
New Insight into Genetic Structure and Diversity of Scots Pine (Pinus sylvestris L.) Populations in Lithuania Based on Nuclear, Chloroplast and Mitochondrial DNA Markers


Abstract
1. Introduction
2. Materials and Methods
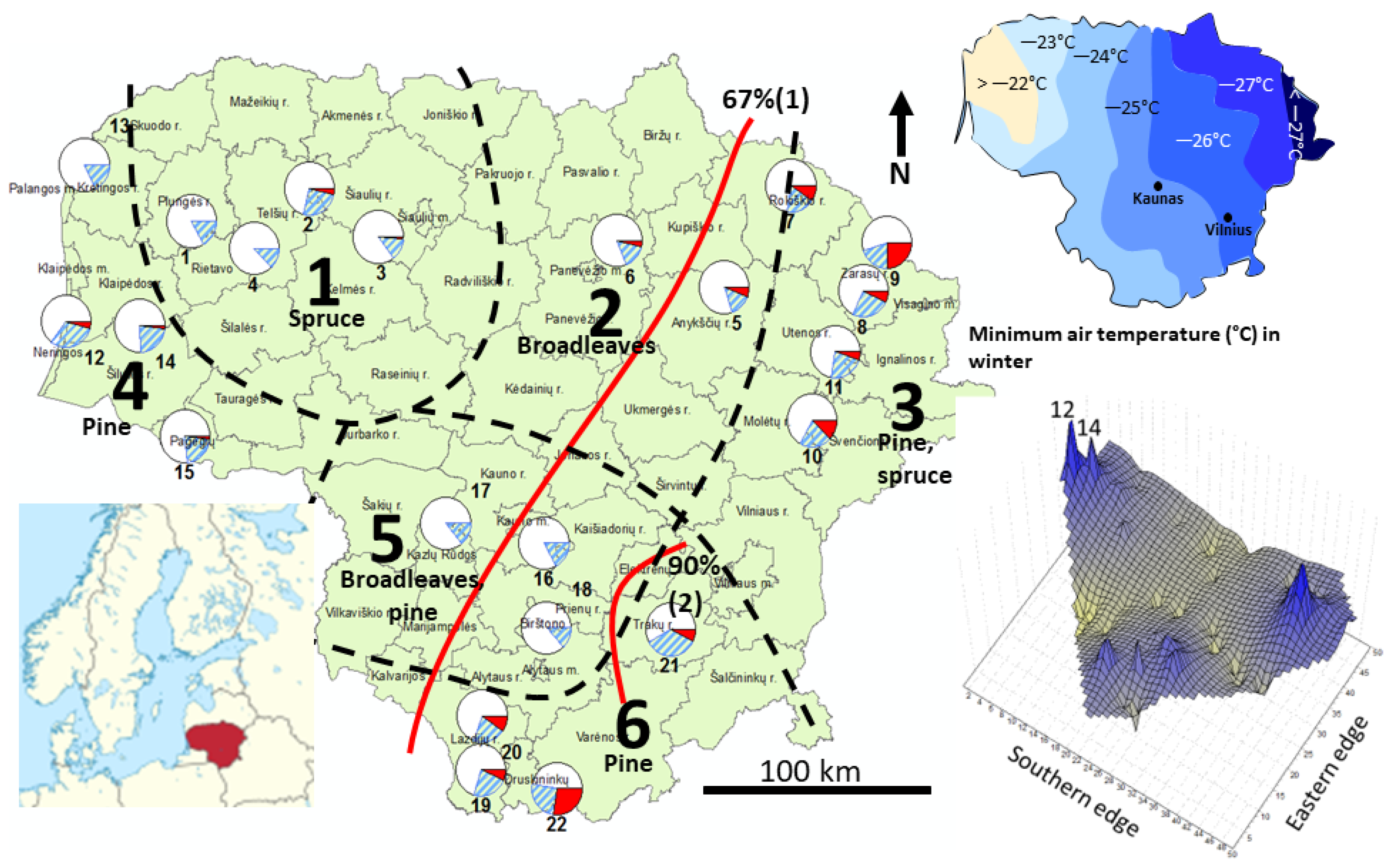
2.1. Study Sites and Sampling
2.2. DNA Analysis
2.3. Molecular Data Analysis
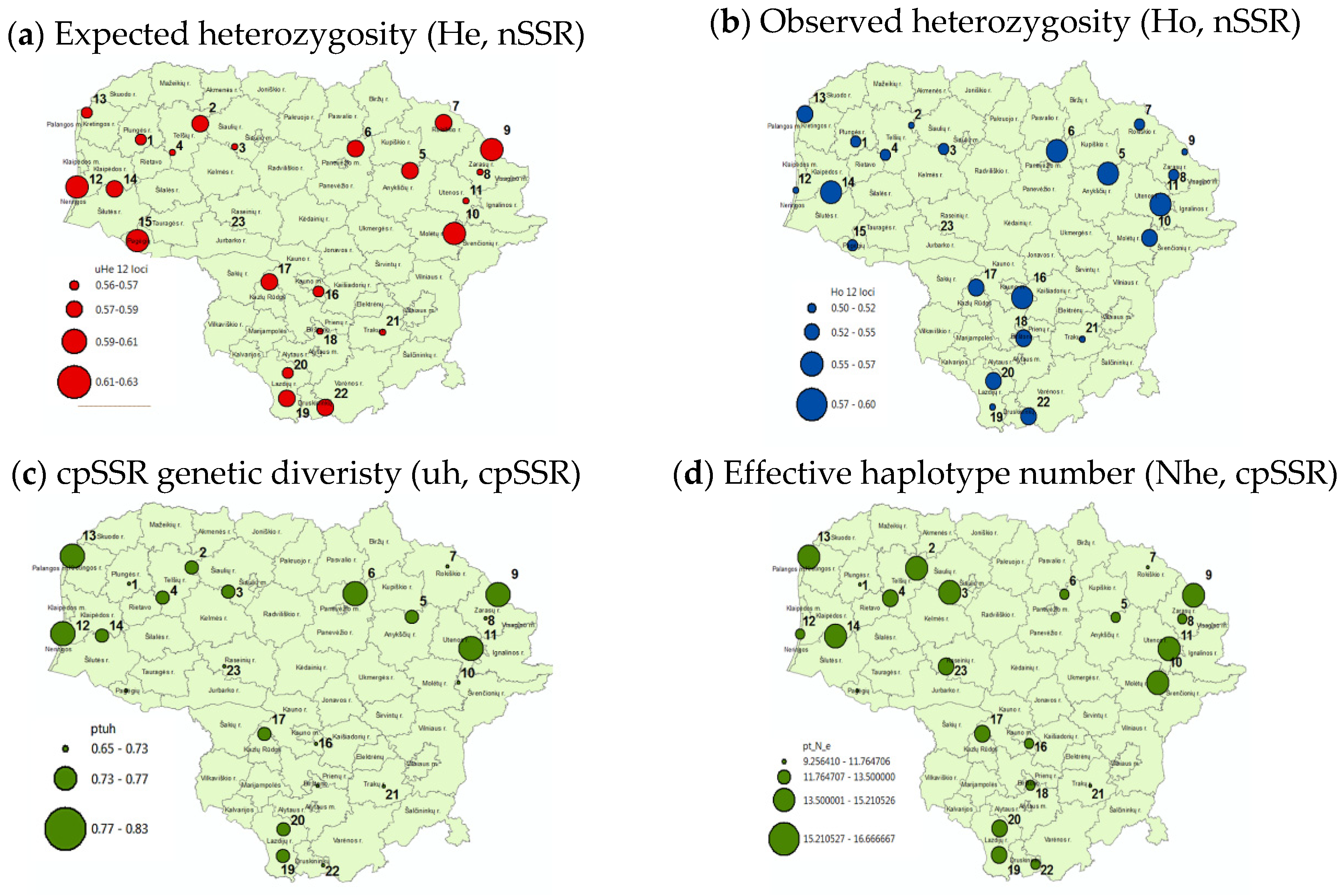
- (a)
- (b)
- The Bayesian clustering approach as the model-based clustering algorithm implemented in STRUCTURE 2.3.3 software [87] and the empirical statistic DeltaK [88] implemented in STRUCTURE HARVESTER [89] to determine the number of clusters/distinct populations (K), from which samples have been drawn based on microsatellite genotypes at multiple loci. STRUCTURE parameters were: the burn-in period of 100,000, 100,000 replications, 20 runs for each the clusters, and with the LOCPRIOR function, allele frequencies were assumed to be correlated, while no admixture model was assumed. The most likely number of clusters (1 to 3) was estimated by submitting the STRUCTURE output to the STRUCTURE HARVESTER [89].
- (c)
- The software BARRIER 2.2 [90] used the Monmonier’s algorithm applied on a Delanaunay triangulation to identify the barriers along which a significant change in the population allele frequency occurs within the investigated geographical range. The statistical confidence of the predicted barriers was tested by bootstrapping over individuals and submitting the 1000 bootstrapped matrices of Goldstein’s pairwise genetic distances (dm2) calculated with the MSA software.
- (d)
3. Results
3.1. Characteristics of the Microsatellite Loci
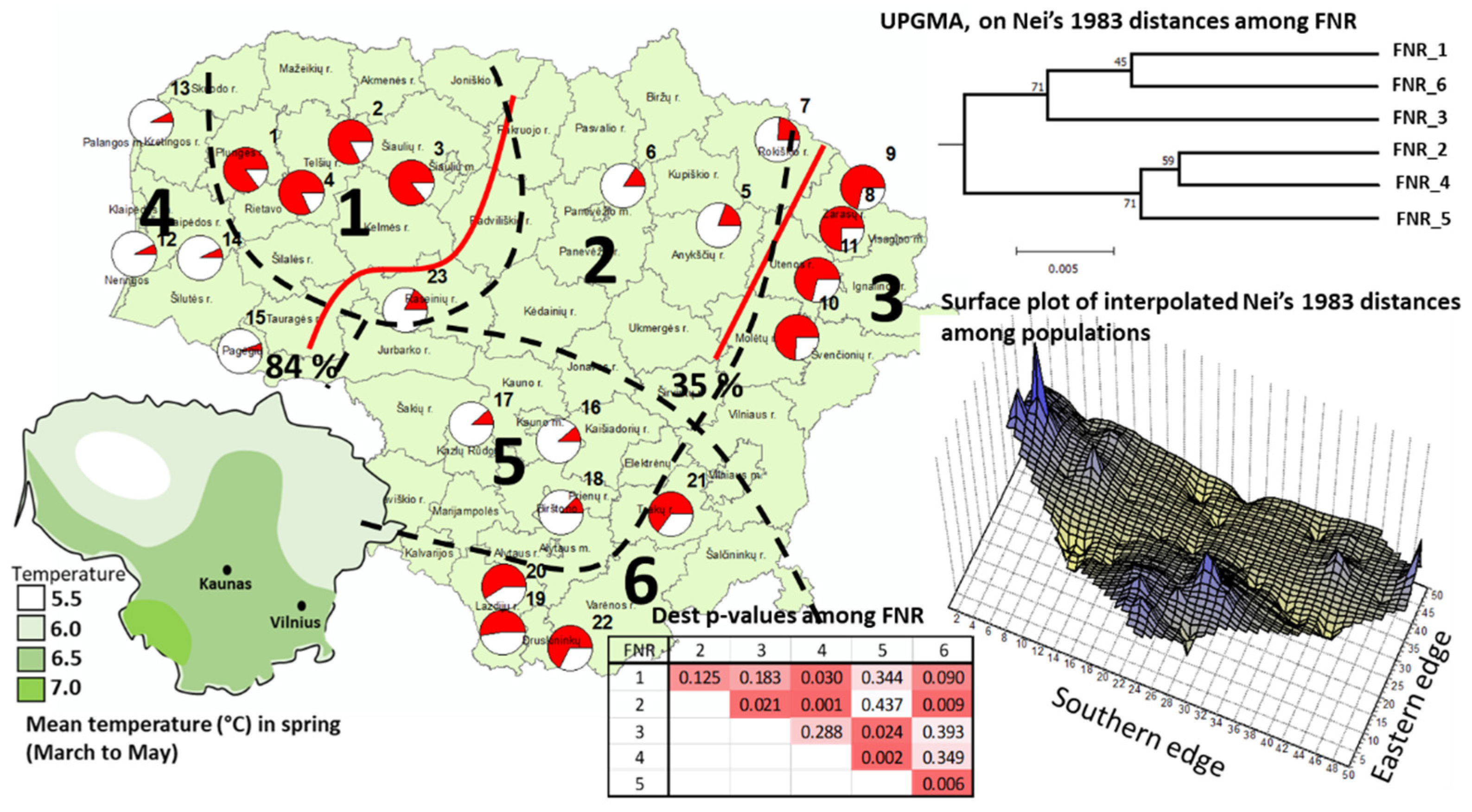
3.2. Genetic Differentiation and Structure
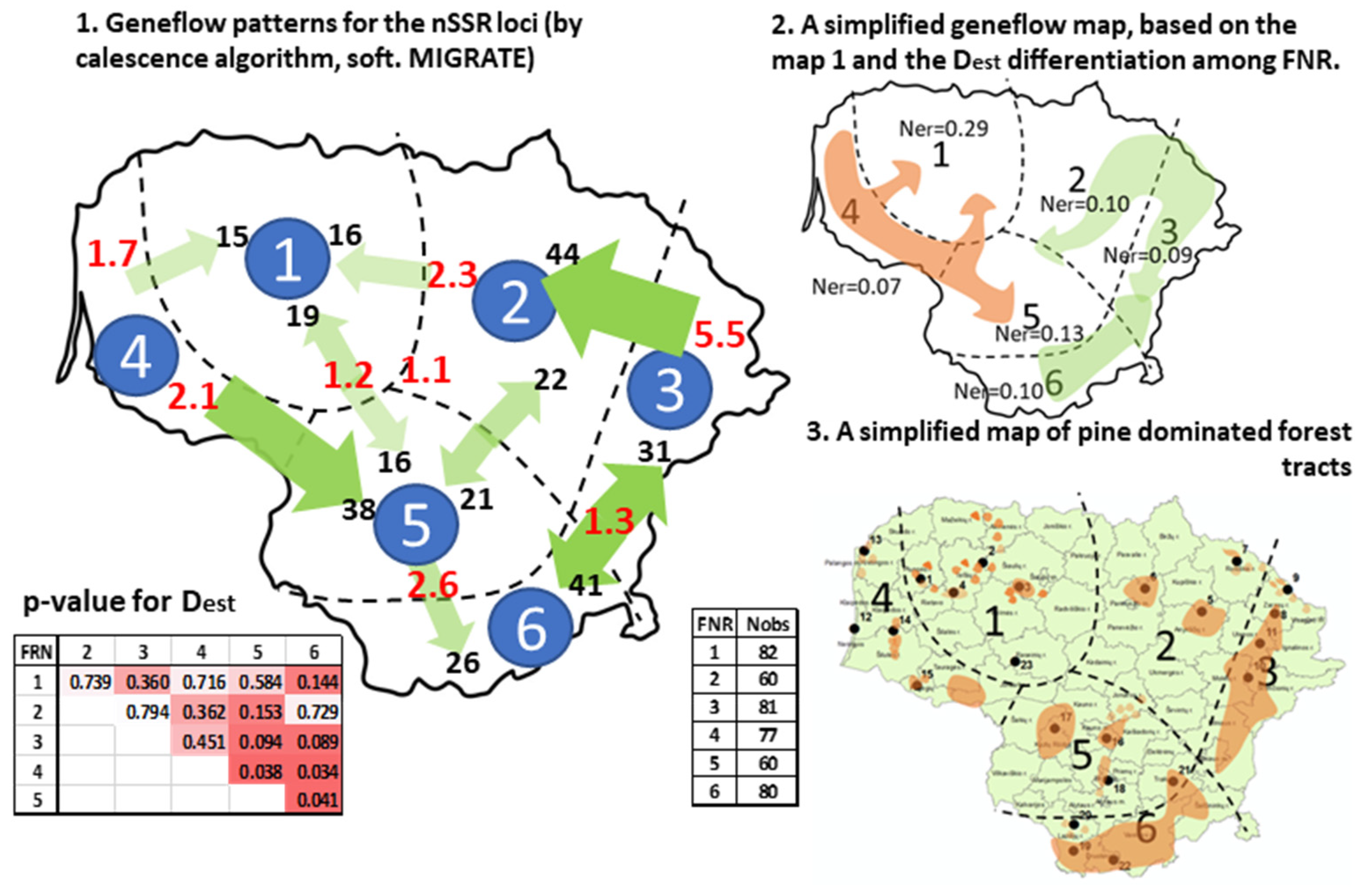
3.2.1. nSSR Loci

- (1)
- The cooler part of the country: the eastern highlands (FNRs 6 and 3) and the northern part of the midland lowlands (FNR 2); a bilateral exchange of similar magnitude between the largest pine-dominated forest tracts in the country in the eastern highlands of FNRs 6 and 3 and strong flow from northeastern highlands (FNR 3) into the adjacent midland lowland (FNR 2).
- (2)
- The milder part of the country, where major flow is directed out of the seaside lowlands (FNR 4) into the southern part of the midland lowlands (FNR 5), with a weaker genetic exchange between the FNRs 4, 5, and 1. The lowlands in FNRs 4 and 2 represent the warmest part of the country (Figure S1).

3.2.2. The Chloroplast SSR Loci
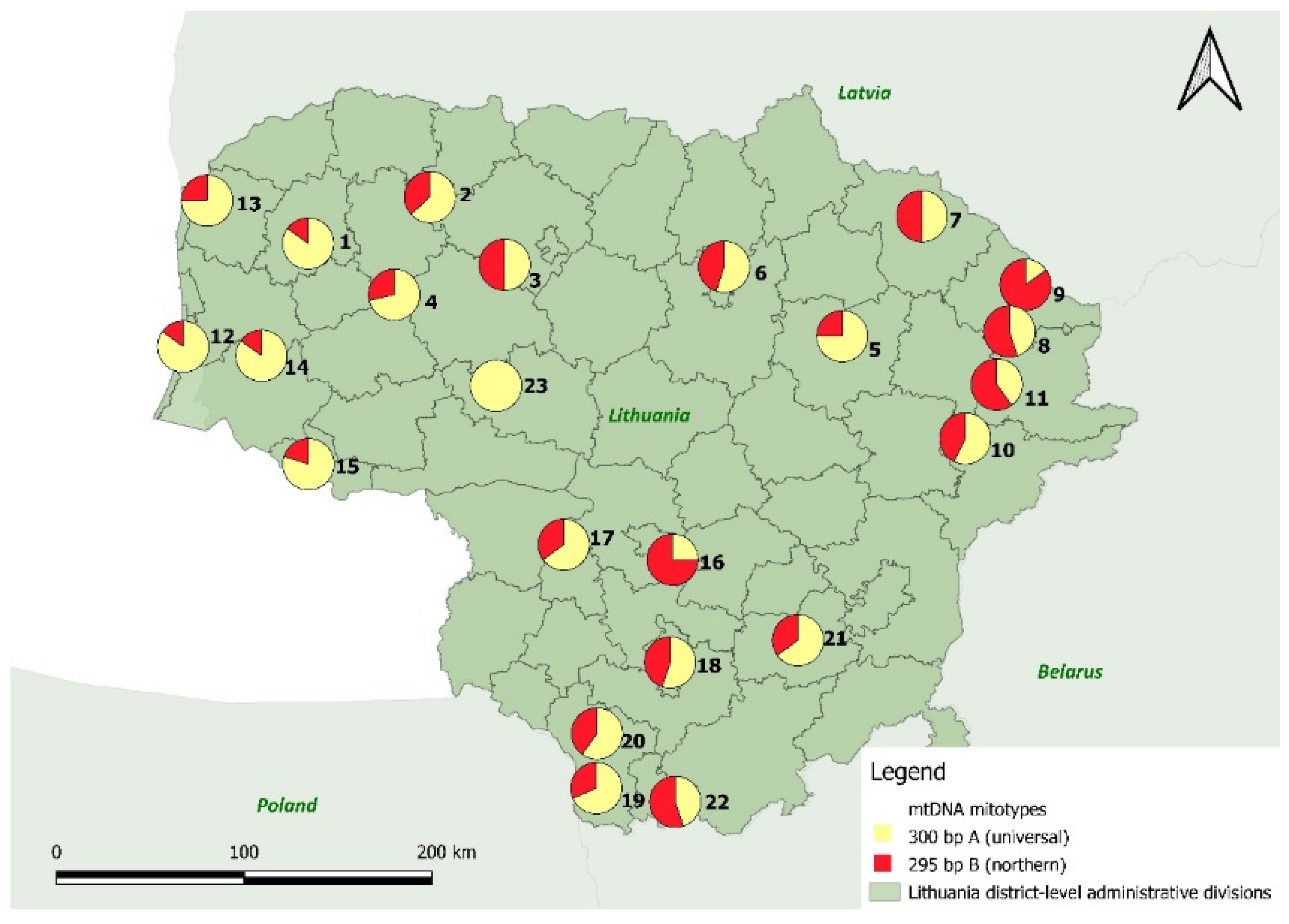
3.2.3. The Mitochondrial DNA Locus
3.3. Within-Population Genetic Diversity
4. Discussion
4.1. Evolutionary Origin
4.2. Population Genetic Structure
4.3. Within-Population Genetic Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gardner, M. Pinus sylvestris. In The IUCN Red List of Threatened Species; IUCN: Gland, Switzerland, 2013; E. T42418A2978732. [Google Scholar]
- Durrant, T.H.; De Rigo, D.; Caudullo, G. Pinus sylvestris in Europe: Distribution, habitat, usage and threats. Eur. Atlas For. Tree Spec. 2016, 132–133. [Google Scholar]
- Mason, W.L.; Alía, R. Current and future status of Scots pine (Pinus sylvestris L.) forests in Europe. For. Syst. 2000, 9, 317–335. [Google Scholar]
- Högberg, P.; Nordgren, A.; Buchmann, N.; Taylor, A.F.S.; Ekblad, A.; Högberg, M.N.; Read, D.J. Large-scale forest girdling shows that current photosynthesis drives soil respiration. Nature 2001, 411, 789–793. [Google Scholar] [CrossRef]
- Lindén, A.; Heinonsalo, J.; Buchmann, N.; Oinonen, M.; Sonninen, E.; Hilasvuori, E.; Pumpanen, J. Contrasting effects of increased carbon input on boreal SOM decomposition with and without presence of living root system of Pinus sylvestris L. Plant Soil. 2014, 377, 145–158. [Google Scholar] [CrossRef]
- Tyrmi, J.S.; Vuosku, J.; Acosta, J.J.; Li, Z.; Sterck, L.; Cervera, M.T.; Pyhäjärvi, T. Genomics of clinal local adaptation in Pinus sylvestris under continuous environmental and spatial genetic setting. Genes Genomes Genet. 2020, 10, 2683–2696. [Google Scholar] [CrossRef]
- Pravdin, L.F. Scots Pine: Variation, Intraspecific Taxonomy and Selection; Annarbor Humphrey Science Publishers Ltd.: London, UK, 1969. [Google Scholar]
- Giertych, M.; Giertych, M.; Mátyás, C. Inheritance of Tree Form; Elsevier: Amsterdam, The Netherlands, 1991; pp. 243–254. [Google Scholar]
- Shutyaev, A.M.; Giertych, M. Genetic subdivisions of the range of Scots pine (Pinus sylvestris L.). Silvae Genet. 2000, 3, 137–151. [Google Scholar]
- Danusevičius, D. Hybrid vigour from intra-specific crosses of Scots pine. Balt. For. 2008, 14, 2–6. [Google Scholar]
- Pyhäjärvi, T.; Kujala, S.T.; Savolainen, O. 275 years of forestry meets genomics in Pinus sylvestris. Evol. Appl. 2020, 13, 11–30. [Google Scholar] [CrossRef]
- Ulaszewski, B.K. Neutralna i Adaptacyjna Zmienność Genetyczna Buka Zwyczajnego Fagus sylvatica L. na Podstawie Analiz Genomowych (Neutral and Adaptive Genetic Diversity of European Beech Fagus sylvatica L. Based on Genomic Analyses). Master’s Thesis, Kazimierz the Great University, Bydgoszcz, Poland, 2018; 185p. (In Polish with extended English summary). [Google Scholar]
- Gugerli, F.; Parducci, L.; Petit, R.J. Ancient plant DNA: Review and prospects. New Phytol. 2005, 166, 409–418. [Google Scholar] [CrossRef]
- Gutaker, R.M.; Burbano, H.A. Reinforcing plant evolutionary genomics using ancient DNA. Curr. Opin. Plant Biol. 2017, 36, 38–45. [Google Scholar] [CrossRef]
- Hattemer, H.H. Concepts and requirements in the conservation of forest genetic resources. For. Genet. 1995, 2, 125–134. [Google Scholar]
- Petit, R.J.; Elmousadik, A.; Pons, O. Identifying populations for conservation on the basis of genetic markers. Conserv. Biol. 1998, 12, S844–S855. [Google Scholar] [CrossRef]
- Rajora, O.P.; Mosseler, A. Challenges and opportunities for conservation of forest genetic resources. Euphytica 2001, 118, 197–212. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Nuclear DNA analyses in genetic studies of populations: Practice, problems and prospects. Mol. Ecol. 2003, 12, 563–584. [Google Scholar] [CrossRef]
- Oliveira, E.J.; Pádua, J.G.; Zucchi, M.I.; Vencovsky, R.; Vieira, M.L.C. Origin, evolution and genome distribution of microsatellites. Genet. Mol. Biol. 2006, 29, 294–307. [Google Scholar] [CrossRef]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; Barkley, A.N.; Jenkins, M.T. Microsatellite Markers in Plants and Insects. In Part I: Applications of Biotechnology. Genes, Genomes and Genomics; Global Science Books: Kagawa ken, Japan, 2009. [Google Scholar]
- Ennos, R.A.; Sinclair, W.T.; Hu, X.S.; Langdon, A. Using organelle markers to elucidate the history, ecology and evolution of plant populations. Mol. Syst. Plant Evolut. 1999, 57, 1–19. [Google Scholar]
- Petit, R.J.; Kremer, A.; Wagner, D.B. Geographic structure of chloroplast DNA polymorphisms in European oaks. Theor. Appl. Genet. 1993, 87, 122–128. [Google Scholar] [CrossRef]
- Hipkins, V.D.; Krutovskii, K.V.; Strauss, S.H. Organelle genomes in conifers: Structure, evolution, and diversity. For. Genet. 1994, 1, 179–189. [Google Scholar]
- Provan, J.; Soranzo, N.; Wilson, N.J.; McNicol, J.W.; Forrest, G.I.; Cottrell, J.; Powell, W. Gene-pool variation in Caledonian and European Scots pine (Pinus sylvestris L.) revealed by chloroplast simple-sequence repeats. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1998, 265, 1697–1705. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolutions. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Petit, R.J.; Aguinagalde, I.; de Beaulieu, J.L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, G.; et al. Glacial refugia: Hotspots but not melting pots of genetic diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.S.; Wagner, D.B. Taxonomic and Population differentiation of mitochondrial diversity in Pinus banksiana and Pinus contorta. Theor. Appl. Genet. 1993, 86, 573–578. [Google Scholar] [CrossRef]
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.G.; Cosson, J.F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Krutovskii, K.V.; Strauss, S.H. Abundant mitochondrial genome diversity, population differentiation and convergent evolution in pines. Genetics 1998, 150, 1605–1614. [Google Scholar] [CrossRef]
- Jeandroz, S.; Bastien, D.; Chandelier, A.; Du Jardin, P.; Favre, J.M. A set of primers for amplification of mitochondrial DNA in Picea abies and other conifer species. Mol. Ecol. Notes 2002, 2, 389–392. [Google Scholar] [CrossRef]
- Godbout, J.; Jaramillo-Correa, J.P.; Beaulieu, J.; Bousquet, J. A mitochondrial DNA minisatellite reveals the postglacial history of jack pine (Pinus banksiana), a broad-range North American conifer. Mol. Ecol. 2005, 14, 3497–3512. [Google Scholar] [CrossRef]
- Soranzo, N.; Alia, R.; Provan, J.; Powell, W. Patterns of variation at a mitochondrial sequence-tagged-site locus provides new insights into the postglacial history of European Pinus sylvestris populations. Mol. Ecol. 2000, 9, 1205–1211. [Google Scholar] [CrossRef]
- Naydenov, K.; Senneville, S.; Beaulieu, J.; Tremblay, F.; Bousquet, J. Glacial vicariance in Eurasia: Mitochondrial DNA evidence from Scots pine for a complex heritage involving genetically distinct refugia at mid-northern latitudes and in Asia Minor. BMC Evol. Biol. 2007, 7, 233. [Google Scholar] [CrossRef]
- Pyhäjärvi, T.; Salmela, M.J.; Savolainen, O. Colonization routes of Pinus sylvestris inferred from distribution of mitochondrial DNA variation. Tree Genet. Genomes 2008, 4, 247–254. [Google Scholar] [CrossRef]
- Dering, M.; Kosinski, P.; Wyka, T.P.; Pers-Kamczyc, E.; Boratynski, A.; Boratynska, K.; Reich, P.B.; Romo, A.; Zadworny, M.; Zytkowiak, R.; et al. Tertiary remnants and Holocene colonizers: Genetic structure and phylogeography of Scots pine reveal higher genetic diversity in young boreal than in relict Mediterranean populations and a dual colonization of Fennoscandia. Divers. Distrib. 2017, 23, 540–555. [Google Scholar] [CrossRef]
- Buchovska, J.; Danusevičius, D.; Stanys, V.; Šikšnianienė, J.B.; Kavaliauskas, D. The location of the northern glacial refugium of Scots pine based on mitochondrial DNA markers. Balt. For. 2013, 19, 2–12. [Google Scholar]
- Neale, D.B.; Sederoff, R.R. Paternal inheritance of chloroplast DNA and maternal inheritance of mitochondrial DNA in loblolly pine. Theor. Appl. Genet. 1989, 77, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Csaikl, U.M.; Glaz, I.; Baliuckas, V.; Petit, R.J.; Jensen, J.S. Chloroplast DNA variation of white oak in the Baltic countries and Poland. For. Ecol. Manag. 2002, 156, 211–222. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef]
- Dumolin, S.; Demesure, B.; Petit, R.J. Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor. Appl. Genet. 1995, 91, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, G.G.; Lelli, L.; Rossi, P.; Morgante, M. A set of primers for the amplification of 20 chloroplast microsatellites in Pinaceae. Mol. Ecol. 1996, 5, 595–598. [Google Scholar] [CrossRef]
- Kavaliauskas, D.; Danusevičius, D.; Baliuckas, V.; Baranov, O. Paprastosios pušies populiacijų genetinė struktūra Lietuvoje pagal chloroplastų DNR žymenis. [Genetic structure of Scots pine populations in Lithuania according to chloroplast DNA markers]. Miškininkystė 2015, 1, 45–55. [Google Scholar]
- Belletti, P.; Ferrazzini, D.; Piotti, A.; Monteleone, I.; Ducci, F. Genetic variation and divergence in Scots pine (Pinus sylvestris L.) within its natural range in Italy. Eur. J. For. Res. 2012, 131, 1127–1138. [Google Scholar] [CrossRef]
- Pyhäjärvi, T.; Garcia-Gil, M.R.; Knürr, T.; Mikkonen, M.; Wachowiak, W.; Savolainen, O. Demographic history has influenced nucleotide diversity in European Pinus sylvestris populations. Genetics 2007, 177, 1713–1724. [Google Scholar] [CrossRef]
- Wachowiak, W.; Wόjkiewicz, B.; Cavers, S.; Lewandowski, A. High genetic similarity between Polish and North European Scots pine (Pinus sylvestris L.) populations at nuclear gene loci. Tree Genet. Genom. 2014, 10, 1015–1025. [Google Scholar] [CrossRef][Green Version]
- Hebda, A.; Wójkiewicz, B.; Wachowiak, W. Genetic characteristics of Scots pine in Poland and reference populations based on nuclear and chloroplast microsatellite markers. Silva Fenn. 2017, 51, 1721. [Google Scholar] [CrossRef][Green Version]
- Langlet, O. Two hundred years genecology. Taxon 1971, 20, 653–721. [Google Scholar] [CrossRef]
- Persson, B.; Beuker, E. Distinguishing between the effects of changes in temperature and light climate using provenance trials with Pinus sylvestris in Sweden. Can. J. For. Res. 1997, 27, 572–579. [Google Scholar] [CrossRef]
- Persson, B. Will climate change affect the optimal choice of Pinus sylvestris provenances? Silva Fenn. 1998, 32, 121–128. [Google Scholar] [CrossRef][Green Version]
- Clapham, D.H.; Ekberg, I.; Eriksson, G.; Norell, L.; Vince-Prue, D. Requirement for far-red light to maintain secondary needle extension growth in northern but not southern populations of Pinus sylvestris (Scots pine). Physiol Plant. 2002, 114, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, G. Evolutionary forces influencing variation among populations of Pinus sylvestris. Silva Fenn. 1998, 32, 694. [Google Scholar] [CrossRef]
- Sarvas, R. Investigations on the flowering and seed crop of Pinus silvestris. Metsat. Tutk. Julk. 1962, 53, 198. [Google Scholar]
- Koski, V. A study of pollen dispersal as a mechanism of gene flow in conifers. Commun. Inst. Fenn. 1970, 70, 1–78. [Google Scholar]
- Varis, S.; Pakkanen, A.; Galofré, A.; Pulkkinen, P. The extent of south-north pollen transfer in finnish Scots pine. Silva Fenn. 2009, 43, 717–726. [Google Scholar] [CrossRef]
- Torimaru, T.; Wang, X.R.; Fries, A.; Andersson, B.; Lindgren, D. Evaluation of pollen contamination in an advanced scots pine seed orchard. Silvae Genet. 2009, 58, 262–269. [Google Scholar] [CrossRef]
- Robledo-Arnuncio, J.J.; Collada, C.; Alia, R.; Gil, L. Genetic structure of montane isolates of Pinus sylvestris L. in a Mediterranean refugial area. J. Biogeogr. 2005, 32, 595–605. [Google Scholar] [CrossRef]
- Sirgėdienė, M.; Danusevičius, D. Morphological differences of cones of Scots pine populations growing in bogs and dry sites [Pelkinių ir normalaus drėgnumo augaviečių paprastųjų pušų kankorėžių morfologiniai skirtumai]. Miškininkystė 2019, 1, 49–67. [Google Scholar]
- Ledig, F.T. Human Impacts on Genetic Diversity in Forest Ecosystems. Oikos 1992, 63, 87–108. [Google Scholar] [CrossRef]
- Lefèvre, F. Human impacts on forest genetic resources in the temperate zone: An updated review. For. Ecol. Manag. 2004, 197, 257–271. [Google Scholar] [CrossRef]
- Finkeldey, R.; Ziehe, M. Genetic implications of silvicultural regimes. For. Ecol. Manag. 2004, 197, 231–244. [Google Scholar] [CrossRef]
- Naydenov, K.D.; Naydenov, M.K.; Tremblay, F.; Alexandrov, A.; Aubin-Fournier, L.D. Patterns of genetic diversity that result from bottlenecks in Scots Pine and the implications for local genetic conservation and management practices in Bulgaria. New For. 2011, 42, 179–193. [Google Scholar] [CrossRef]
- Ratnam, W.; Rajora, P.O.; Finkeldey, R.; Aravanopoulos, F.; Bouvet, J.M.; Vaillancourt, R.E.; Kanashiro, M.; Fady, B.; Tomita, M.; Vinson, C. Genetic effects of forest management practices: Global synthesis and perspectives. For. Ecol. Manag. 2014, 333, 52–65. [Google Scholar] [CrossRef]
- Gil, M.R.G.; Floran, V.; Östlund, L.; Gull, B.A. Genetic diversity and inbreeding in natural and managed populations of Scots pine. Tree Genet. Genomes 2015, 11, 28. [Google Scholar]
- Myking, T.; Rusanen, M.; Steffenrem, A.; Kjær, E.D.; Jansson, G. Historic transfer of forest reproductive material in the Nordic region: Drivers, scale and implications. Forestry 2016, 89, 325–337. [Google Scholar] [CrossRef]
- Danusevičius, D.; Kavaliauskas, D.; Fussi, B. Optimum Sample Size for SSR-based Estimation of Representative Allele Frequencies and Genetic Diversity in Scots Pine Populations. Balt. For. 2016, 22, 194–202. [Google Scholar]
- Aravanopoulos, F.A. Do silviculture and forest management affect the genetic diversity and structure of long-impacted forest tree populations? Forests 2018, 9, 355. [Google Scholar] [CrossRef]
- Mátyás, C.; Ackzell, L.; Samuel, C.J.A. EUFORGEN Technical Guidelines for Genetic Conservation and Use for Scots pine (Pinus sylvestris); International Plant Genetic Resources Institute: Rome, Italy, 2004. [Google Scholar]
- Floran, V.; Sestras, R.E.; Gil, M.R.G. Organelle genetic diversity and phylogeography of Scots pine (Pinus sylvestris L.). Not. Bot. Horti. Agrobot. Cluj Nap. 2011, 39, 317–322. [Google Scholar] [CrossRef][Green Version]
- Metzger, M.J.; Bunce, R.G.H.; Jongman, R.H.; Mücher, C.A.; Watkins, J.W. A climatic stratification of the environment of Europe. Glob. Ecol. Biogeogr. 2005, 14, 549–563. [Google Scholar] [CrossRef]
- Lithuanian State Forest Service Yearbook. 2020. Available online: https://amvmt.lrv.lt/uploads/amvmt/documents/files/Statistika/MiskuStatistika/2020/01%20Misku%20ukio%20statistika%202020_m.pdf (accessed on 1 February 2022).
- Danusevičius, J. Genetics and Breeding of Pine in Lithuania (Monograph); Lithuanian Forest Research Institute: Kaunas, Lithuania, 2001; 234p, (in Lithuanian with English headings of tables and figures). [Google Scholar]
- Ramanauskas, V. Lietuvos pušynų kankorėžiai ir sėklos. Girios 1978, 3, 3–5. [Google Scholar]
- Karazija, S. Lietuvos Miškų Tipai; Mokslas: Vilnius, Lithuania, 1998. [Google Scholar]
- Sebastiani, F.; Pinzauti, F.; Kujala, S.T.; González-Martínez, S.C.; Vendramin, G.G. Novel polymorphic nuclear microsatellite markers for Pinus sylvestris L. Conserv. Genet. Resour. 2012, 4, 231–234. [Google Scholar] [CrossRef]
- Soranzo, N.; Provan, J.; Powell, W. Characterization of microsatellite loci in Pinus sylvestris L. Mol. Ecol. 1998, 7, 1260–1261. [Google Scholar]
- Auckland, L.D.; Bui, T.; Zhou, Y.; Shepherd, M.; Williams, C.G. Conifer Microsatellite Handbook; A&M University: College Station, TX, USA, 2002; 57p. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, G.G.; Anzidei, M.; Madaghiele, A.; Bucci, G. Distribution of genetic diversity in Pinus pinaster Ait. as revealed by chloroplast microsatellites. Theor. Appl. Genet. 1998, 97, 456–463. [Google Scholar] [CrossRef]
- Eliades, N.G.; Eliades, D.G. HAPLOTYPE ANALYSIS: Software for Analysis of Haplotype Data; Genetics and Forest Tree Breeding, Georg-August University: Goettingen, Germany, 2009. [Google Scholar]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9.3). 2001. Available online: http://www.unil.ch/izea/softwares/fstat.html. (accessed on 12 October 2021).
- Nei, M. Genetic polymorphism and the role of mutation in evolution. Evol. Genes Prot. 1983, 71, 165–190. [Google Scholar]
- Peakall, R.O.D.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes. 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Takezaki, N.; Nei, M.; Tamura, K. POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol. Biol. Evol. 2010, 27, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; Von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Manni, F.; Guérard, E.; Heyer, E. Geographic patterns of (genetic, morphologic, linguistic) variation: How barriers can be detected by using Monmonier’s algorithm. Hum. Biol. 2004, 76, 173–190. [Google Scholar] [CrossRef]
- Miller, M.P. Alleles in Space (AIS): Computer software for the joint analysis of interindividual spatial and genetic information. J. Hered. 2005, 96, 722–724. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Nei, M.; Tajima, F.; Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef]
- Beerli, P. How to use MIGRATE or why are Markov chain Monte Carlo programs difficult to use. Popul. Genet. Anim. Conserv. 2009, 17, 42–79. [Google Scholar] [CrossRef]
- Galvonaitė, A.; Valiukas, D.; Kilpys, J.; Kitrienė, Z.; Misiūnienė, M. Climate Atlas of Lithuania; Lithuanian Hydrometeorological Service under Ministry of Environment: Vilnius, Lithuania, 2013. [Google Scholar]
- Kujala, S. Dissecting genetic variation in European Scots pine (Pinus sylvestris L.): Special emphasis on polygenic adaptation. Acta Univ. Oul. A 2015, 661, 75. [Google Scholar]
- Tóth, E.G.; Vendramin, G.G.; Bagnoli, F.; Cseke, K.; Höhn, M. High genetic diversity and distinct origin of recently fragmented Scots pine (Pinus sylvestris L.) populations along the Carpathians and the Pannonian Basin. Tree Genet. Genomes 2017, 13, 1–12. [Google Scholar] [CrossRef]
- Nowakowska, J.A. ZmiennoÊç genetyczna polskich wybranych populacji sosny zwyczajnej (Pinus sylvestris L.) na podstawie analiz polimorfizmu DNA. Rozpr. I Monogr. 2007, 9, 1–118. [Google Scholar]
- Danusevičius, D.; Buchovska, J.; Žulkus, V.; Daugnora, L.; Girininkas, A. DNA Markers Reveal Genetic Associations among 11,000-Year-Old Scots Pine (Pinus sylvestris L.) Found in the Baltic Sea with the Present-Day Gene Pools in Lithuania. Forests 2021, 12, 317. [Google Scholar] [CrossRef]
- Karhu, A.; Hurme, P.; Karjalainen, M.; Karvonen, P.; Kärkkäinen, K.; Neale, D.; Savolainen, O. Do molecular markers reflect patterns of differentiation in adaptive traits of conifers? Theor. Appl. Genet. 1996, 93, 215–221. [Google Scholar] [CrossRef]
- Kujala, S.T.; Savolainen, O. Sequence variation patterns along a latitudinal cline in Scots pine (Pinus sylvestris): Signs of clinal adaptation? Tree Genet. Genomes 2012, 8, 1451–1467. [Google Scholar] [CrossRef]
- Prus-Głowacki, W.; Urbaniak, L.; Bujas, E.; Curtu, A.L. Genetic variation of isolated and peripheral populations of Pinus sylvestris (L.) from glacial refugia. Flora: Morphol. Distrib. Funct. Ecol. Plants 2012, 207, 150–158. [Google Scholar] [CrossRef]
- Bilgen, B.B.; Kaya, N. Genetic diversity among Pinus sylvestris L. populations and its implications for genetic conservation: Comparison of nuclear and chloroplast microsatellite markers. Fresenius Environ. Bull. 2017, 26, 6873. [Google Scholar]
- Persson, A.; Persson, B. Survival, Growth and Quality of Norway Spruce (Picea abies (L.) Karst.) Provenances at the Three Swedish Sites of the IUFRO 1964/68 Provenance Experiment; Rapport-Sveriges Lantbruksuniversitet: Uppsala, Sweden, 1992; Report 29; p. 67. [Google Scholar]
- Danusevičius, D.; Gabrilavičius, R. Variation in juvenile growth rhythm among Picea abies provenances from the Baltic states and adjacent regions. Scand. J. For. Res. 2001, 16, 305–317. [Google Scholar] [CrossRef]
- Eriksson, G. Pinus sylvestris Recent Genetic Research; Department of Plant Biology and Forest Genetics, SLU: Uppsala, Sweden, 2008; 111p, ISBN 978-91-85911-90-5. [Google Scholar]
- Baskerville, G.L.; Emin, P. Rapid estimation of heat accumulation from maximum and minimum temperatures. Ecology 1969, 50, 514–517. [Google Scholar] [CrossRef]
- Sarvas, R. Investigations on the annual cycle of development of forest trees. Autumn dormancy and winter dormancy. Commun. Linstinstituti For. Fenn. 1974, 84, 1–101. [Google Scholar]
- Lechowicz, M.J. Why do temperate deciduous trees leaf out at different times? Adaptation and ecology of forest communities. Am. Nat. 1984, 124, 821–842. [Google Scholar] [CrossRef]
- Badeck, F.W.; Bondeau, A.; Böttcher, K.; Doktor, D.; Lucht, W.; Schaber, J.; Sitch, S. Responses of spring phenology to climate change. New Phytol. 2004, 162, 295–309. [Google Scholar] [CrossRef]
- Antonucci, S.; Rossi, S.; Deslauriers, A.; Lombardi, F.; Marchetti, M.; Tognetti, R. Synchronisms and correlations of spring phenology between apical and lateral meristems in two boreal conifers. Tree Physiol. 2015, 35, 1086–1094. [Google Scholar] [CrossRef]
- Fu, Y.H.; Liu, Y.; De Boeck, H.J.; Menzel, A.; Nijs, I.; Peaucelle, M.; Janssens, I.A. Three times greater weight of daytime than of night-time temperature on leaf unfolding phenology in temperate trees. New Phytol. 2016, 212, 590–597. [Google Scholar] [CrossRef]
- Dai, W.; Jin, H.; Zhang, Y.; Liu, T.; Zhou, Z. Detecting temporal changes in the temperature sensitivity of spring phenology with global warming: Application of machine learning in phenological model. Agric. For. Meteorol. 2019, 279, 107702. [Google Scholar] [CrossRef]
- Wang, H.; Wu, C.; Ciais, P.; Penuelas, J.; Dai, J.; Fu, Y.; Ge, Q. Overestimation of the effect of climatic warming on spring phenology due to misrepresentation of chilling. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Hannerz, M. Genetic and Seasonal Variation in Hardiness and Growth Rhythm in Boreal and Temperate Conifers; Skogforsk: Uppsala, Sweden, 1998; Report No. 2; pp. 1–140. [Google Scholar]
- McKown, A.D.; Guy, R.D.; Azam, M.S.; Drewes, E.C.; Quamme, L.K. Seasonality and phenology alter functional leaf traits. Oecologia 2013, 172, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Delpierre, N.; Guillemot, J.; Dufrêne, E.; Cecchini, S.; Nicolas, M. Tree phenological ranks repeat from year to year and correlate with growth in temperate deciduous forests. Agric. For. Meteorol. 2017, 234, 1–10. [Google Scholar] [CrossRef]
- Danusevičius, D.; Kembrytė, R.; Buchovska, J.; Baliuckas, V.; Kavaliauskas, D. Genetic signature of the natural genepool of Tilia cordata Mill. in Lithuania: Compound evolutionary and anthropogenic effects. Ecol. Evol. 2021, 11, 6260–6275. [Google Scholar] [CrossRef]
- Sheller, M.; Ciocîrlan, E.; Mikhaylov, P.; Kulakov, S.; Kulakova, N.; Ibe, A.; Sukhikh, T.; Curtu, A.L. Chloroplast DNA Diversity in Populations of P. sylvestris L. from Middle Siberia and the Romanian Carpathians. Forests 2021, 12, 1757. [Google Scholar] [CrossRef]
- Urbaniak, L.; Wojnicka-Półtorak, A.; Celiński, K.; Lesiczka, P.; Pawlaczyk, E.; Aučina, A. Genetic resources of relict populations of Pinus sylvestris (L.) in Western Carpathians assessed by chloroplast microsatellites. Biologia 2019, 74, 1077–1086. [Google Scholar] [CrossRef]
- Kling, M.M.; Ackerly, D.D. Global wind patterns shape genetic differentiation, asymmetric gene flow, and genetic diversity in trees. Proc. Natl. Acad. Sci. USA 2021, 118, e2017317118. [Google Scholar] [CrossRef] [PubMed]
- Slatkin, M. Rare alleles as indicators of gene flow. Evolution 1985, 39, 53–65. [Google Scholar] [CrossRef]
- Helenurm, K.; Parsons, L.S. Genetic Variation and the Reproduction of Cordylanthus maritimus ssp. maritimus to Sweetwater Marsh, California. Restor. Ecol. 1997, 5, 236–244. [Google Scholar] [CrossRef]
- Menges, E.S. Restoration demography and genetics of plants: When is a translocation successful? Aust. J. Bot. 2008, 56, 187–196. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus * | Na | Null Allele Freq. | Ho | uHe | FIS | p-Value Dest |
|---|---|---|---|---|---|---|
| nSSRs | ||||||
| Psyl16 | 13 | 0.02 | 0.78 | 0.81 | 0.01 | 0.179 |
| Psyl18 | 7 | 0.02 | 0.12 | 0.12 | 0.06 | 0.614 |
| Psyl2 | 14 | 0.07 | 0.36 | 0.41 | 0.09 | 0.029 |
| Psyl25 | 8 | 0.04 | 0.08 | 0.09 | 0.07 | 0.156 |
| Psyl42 | 6 | 0.01 | 0.69 | 0.70 | −0.01 | 0.773 |
| Psyl44 | 6 | 0.15 | 0.06 | 0.12 | 0.39 | 0.003 |
| Psyl57 | 10 | 0.07 | 0.52 | 0.61 | 0.12 | 0.915 |
| PtTX4001 | 18 | −0.03 | 0.81 | 0.77 | −0.07 | 0.346 |
| PtTX4011 | 9 | 0.10 | 0.53 | 0.67 | 0.18 | 0.234 |
| Spac11.4 | 19 | 0.01 | 0.85 | 0.87 | −0.01 | 0.180 |
| Spac12.5 | 35 | 0.02 | 0.91 | 0.95 | 0.01 | 0.263 |
| Spac7.14 | 36 | 0.03 | 0.90 | 0.95 | 0.03 | 0.386 |
| Mean | 7.5 | - | 0.55 | 0.59 | 0.07 | 0.075 |
| St. error | 0.35 | - | 0.02 | 0.02 | 0.01 | - |
| cpSSRs ** | ||||||
| Pt71963 | 8 | - | - | 0.71 | - | 0.050 |
| Pt15169 | 9 | - | - | 0.78 | - | 0.975 |
| Pt30204 | 7 | - | - | 0.75 | - | 0.001 |
| Source | df | Var Comp% | Differ. Statistics | Value |
|---|---|---|---|---|
| cpSSR (3 Loci) | ||||
| Among regions | 5 | 6% | PhiRT | 0.065 *** |
| Among pops. within region | - | - | PhiSR | 0.187 *** |
| Among populations | 22 | 17% | PhiST | 0.240 *** |
| Within populations | 339 | 76% | - | - |
| nSSR (12 loci) | ||||
| Among regions | 5 | 0.1% | FRT | 0.001 ns |
| Among pops. within region | - | - | FSR | 0.005 ** |
| Among populations | 21 | 0.4% | FST | 0.005 ** |
| Within populations | 852 | 99.5% | - | - |
| Pop ID | Pop | Lat. | Long. | Alt. | nSSR | cpSSR | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Na | Ar | uHe | Ho | FIS | N | Nh | Nhr | uh | A | Rh | Nhe | Fpr | |||||
| 1 | PLUN | 55.58 | 21.53 | 122 | 20 | 8.3 | 7.3 | 0.57 | 0.53 | 0.11 | 20 | 17 | 4 | 0.72 | 16 | 12.9 | 11.8 | 0.05 |
| 2 | TRYS | 56.02 | 22.35 | 118 | 19 | 7.4 | 6.8 | 0.59 | 0.51 | 0.11 | 19 | 16 | 3 | 0.74 | 17 | 14.4 | 15.7 | 0.00 |
| 3 | KURT | 55.50 | 23.04 | 108 | 22 | 7.9 | 6.9 | 0.58 | 0.54 | 0.05 | 22 | 16 | 4 | 0.77 | 19 | 14.1 | 16.1 | 0.09 |
| 4 | VARN | 55.51 | 22.16 | 169 | 21 | 7.3 | 6.5 | 0.56 | 0.55 | 0.05 | 20 | 18 | 5 | 0.77 | 17 | 13.7 | 14.3 | 0.10 |
| Mean FNR 1 | 82 | 7.7 | 6.9 | 0.58 | 0.53 | 0.08 | 81 | 16.8 | 4.0 | 0.75 | 17.3 | 13.8 | 14.5 | 0.06 | ||||
| 5 | MIKE | 55.4 | 25.1 | 80 | 20 | 7.5 | 6.7 | 0.59 | 0.58 | 0.03 | 20 | 17 | 4 | 0.750 | 16 | 13.0 | 13.3 | 0.00 |
| 6 | GEGU | 55.51 | 24.29 | 60 | 20 | 7.7 | 7.0 | 0.59 | 0.57 | 0.03 | 20 | 17 | 4 | 0.79 | 16 | 13.0 | 13.3 | 0.15 |
| 7 | ROKI | 56.01 | 25.39 | 127 | 20 | 7.8 | 7.1 | 0.60 | 0.54 | 0.15 | 20 | 15 | 2 | 0.73 | 15 | 12.2 | 11.8 | 0.00 |
| Mean FNR 2 | 60 | 7.6 | 6.9 | 0.60 | 0.60 | 0.07 | 60 | 16.3 | 3.3 | 0.8 | 15.7 | 12.7 | 12.8 | 0.05 | ||||
| 8 | SALA | 55.35 | 25.51 | 164 | 20 | 7.8 | 7.0 | 0.57 | 0.53 | 0.08 | 17 | 17 | 5 | 0.72 | 14 | 13.0 | 12.6 | 0.06 |
| 9 | GRAZ | 55.59 | 26.08 | 183 | 20 | 7.1 | 6.6 | 0.61 | 0.52 | 0.25 | 18 | 17 | 4 | 0.81 | 17 | 15.1 | 16.2 | 0.11 |
| 10 | LABA | 55.16 | 25.47 | 174 | 21 | 7.9 | 7.0 | 0.61 | 0.56 | 0.13 | 21 | 17 | 5 | 0.7 | 18 | 14.1 | 16.3 | 0.05 |
| 11 | AZVI | 55.23 | 26.29 | 177 | 20 | 7.1 | 6.5 | 0.58 | 0.58 | 0.14 | 20 | 17 | 4 | 0.79 | 18 | 14.6 | 16.7 | 0.00 |
| Mean FNR 3 | 81 | 7.5 | 6.8 | 0.59 | 0.55 | 0.15 | 76 | 17.0 | 4.5 | 0.755 | 16.8 | 14.2 | 15.4 | 0.05 | ||||
| 12 | JUOD | 55.31 | 21.06 | 27 | 20 | 7.7 | 7.1 | 0.63 | 0.52 | 0.29 | 18 | 19 | 6 | 0.83 | 15 | 13.3 | 13.5 | 0.11 |
| 13 | DARB | 56.01 | 21.16 | 40 | 17 | 7.1 | 6.8 | 0.58 | 0.55 | 0.04 | 20 | 17 | 4 | 0.81 | 17 | 13.9 | 15.4 | 0.10 |
| 14 | SVEK | 55.31 | 21.45 | 38 | 20 | 7.8 | 7.1 | 0.6 | 0.58 | 0.01 | 20 | 15 | 3 | 0.75 | 18 | 14.6 | 16.7 | 0.05 |
| 15 | PAGE | 55.1 | 22.28 | 25 | 20 | 7.9 | 7.1 | 0.61 | 0.54 | 0.16 | 19 | 13 | 1 | 0.65 | 12 | 10.2 | 9.3 | 0.00 |
| Mean FNR 4 | 77 | 7.6 | 7.0 | 0.61 | 0.55 | 0.12 | 77 | 16.0 | 3.5 | 0.76 | 15.5 | 13.0 | 13.7 | 0.07 | ||||
| 16 | VAIS | 54.56 | 24.04 | 87 | 20 | 7.8 | 7.0 | 0.59 | 0.6 | 0.00 | 19 | 16 | 5 | 0.7 | 15 | 12.8 | 13.4 | 0.11 |
| 17 | BRAZ | 54.54 | 23.27 | 82 | 20 | 7.3 | 6.7 | 0.59 | 0.56 | 0.02 | 20 | 18 | 5 | 0.75 | 17 | 13.7 | 14.3 | 0.10 |
| 18 | PUNI | 54.28 | 24.03 | 81 | 20 | 7.2 | 6.6 | 0.58 | 0.57 | 0.00 | 20 | 16 | 3 | 0.7 | 16 | 13.0 | 13.3 | 0.05 |
| Mean FNR 5 | 60 | 7.4 | 6.8 | 0.6 | 0.6 | 0.00 | 59 | 16.7 | 4.3 | 0.7 | 16.0 | 13.2 | 13.7 | 0.09 | ||||
| 19 | ANCI | 54.04 | 23.53 | 122 | 20 | 7.3 | 6.7 | 0.59 | 0.5 | 0.14 | 17 | 18 | 6 | 0.77 | 16 | 15.0 | 15.2 | 0.29 |
| 20 | VEIS | 54.02 | 23.12 | 138 | 20 | 7.2 | 6.8 | 0.58 | 0.57 | 0.03 | 20 | 16 | 3 | 0.76 | 17 | 13.7 | 14.3 | 0.10 |
| 21 | TRAK | 53.57 | 23.47 | 122 | 20 | 7.3 | 6.7 | 0.57 | 0.52 | 0.12 | 18 | 18 | 5 | 0.72 | 13 | 11.5 | 10.8 | 0.11 |
| 22 | DRUS | 54.28 | 24.51 | 153 | 20 | 7.8 | 7.1 | 0.6 | 0.55 | 0.13 | 18 | 14 | 2 | 0.71 | 15 | 13.3 | 12.5 | 0.06 |
| Mean FNR 6 | 80 | 7.4 | 6.8 | 0.59 | 0.54 | 0.10 | 73 | 16.5 | 4.0 | 0.74 | 15.3 | 13.4 | 13.2 | 0.14 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kavaliauskas, D.; Danusevičius, D.; Baliuckas, V. New Insight into Genetic Structure and Diversity of Scots Pine (Pinus sylvestris L.) Populations in Lithuania Based on Nuclear, Chloroplast and Mitochondrial DNA Markers. Forests 2022, 13, 1179. https://doi.org/10.3390/f13081179
Kavaliauskas D, Danusevičius D, Baliuckas V. New Insight into Genetic Structure and Diversity of Scots Pine (Pinus sylvestris L.) Populations in Lithuania Based on Nuclear, Chloroplast and Mitochondrial DNA Markers. Forests. 2022; 13(8):1179. https://doi.org/10.3390/f13081179
Chicago/Turabian StyleKavaliauskas, Darius, Darius Danusevičius, and Virgilijus Baliuckas. 2022. "New Insight into Genetic Structure and Diversity of Scots Pine (Pinus sylvestris L.) Populations in Lithuania Based on Nuclear, Chloroplast and Mitochondrial DNA Markers" Forests 13, no. 8: 1179. https://doi.org/10.3390/f13081179
APA StyleKavaliauskas, D., Danusevičius, D., & Baliuckas, V. (2022). New Insight into Genetic Structure and Diversity of Scots Pine (Pinus sylvestris L.) Populations in Lithuania Based on Nuclear, Chloroplast and Mitochondrial DNA Markers. Forests, 13(8), 1179. https://doi.org/10.3390/f13081179