 2022. "Incompatibility Phylogenetic Signals between Double-Digest Restriction Site-Associated DNA Sequencing and Plastid Genomes in Chinese Curcuma (Zingiberaceae)—A Recent Qinghai–Tibetan Plateau Diversification Genera" Forests 13, no. 2: 280. https://doi.org/10.3390/f13020280