Abstract
The conservation and diversity of microRNA (miRNA) families provide insights into the evolution of miRNA genes. However, there are few studies to explore the miRNA genes at the genus level in plants. Here, we identified 1194 miRNA loci in four Populus species P. deltoides, P. euphratica, P. tremula, and P. trichocarpa from Aigeiros, Turanga, Populus, and Tacamahaca sections, respectively, by combining de novo and homolog-based approaches. Our results indicated that a similar number of miRNA loci exist in each species (296–301 miRNA loci). Among the identified 143 miRNA families, 68 families are shared by the studied four species, and 31 families are species-specific, which might be related to local adaptation. Additionally, multiple miRNA-related single nucleotide polymorphisms (SNPs) were found, indicating that polymorphisms in pre-miRNA hairpins were likely to affect miRNA biogenesis. This study expanded the breadth and depth of miRNA annotations and provided valuable resources for further exploring the diversity and function of poplar miRNAs.
1. Introduction
The genus Populus receives increasing attention as a source of fiber, lumber, windbreaks, and biomass energy, ideally suited for supporting rural livelihoods, alleviating poverty, and contributing to sustainable development [1]. The genus appears to have relatively low species diversity compared to other tree genera, with 30 species subdivided into six sections (Abaso, Aigeiros, Leucoides, Populus, Tacamahaca, and Turanga) [2]. Nevertheless, poplars show extensive and varied ranges of habitats; for example, species from Aigeiros and Tacamahaca sections are typically riparian, P. tremuloides and the closely-related P. tremula occur in mountainous or upland habitats, and P. euphratica grows in extremely hot and dry environments [3].
Poplars have become one of the most widely studied model organisms, mainly because of their relatively short reproductive cycle; fast growth; small genome size; and ease of transformation, regeneration, and asexual reproduction [3,4,5]. More recently, the development of a series of technologies, such as single-molecule sequencing, chromosome conformation capture (such as Hi-C), and optical mapping, have greatly improved genome sequence completeness and contiguity of poplars [6,7,8]. Additionally, extensive poplar transcriptomes, small RNAomes, degradome, and whole-genome resequencing knowledge are available [9,10,11]. The in-depth excavation of these genomic resources would enhance our understanding of poplars’ diversity and habitat adaptability.
MicroRNAs (miRNAs) are endogenous non-coding RNA gene products [12], which associate with argonaute (AGO) proteins to negatively regulate gene expression through cleavage and/or translational inhibition of target mRNAs [13]. MiRNAs have been demonstrated to have an influential role in response to extrinsic stress (such as drought, temperature, salinity, and exposure to UV radiation) as well as developmental processes, including root initiation and development of leaf, vascular, flower, and seed. For example, the overexpression of miR167a inhibited target transcripts (PeARF8.1) and improved lateral root development in poplar [13]. Further, numerous biological processes, including the maintenance of genome integrity, metabolism, and immunity against pathogens, are also regulated by miRNAs [14]. As an important regulatory factor, miRNA biogenesis and mechanism of action have become an intense focus of research.
In the past two decades, active research on the specific post-transcriptional regulation of miRNAs in Populus has taken place. For example, miR476a has been shown to be a regulator of wound-induced adventitious rooting in P. tomentosa [15]. The origin, evolution, and potential roles in the local adaptation of Populus-specific miRNAs were examined [16]. Furthermore, considerable efforts were made to explore the role of miRNAs during stress response in P. euphratica [17], P. cathayana [18], and P. trichocarpa [19]. However, although guidelines and specific criteria for accurate annotation of miRNA are unambiguous [20], adherence to the established annotation practices varies among scientists [21]. A synthetic understanding of miRNA diversity and function in Populus will be obscured by specious annotation of putative miRNA loci [22]. Hence, it is urgent to fully and accurately identify miRNA loci and analyze their relationships among sections in the genus Populus.
Here, combining de novo and homolog-based genome-wide identification, we comprehensively annotated miRNAs of four poplar species (P. deltoides, P. euphratica, P. tremula, and P. trichocarpa) (Figure 1). Our specific objectives were to: (1) identify known and novel miRNA loci according to stringent criteria; (2) reveal the interspecific variation of miRNA families and miRNA sequences among different poplar species; and (3) discover miRNA-related single nucleotide polymorphisms (SNPs). This study performed a broad and deep annotation of miRNAs in the Populus genus, which is expected to provide valuable resources for further exploring the biogenesis, diversity, and function of miRNAs.
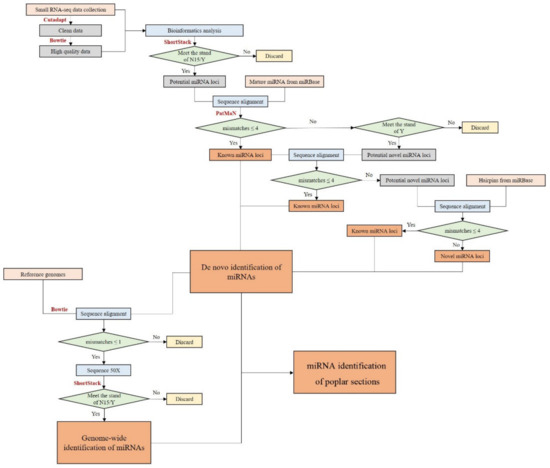
Figure 1.
Flow chart of miRNA loci identification.
2. Materials and Methods
2.1. De Novo Identification of miRNAs Using Small RNA-Seq Data
Small RNA (sRNA) data of P. deltoides (Aigeiros section), P. euphratica (Turanga section), P. tremula (Populus section), and P. trichocarpa (Tacamahaca section) were downloaded from NCBI Sequence Read Archive (SRA). The origins and accession numbers of each sample are shown in Table S1, with a total of 107 small RNA-seq libraries from 8 tissues. The raw data were processed by 3′ adapter trimming using Cutadapt (version 2.10) [23], discarding reads that were <18 nt or >33 nt. The cleaned reads were mapped to the Rfam database (version 11.0) [24] to remove non-coding RNAs (rRNA, tRNA, snRNA, scRNA, and snoRNA) using Bowtie (version 1.3.0) [25] (mismatch ≤ 1) to obtain high quality reads.
ShortStack program (version 3.3.3) [26] was applied to identify potential miRNA loci of the four poplar species using preprocessed sRNA reads and the corresponding reference genomes [6,7,8,10]. The parameter settings of the ShortStack program were: foldsize = 500; mincov = 2; ranmax = 35; and other parameters remained as the default settings. Sequences that met the criteria of N15 (passed all tests except that the miRNA-star was not sequenced) or Y (passed all tests including sequencing of the exact miRNA-star) were reported.
The obtained mature miRNA candidates were aligned with mature miRNA sequences in miRBase (version 22.1) [27] by PatMaN (version 1.2) [28] (mismatches ≤ 4) to identify known miRNAs. The remaining sequences were mapped to sequences of known miRNA precursors to identify potential miRNA-stars. The other candidates meeting the standard of Y were considered novel miRNAs. The scripts for miRNA annotation were written in Perl and Python (Figure 1).
2.2. Homolog-Based Genome-Wide Identification of miRNAs
We aligned the de novo identified miRNA sequences to the reference genomes of the four-poplar species by Bowtie. After this, the alignment sequences were extracted by Samtools (version 1.9) [29] and python scripts (mismatches ≤ 1), which were amplified 50 times and submitted to the ShortStack program to find potential miRNAs. The parameter settings of the ShortStack program were as follows: mismatches = 0; foldsize = 500; mincov = 2; ranmax = 35; and other parameters remained as the default settings. Sequences that met the criteria of N15 or Y were annotated as miRNA loci (Figure 1).
2.3. Analysis of the Conservation of miRNA Loci in Each Species
For P. euphratica, P. tremula, and P. trichocarpa, 50 whole-genome resequencing data of each species were downloaded from NCBI SRA. For P. deltoides, 50 samples were randomly selected from the previous publication of our lab [30]. After removing low-quality reads and trimming of adapter sequences by Trimmomatic (version 0.39), the clean reads were aligned to the corresponding reference genome using Burrows-Wheeler Aligner (BWA, version 0.7.17, http://bio-bwa.sourceforge.net/, accessed on 10 November 2021) with default parameters. Samtools (version 1.9) [29] was further applied to screen primary uniquely mapped reads. BEDTools (version 2.30.0) [31] was applied to calculate the depth and breadth of coverage at the genomic ranges of each miRNA loci. When the average depth was over 2 and the breadth was over 80%, we considered the miRNA locus detected in the sample.
2.4. Analysis of miRNA Families among Different Species
We compared the similarities and differences of miRNA families among the four different poplar species. Next, we examined base variations among all miRNA sequences in the same miRNA family shared by the four-poplar species through python scripts and counted the unique sequences in each miRNA family. Furthermore, based on the alignment of nucleotide sequences in the same miRNA family by ClustalW, a phylogenetic tree was established to understand their clustering relations by MEGA X (available at https://www.megasoftware.net, accessed on 10 November 2021), which was constructed using the maximum composite likelihood method with 1000 bootstrap replicates.
We explored the reason for the miRNA family absence in each species through comparison with the other three species. If one certain miRNA family was absent in a poplar species, the sequences (hairpin and mature miRNA sequences) of the miRNA families in other species were used to search against the genome sequence without this miRNA family. Firstly, the hairpin sequence was aligned to the genome using Blast. If the percentage sequence identity was less than 0.9 or the aligned length was less than 90% of the hairpin length, it indicated that the hairpin sequence was the reason for the absence of this miRNA family. Secondly, the mature miRNA sequences were aligned to the filtered hairpin sequence by Bowtie. If the mismatch number of sequences in a miRNA family was greater than 1, it suggested that the mature miRNA sequence was the reason for the miRNA family’s absence. However, if the number of mismatches was less than or equal to 1, the cause of the miRNA family absence was a hairpin structural failure.
2.5. SNP Calling
The genomic variant information of 549 P. trichocarpa individuals was downloaded from Phytozome. The whole-genome resequencing data of P. deltoides (97 samples) were from our previous publication [30]. The QC and alignment steps were performed as described above. We corrected mate-pair alignments and marked duplicate molecules using the Samtools (version 1.9) [29]. Freebayes (version 0.9.21) [32] was applied to call SNPs. We counted the SNPs in miRNAs loci (including pre-miRNAs, mature miRNAs, and miRNA seed regions (2–7 nt)) to calculate the SNP densities. As shown in Formula (1), the SNP densities of miRNA were defined as the number of SNPs in miRNAs at the site per 1000 nt.
where Yn was the SNP density; Nsnp was the SNP number; and Pn was the base length; nt was nucleotide (n = pre-miRNA, mature miRNA, or miRNA seed region).
2.6. Prediction of miRNA Targets and GO Enrichment Analysis
For each species, the sequences of mature miRNAs and the corresponding transcript sequences were applied for target prediction using the online tool psRNATarget [33] with the default parameters. Specifically, the maximum cutoff of score based on given scoring schema = 3; penalty for G: U pair = 0.5; penalty for other mismatched = 1; extra penalty weight for mismatched in seed region; HSP size = 19; penalty for opening gap = 2; and penalty for extending gap = 0.5. Gene Ontology (GO) [34] terms of the miRNA targets were annotated using Diamond software (version 0.9.19) with information from the Swiss-Prot database, and the E-value was set to ≤10−5. GO enrichment analysis (FDR ≤ 0.05) was performed through the R package clusterprofiler (version 3.18.1) [35].
3. Results
3.1. Identification of miRNA Loci in Each Poplar Species
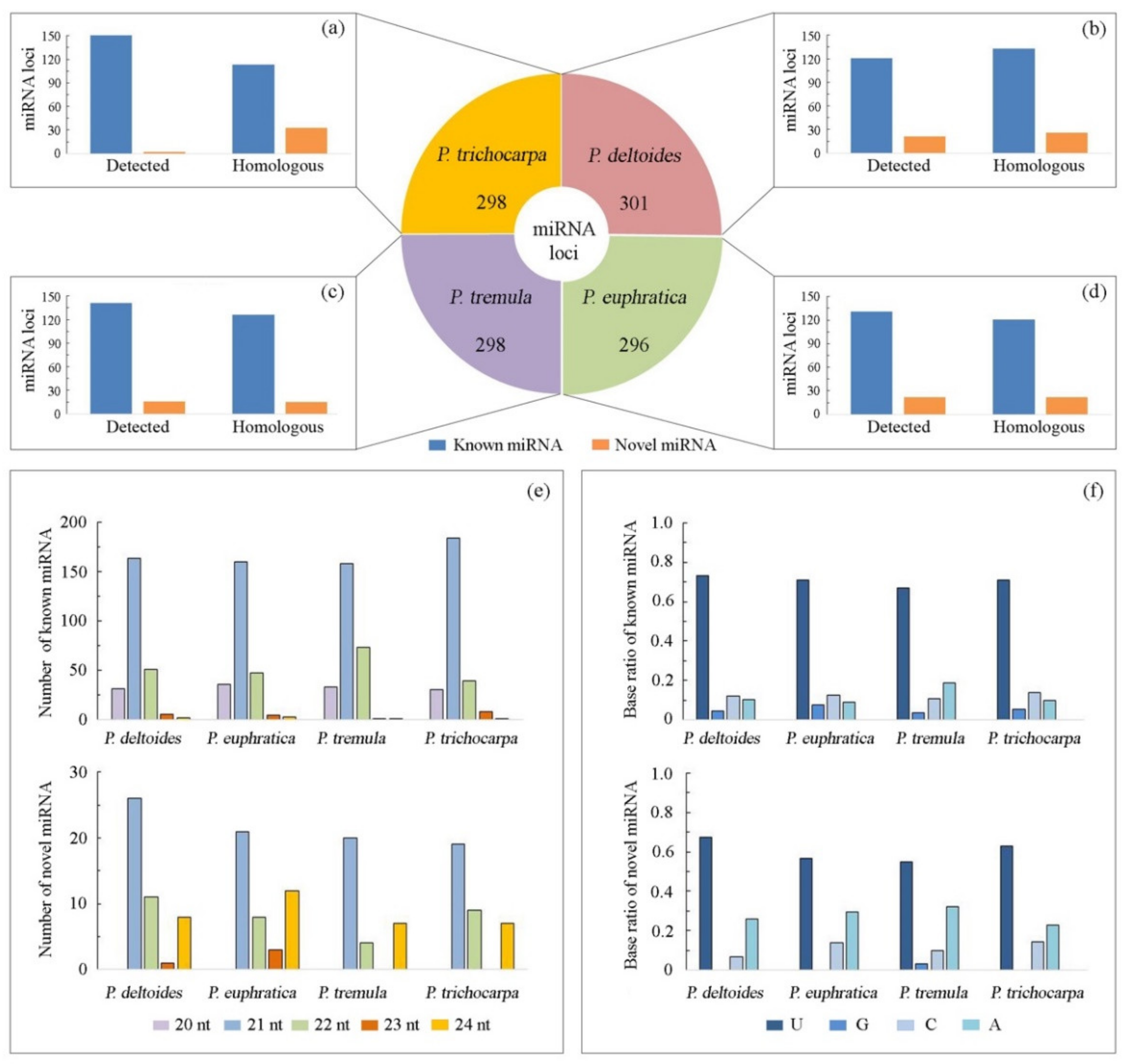
A total of 107 small RNA-seq libraries of four species (P. deltoides, P. euphratica, P. tremula, and P. trichocarpa) from different poplar sections were comprehensively analyzed using our bioinformatic pipeline (Figure 1). Initially, 605 miRNA loci were annotated in all libraries, including 544 known and 61 novel miRNAs (Table S2). As shown in Figure 2a–d, 142, 153, 157, and 153 miRNA loci were de novo identified successively in P. deltoides, P. euphratica, P. tremula, and P. trichocarpa, respectively. Furthermore, these 605 annotated miRNA sequences were successively aligned to the reference genome of the four-poplar species to look for potential miRNA loci that were not detected in the small RNA-seq libraries (Table S3). Compared with the de novo identification results, an additional 159, 143, 141, and 146 miRNA loci were found, respectively, in P. deltoides, P. euphratica, P. tremula, and P. trichocarpa by genome-scale homology search (Figure 2a–d). The length of miRNAs ranged from 20 nt to 24 nt, and the 21 nt miRNAs were the most dominant (Figure 2e). A significant bias towards U was observed in the first nucleotides of miRNA sequences (Figure 2f). To check whether these identified miRNAs are representative of each species, we evaluate the presence of miRNAs in individual natural poplar populations using public and in-house data. By mapping the whole-genome resequencing data onto the genome, we found that 274 (91.0%), 248 (83.8%), 266 (89.3%), and 281 (94.0%) pre-miRNA loci were detected in more than 80% of individuals in P. deltoides, P. euphratica, P. tremula, and P. trichocarpa, respectively, recovering most of the miRNA loci identified in a single reference genome (Table S4).
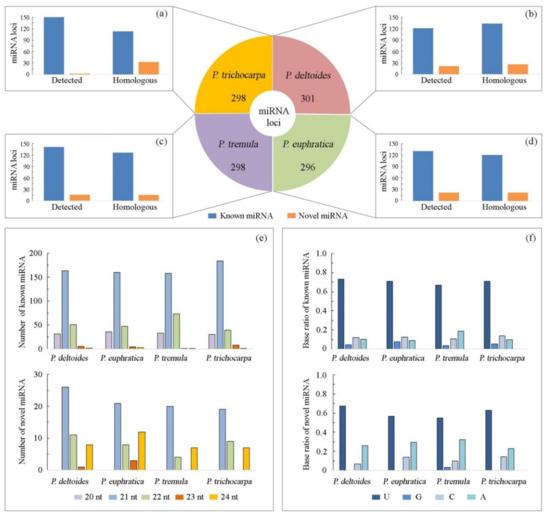
Figure 2.
miRNA annotation of the four-poplar species by combining de novo and homolog-based genome-wide identification. The number of miRNA loci in P. deltoides, P. euphratica, P. tremula, and P. trichocarpa, respectively (a–d). The length of miRNAs (e) and distribution of bases at the 5′ end of miRNAs (f) in the four species.
3.2. Distributions of miRNAs Families in Poplar Species
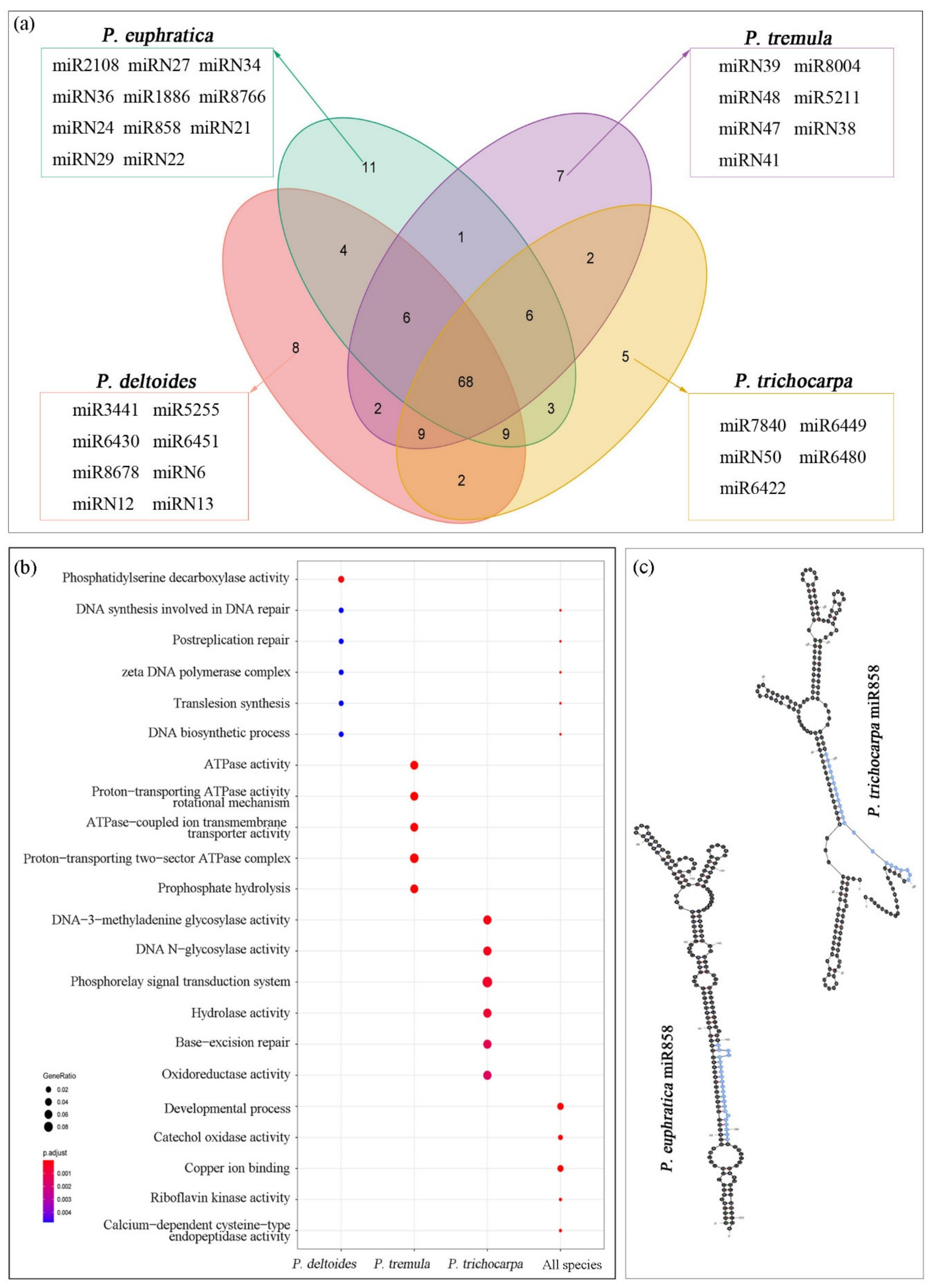
This study identified 143 miRNA families in total, including 92 known and 51 novel miRNA families (Figure 3). For each species, 108, 108, 101, and 104 miRNA families in P. deltoides, P. euphratica, P. tremula, and P. trichocarpa were annotated, respectively, of which 68 miRNA families were shared by all species. Due to the lack of hairpin precursor sequence, improper hairpin structure, or no mature miRNA sequence, 35, 35, 42, and 39 miRNA families were not detected in P. deltoides, P. euphratica, P. tremula, and P. trichocarpa, respectively (Figure 4a). Furthermore, 8, 11, 7, and 5 miRNA families were specific to P. deltoides, P. euphratica, P. tremula, and P. trichocarpa, respectively (Figure 4a). GO enrichment analysis found that targets of miRNAs specific to P. deltoides, P. tremula, and P. trichocarpa are most significantly enriched in the pathways of phosphatidylserine decarboxylase activity, ATPase activity, and phosphorelay signal transduction system, respectively, while target genes of P. euphratica miRNAs are not significantly enriched in any pathway (Figure 4b). The target genes of miRNAs shared by the four species were mainly enriched in the pathways of developmental processes, copper ion binding, and catechol oxidase activity. Moreover, the additional GO enrichment results of different sets are shown in Figure S1. It is noteworthy that the miR858 was observed only in P. euphratica, which was due to the lack of hairpin precursor sequence in P. deltoides and P. tremula and the hairpin structural failure in P. trichocarpa (Figure 4c). Here, we suggested that the hairpin precursor sequence missing from the genome was the main reason for the difference in miRNA families among the four-poplar species.

Figure 3.
miRNA family alignments and annotations of four poplar species.
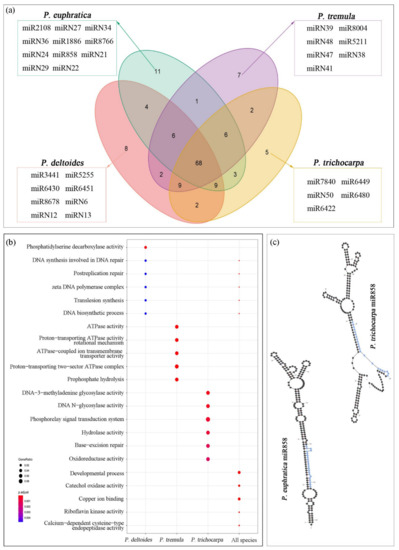
Figure 4.
Comparative analysis of miRNA families among four poplar species (a). GO enrichment analysis of miRNAs specific to P. deltoides, P. tremula, and P. trichocarpa and miRNAs shared by the four species (b). Pre-miR858 secondary structure of P. euphratica and P. trichocarpa (c).
3.3. Variations of miRNAs in Shared miRNA Families
As shown in Table 1, each shared miRNA family (68 in total, Figure 3a) contains 4–96 miRNA sequences. There were 16 miRNA families without miRNA sequence variation, such as miR390, miR168, and miR394. Additionally, multiple unique sequences of miRNA in some shared miRNA families were found, such as miR169, miR339, and miR167, which might regulate different miRNA targets. For example, eight sequences in the miR1444 containing four unique sequences were clustered into two groups (Figure S2a). Four miR1444a sequences from different poplar species were grouped and were located on chromosome 8, and the other miR1444d sequences were located on chromosome 10. Furthermore, variations of target genes of miRNA sequences in different groups were observed (Figure S2b).

Table 1.
Multiple unique sequences of miRNA in shared miRNA families.
3.4. Short Genomic Variants in miRNA Loci
We systematically identified and analyzed miRNA-related polymorphisms based on whole-genome resequencing data of P. deltoides and P. trichocarpa (Tables S5 and S6). The short variants include SNPs and short indels, which are called SNPs for briefly. For P. deltoides, we found 90 SNPs in 70 mature miRNA sequences (52 miRNA families) and 1020 SNPs in 256 pre-miRNAs. Among them, three mature miRNAs contained more than three SNPs, such as pdl-miR6450c, ptc-miR6449, and ptc-miR5741c. For P. trichocarpa, 136 SNPs in 84 mature miRNAs of 63 miRNA families and 1316 SNPs in 267 pre-miRNAs were observed. The genome resequencing of 549 P. trichocarpa trees found that the SNP density of miRNA seed regions (20.07 nt/kb) was similar to that in the mature miRNAs (21.46 nt/kb) while lower than that in the pre-miRNAs (30.84 nt/kb). The same trend was observed in P. deltoides (Table 2). The results showed that most miRNAs were conserved (>70% mature miRNAs without SNP), and their target genes were mainly enriched in the developmental processes (Figure S3).
Table 2.
miRNA related polymorphisms of P. deltoides and P. trichocarpa.
4. Discussion
In this study, we comprehensively annotated miRNAs of four poplar species by combining de novo and genome-wide homolog searching methods. A comparative analysis was conducted to reveal the similarities and differences of miRNA families and miRNA sequences among P. deltoides, P. euphratica, P. tremula, and P. trichocarpa. Additionally, the whole-genome sequencing data was used to identify miRNA-related SNPs. These findings are expected to provide a valuable resource for exploring the evolution and regulation mechanisms of miRNAs in poplar trees.
4.1. The Number of miRNA Loci Is Similar in the Four-Poplar Species
Recently, a growing number of miRNAs have been characterized in different plant species [36]; however, some of these miRNA annotations obtained by sequence homology alone and/or predicted from stem-loop structures seem questionable and incomplete [37]. For example, a previous review showed that one-third of the plant miRNA loci in miRBase failed to meet the criteria required for miRNA annotation, as most lack evidence of the miRNA-star sequence expression [22]. Furthermore, sampling of other tissues at different developmental stages or in response to physiological conditions is also likely to isolate more miRNA sequences. Here, miRNAs of the four-poplar species were identified comprehensively and carefully, ensuring that sampling bias does not affect the interpretation of the available data. Firstly, accurate annotations and useful descriptions of miRNA loci were performed by the ShortStack program based on 107 small RNA-seq libraries, which were constructed by high-throughput sequencing. The annotation of miRNAs by ShortStack is conducted on a strict set of structural and expression-based criteria [26]. Secondly, with the availability of high-coverage genomes of Populus trees [6,7,8,10], the miRNA genes were identified by comparative genomics to prevent the loss of miRNAs by the limited sequencing data. Combining the two methods allowed for the most robust identification of miRNAs in the four-poplars species, including 1038 known and 156 novel miRNAs (Tables S2 and S3). Analyses of the secondary structure of genes corresponding to the novel miRNAs identified confirmed that they all contained miRNA-star sequences. Although Populus miRNAs undergo rapid turnover during evolution, our results showed that the number of miRNA loci in the studied four-poplar species was similar, including 296–301 miRNA loci in each genome.
4.2. Most of the Known miRNA Families Are Shared by All Species
The miRNA family is defined on evolutionary conservative principles, which usually includes several mature miRNAs of nearly identical sequences [38]. A total of 143 miRNA families were identified in four selected Populus species, and each species contained at least 100 miRNA families, which was consistent with previous predictions [39]. Among them, over 60% of known miRNA families were shared by all species, of which miRNA sequences in 16 miRNA families were conserved among species (Table 1). Many shared miRNA families were inherited from the ancestral embryophyte and were involved in a wide range of plant development processes [22], such as miR156, miR159, miR390, miR408, miR530, and miR536. Furthermore, some miRNA sequence heterogeneity belonging to the same family was observed, such as miR169, which was represented by 20–27 members, respectively (Table 1). One hypothesis suggested that there were differences in the selection pressure of duplicated miRNAs in poplar and that having more miRNA members might be conducive to adapting to perennial growth [40]. Similarly, we found that multiple unique sequences of miRNA in some shared miRNA families could promote the specific post-transcriptional regulation of miRNAs. Interestingly, we noted that some of the miRNA families unique to each species were novel miRNA families. A previous study showed that novel miRNAs of P. trichocarpa were weakly expressed and had few targets, but their expression and the number of targets increased gradually during evolution, which might be related to local adaptation [9].
4.3. The Loss of the miRNA Families Caused by Variations in Pre-miRNA Hairpins
Some studies showed that most novel miRNA family members were likely to lack any function and that they would soon be lost through neutral selection [41]. Consistent with previous studies, most of these missing miRNA families were novel miRNA families. Compared with that present in the other three species, we suggested that the variation of pre-miRNA hairpin was the main reason for the loss of the miRNA families in the fourth species (Figure 3). In this study, we found 1020 and 1316 SNPs in 271 and 289 pre-miRNAs of P. deltoides and P. trichocarpa, respectively. The SNP density of pre-miRNA hairpin was higher than that of miRNA seed regions and mature miRNAs, results that are consistent with previous studies [42]. Researchers discovered that the SNP in pre-miRNA sequences could decrease the hairpin structure stability to reduce the production of mature miRNAs [43]. Since the processing of miRNA genes depends on their secondary structure, it was observed that a minor amount of miRNA families’ loss is caused by variations in folding (Figure 3). Significantly, the miR858 was only detected in P. euphratica, which had previously been reported as Arabidopsis-specific [44]. We found that miR858 was absent in P. deltoides and P. tremula because of the lack of hairpin sequence and in P. trichocarpa due to hairpin structural failure. Although most miRNAs were conserved, we found 90 and 136 SNPs in 70 and 84 mature miRNAs of P. deltoides and P. trichocarpa, respectively. The changes in miRNA targets caused by these SNPs will be a potentially useful clue for Populus trees to study miRNA function and find SNP-associated phenotypes.
5. Conclusions
We comprehensively annotated miRNAs of four species from different poplar sections by combining methods of de novo identification and genome-wide homolog searching, significantly expanding the breadth and depth of miRNA annotation in Populus. A total of 1194 miRNA loci were identified, indicating that the number of miRNA loci in each species was similar. Most known miRNA families were shared, while a few species-specific had been annotated, which might be related to the various species’ habitat adaptability. Additionally, multiple miRNA-related SNPs were found and could be a useful clue to studying the biosynthesis and regulatory function of Populus miRNAs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/f13060873/s1, Figure S1: The GO enrichment analysis of miRNA target genes in different gene sets; Figure S2: (a) Cluster analysis of miR1444 family members and (b) GO analysis of miR1444 targets; Figure S3: GO analysis of targets of miRNAs without SNP in P. deltoides and P. trichocarpa; Table S1: RNA-seq data of 107 samples downloaded from NCBI SRA and GEO database and used for miRNA analysis; Table S2: De novo identification of miRNAs using small RNA-seq data; Table S3: Homolog-based genome-wide identification of miRNAs; Table S4: Identification of the presence of miRNAs in individual of natural poplar populations; Table S5: SNPs in mature miRNA sequences; Table S6: SNPs in Pre-miRNA hairpins.
Author Contributions
Conceptualization, L.-J.X. and Y.G.; methodology, Y.-L.Q. and Y.A.E.-K.; formal analysis, data curation, and writing original draft preparation, Y.G. and Y.-L.Q.; writing—review and editing, Y.A.E.-K. and L.-J.X.; project administration and funding acquisition, L.-J.X. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of China to L.-J.X. (funding number: 32171826).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anne, M.R.; Nathaniel, R.S.; Rodríguez-Acosta, M. Populus trees. For. Trees 2007, 7, 1–28. [Google Scholar] [CrossRef]
- Eckenwalder, J.E. Systematics and evolution of Populus. Biol. Popul. Its Implic. Manag. Conserv. 1996, 7, 32. [Google Scholar]
- Zhang, B.; Zhu, W.; Diao, S.; Wu, X.; Lu, J.; Ding, C.J. The poplar pangenome provides insights into the evolutionary history of the genus. Commun. Biol. 2019, 2, 215. [Google Scholar] [CrossRef] [PubMed]
- Cervera, M.T.; Storme, V.; Soto, A.; Ivens, B.; Van Montagu, M.; Rajora, O.P.; Boerjan, W. Intraspecific and interspecific genetic and phylogenetic relationships in the genus Populus based on AFLP markers. Theor. Appl. Genet. 2005, 111, 1440–1456. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G. Populus: Arabidopsis for forestry. Do we need a model tree? Ann. Bot. 2002, 90, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Wu, H.; Zhang, J.; Pan, Z.; Zhao, W.; Li, Z.; Tong, C. Genome assembly of Salicaceae Populus deltoides (Eastern Cottonwood) I-69 based on nanopore sequencing and Hi-C technologies. J. Hered. 2021, 112, 303–310. [Google Scholar] [CrossRef]
- Schiffthaler, B.; Delhomme, N.; Bernhardsson, C.; Jenkins, J.; Jansson, S.; Ingvarsson, P.; Schmutz, J.; Street, N. An improved genome assembly of the European aspen Populus tremula. Biorxiv 2019, 805614. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, Y.; Zhang, J.; Ma, X.; Li, Y.; Li, M.; Wang, D.; Kang, M.; Wu, H.; Yang, Y.; et al. Improved genome assembly provides new insights into genome evolution in a desert poplar (Populus euphratica). Mol. Ecol. Resour. 2020, 20, 781–794. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Chen, B.; Quan, M.; Li, Y.; Li, B.; Zhang, D. Genetic variations and miRNA-target interactions contribute to natural phenotypic variations in Populus. New Phytol. 2016, 212, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Tuskan, G.A.; DiFazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef] [Green Version]
- Evans, L.M.; Slavov, G.T.; Rodgers-Melnick, E.; Martin, J.; Ranjan, P.; Muchero, W.; Brunner, A.M.; Schackwitz, W.; Gunter, L.; Chen, J.G.; et al. Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat. Genet. 2014, 46, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- Dezulian, T.; Palatnik, J.F.; Huson, D.H.; Weigel, D. Conservation and divergence of microRNA families in plants. Genome Biol. 2005, 6, 1–25. [Google Scholar] [CrossRef]
- Cai, H.; Yang, C.; Liu, S.; Qi, H.; Wu, L.; Xu, L.A. MiRNA-target pairs regulate adventitious rooting in Populus: A functional role for miR167a and its target Auxin response factor 8. Tree Physiol. 2019, 39, 1922–1936. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, S.; Yu, B. microRNA biogenesis, degradation and activity in plants. Cell. Mol. Life Sci. 2015, 72, 87–99. [Google Scholar] [CrossRef]
- Xu, C.; Tao, Y.; Fu, X.; Guo, L.; Xing, H.; Li, C.; Yang, Z.; Su, H.; Wang, X.; Hu, J.; et al. The microRNA476a-RFL module regulates adventitious root formation through a mitochondria-dependent pathway in Populus. New Phytol. 2021, 230, 2011–2028. [Google Scholar] [CrossRef]
- Xie, J.; Yang, X.; Song, Y.; Du, Q.; Li, Y.; Chen, J.; Zhang, D. Adaptive evolution and functional innovation of Populus-specific recently evolved microRNAs. New Phytol. 2017, 213, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Qin, Y.; Duan, H.; Yin, W.; Xia, X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J. Exp. Bot. 2011, 62, 3765–3779. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, M.; Jiang, J.; Qiao, G.; Lin, S.; Li, H.; Xie, L.; Zhuo, R. Expression profile of miRNAs in Populus cathayana L. and Salix matsudana Koidz under salt stress. Mol. Biol. Rep. 2012, 39, 8645–8654. [Google Scholar] [CrossRef]
- Shuai, P.; Liang, D.; Zhang, Z.; Yin, W.; Xia, X. Identification of drought-responsive and novel Populus trichocarpa microRNAs by high-throughput sequencing and their targets using degradome analysis. BMC Genom. 2013, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.S.; Tarver, J.E.; Foroozani, A.; Donoghue, P.C. MicroRNA annotation of plant genomes—Do it right or not at all. Bioessays 2017, 39, 1600113. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.S.; Tarver, J.E.; Hiscock, S.J.; Donoghue, P.C. Evolutionary history of plant microRNAs. Trends Plant Sci. 2014, 19, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010, 32, 11.7.1–11.7.14. [Google Scholar] [CrossRef]
- MJ, A. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2018, 47, 155–162. [Google Scholar] [CrossRef]
- Prufer, K.; Stenzel, U.; Dannemann, M.; Green, R.E.; Lachmann, M.; Kelso, J. PatMaN: Rapid alignment of short sequences to large databases. Bioinformatics 2008, 24, 1530–1532. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map (SAM) format and SAMtools 1000 genome project data processing subgroup. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Wu, H.; Chen, Y.; Li, X.; Hou, J.; Lu, J.; Wei, S.; Dai, X.; Olson, M.S.; Liu, J.; et al. Evidences for a role of two Y-specific genes in sex determination in Populus deltoides. Nat. Commun. 2020, 11, 5893. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, Y.; Roulin, A.C.; Muller, K.; Ebert, D. SNPs Called with Freebayes. 2017. Available online: https://datadryad.org/stash/dataset/doi:10.5061/dryad.g89m1 (accessed on 20 April 2022).
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, 258–261. [Google Scholar] [CrossRef] [Green Version]
- Yu, G. Statistical analysis and visualization of functional profiles for genes and gene clusters. J. Integr. Biol. 2012, 16, 284–287. [Google Scholar]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [Green Version]
- The International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and functional diversification of miRNA gene. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Barakat, A.; Wall, P.K.; Diloreto, S.; Depamphilis, C.W.; Carlson, J.E. Conservation and divergence of microRNAs in Populus. BMC Genom. 2007, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yin, W.; Xia, X. Identification of microRNAs and their targets from Populus euphratica. Biochem. Biophys. Res. Commun. 2009, 388, 272–277. [Google Scholar] [CrossRef]
- Nozawa, M.; Miura, S.; Nei, M. Origins and evolution of microRNA genes in plant species. Genome Biol. Evol. 2012, 4, 230–239. [Google Scholar] [CrossRef] [Green Version]
- De Wit, E.; Linsen, S.E.; Cuppen, E.; Berezikov, E. Repertoire and evolution of miRNA genes in four divergent nematode species. Genome Res. 2009, 19, 2064–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Tong, Y.; Zhang, H.M.; Wang, K.; Hu, T.; Shan, G.; Sun, J.; Guo, A.Y. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum. Mutat. 2011, 33, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).