Physiological, Transcriptomic and Metabolomic Analyses of Overwintering Cryptomeria fortunei Needles


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Physiological Parameter Determination
2.3. RNA Extraction and cDNA Library Construction
2.4. Transcriptomic Analysis
2.5. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Validation
2.6. Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
2.7. Statistical Analysis
3. Results
3.1. Changes in Physiological Indicators in Overwintering C. fortunei
3.2. De novo Assembly and Annotation of the C. fortunei Transcriptome
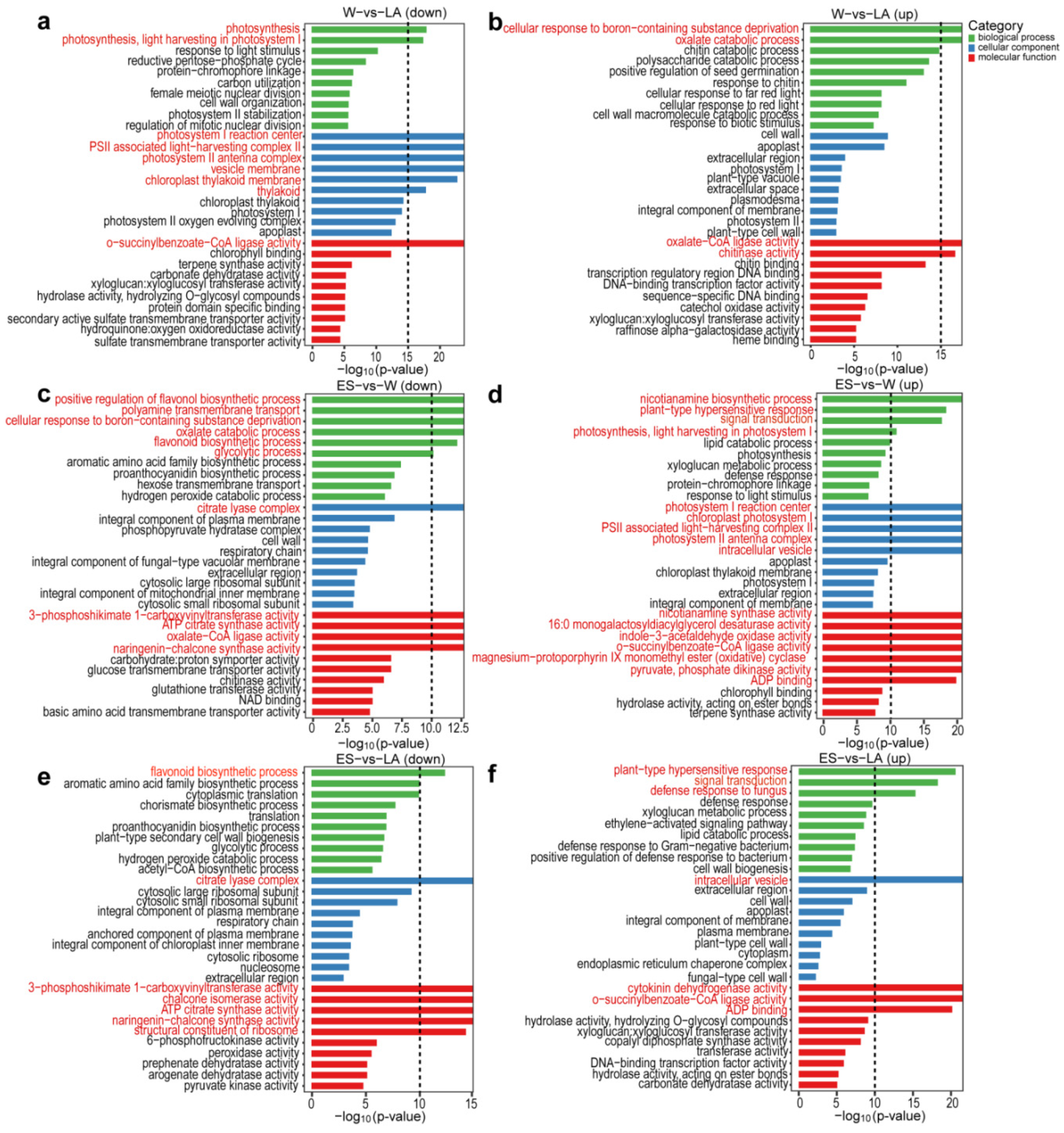
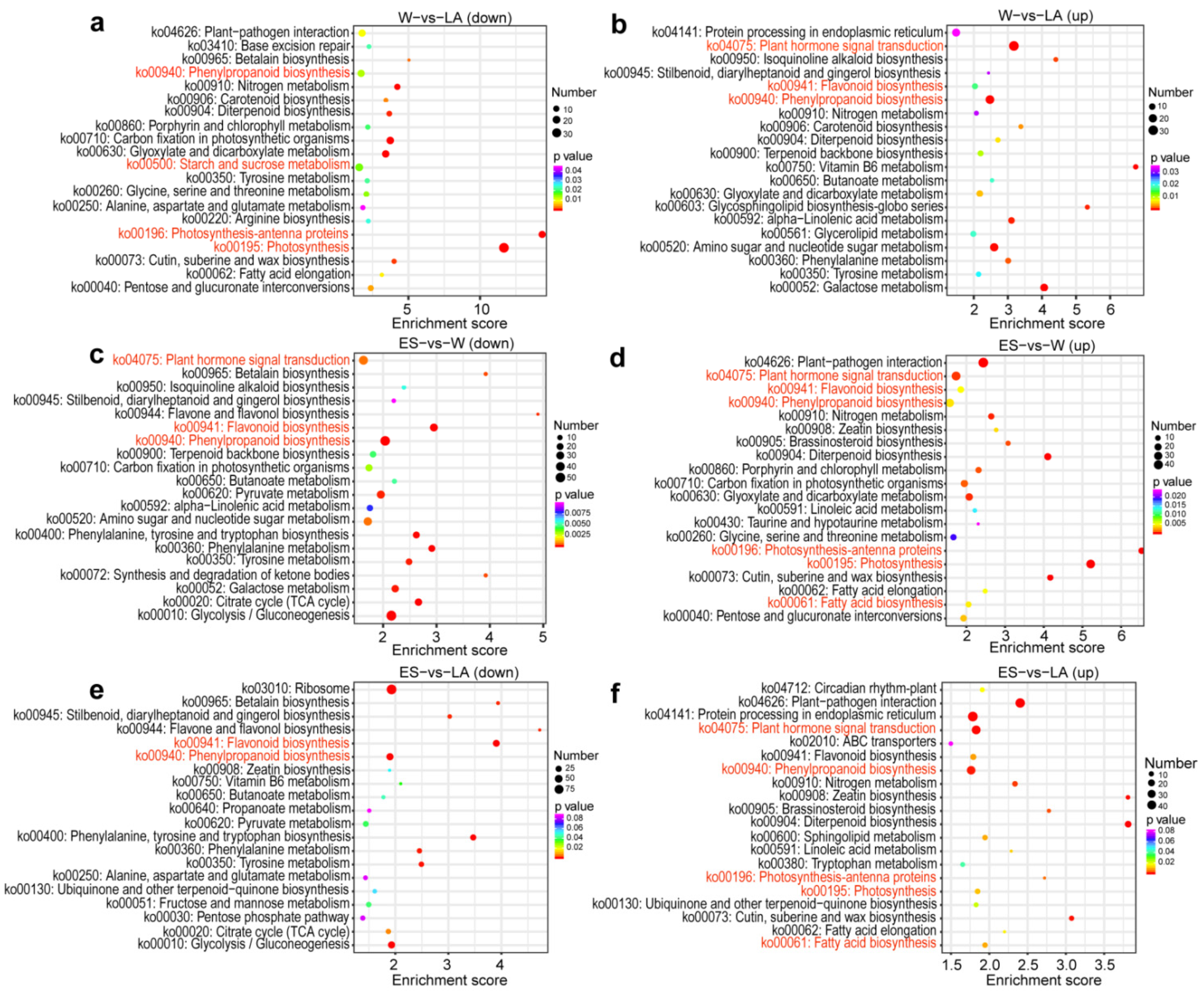
3.3. Functional Enrichment Analysis of DEGs
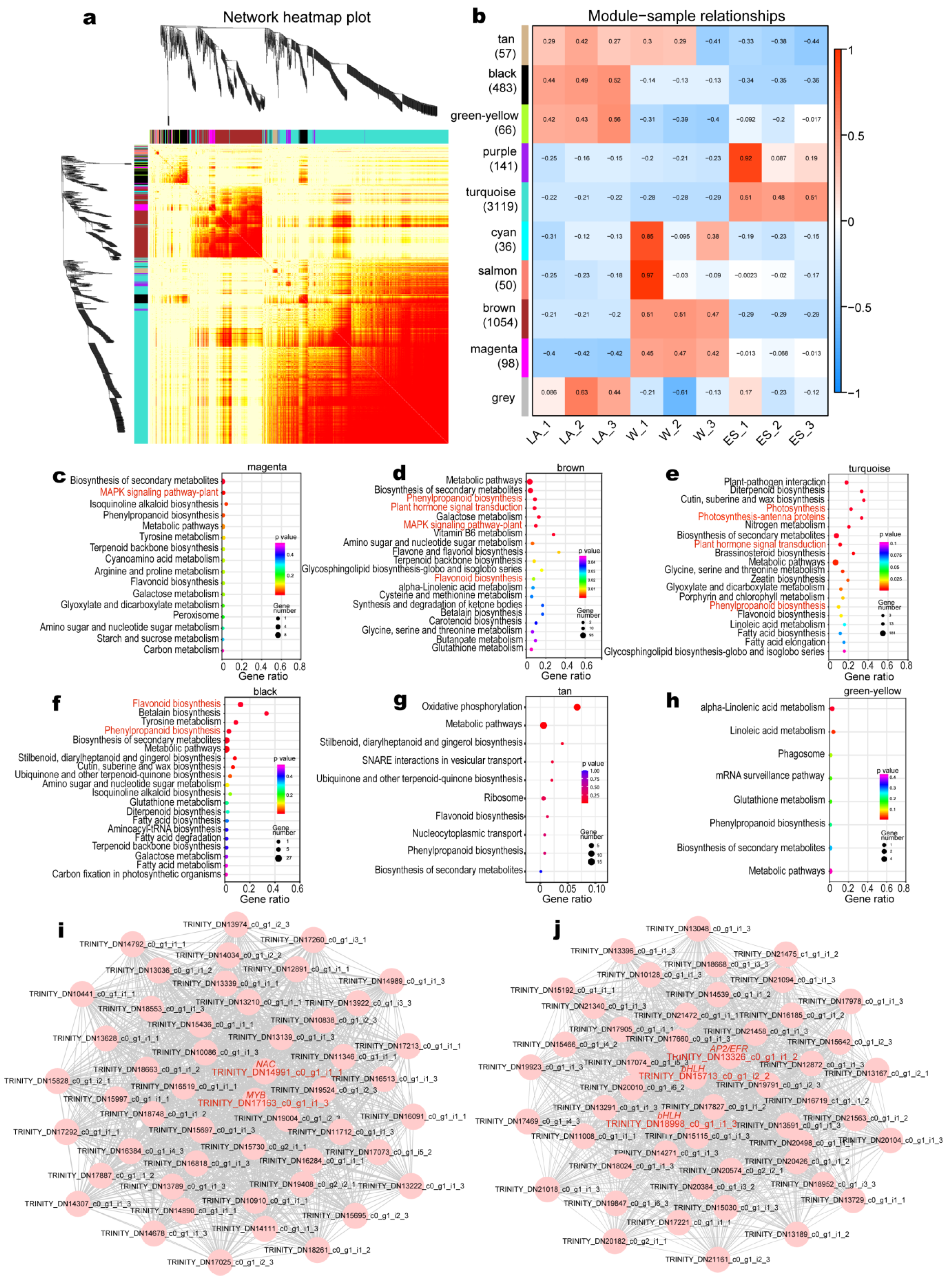
3.4. WGCNA
3.5. Metabolomic Analysis
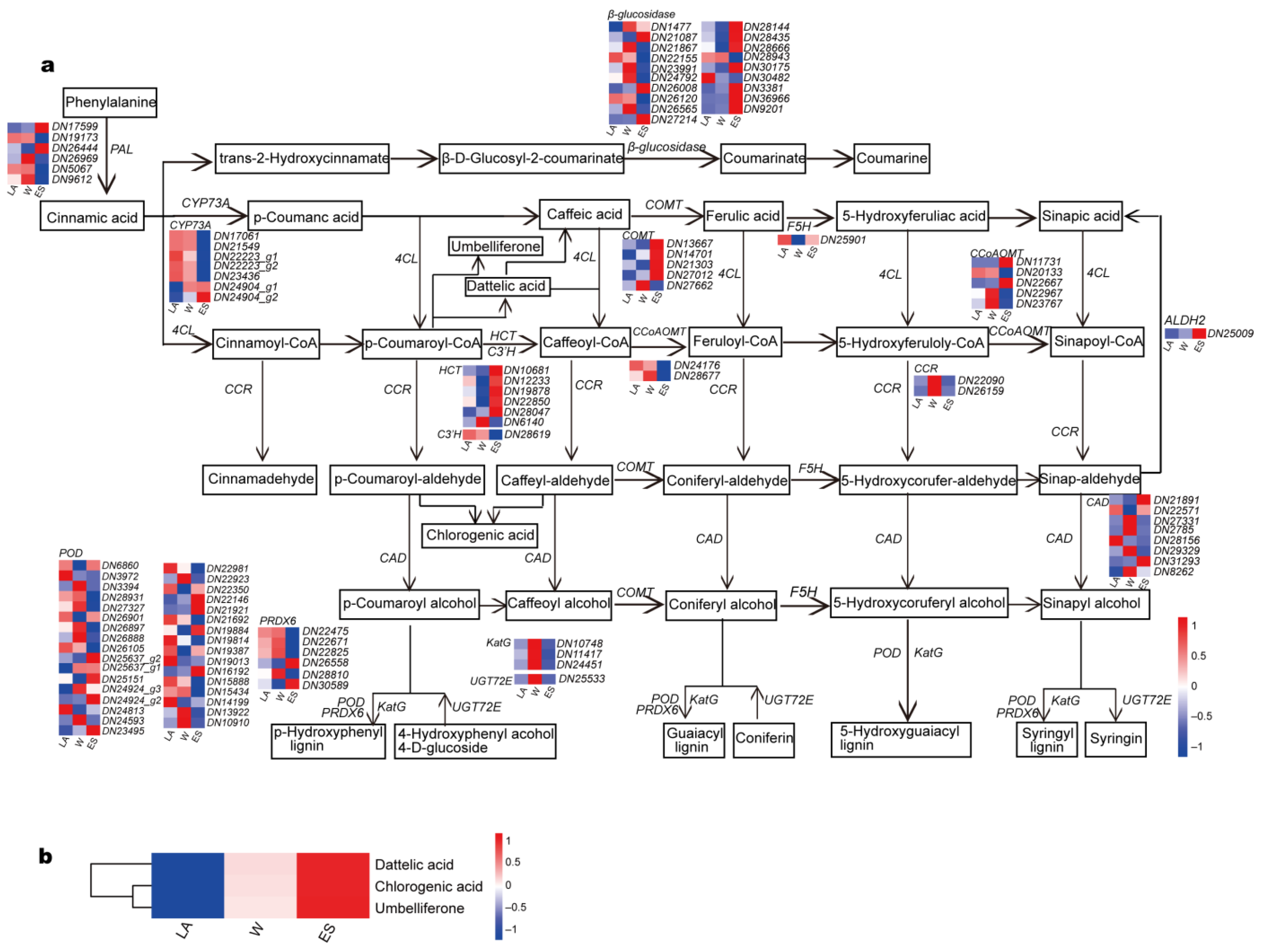
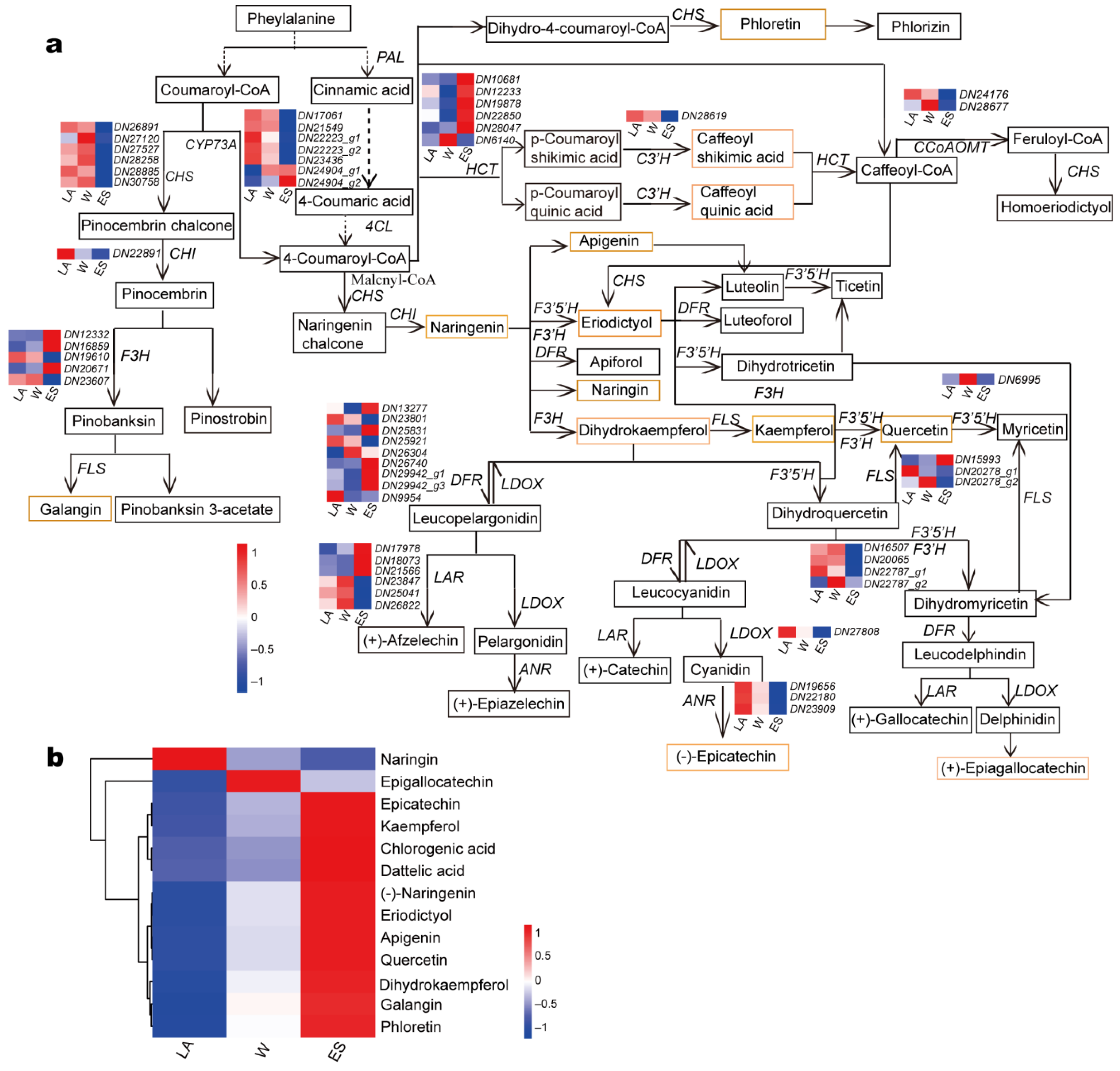
3.6. Identification of DEGs Involved in Phenylpropanoid Biosynthesis and Flavonoid Biosynthesis Pathways
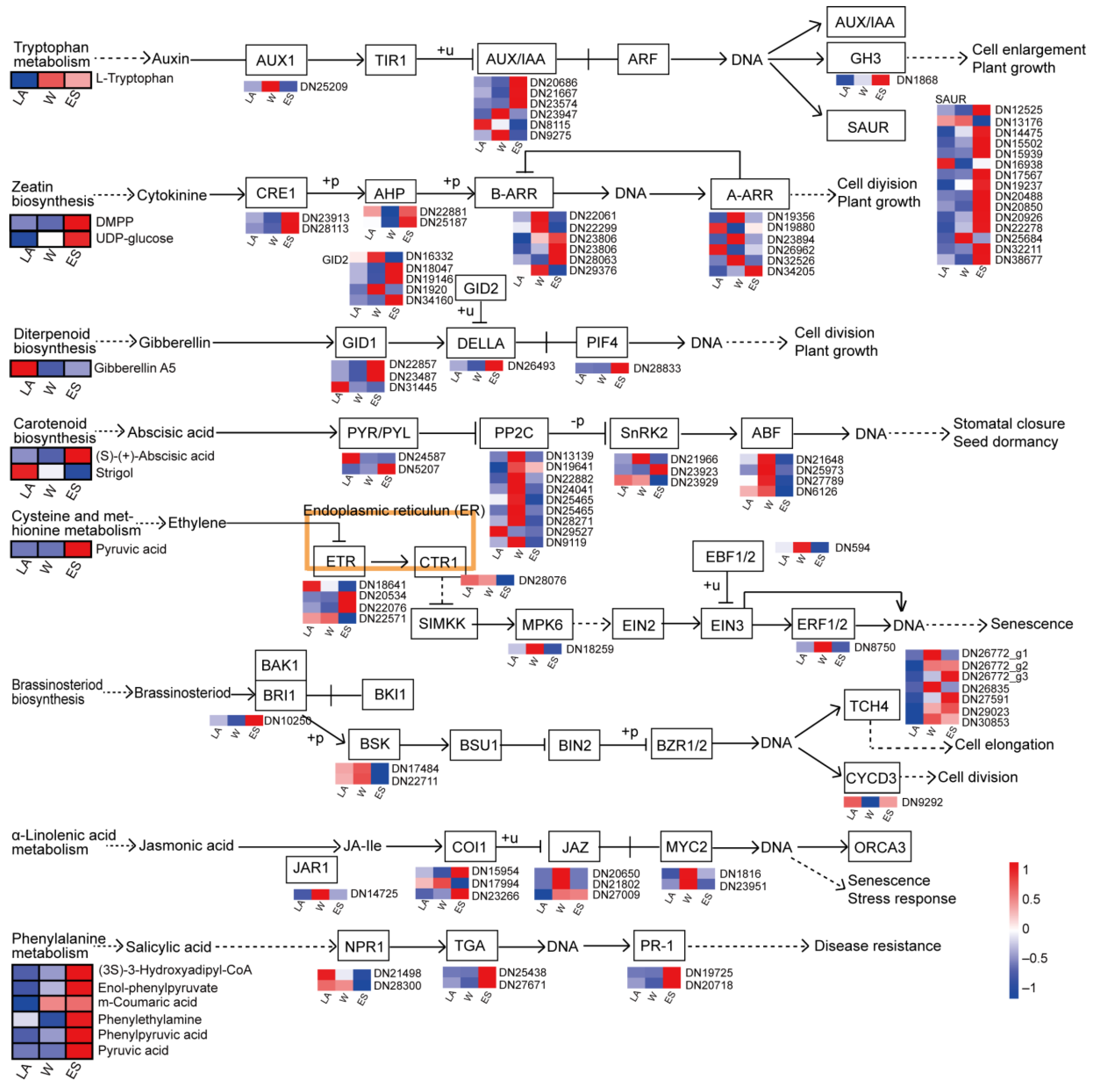
3.7. Identification of DEGs Involved in Plant Hormone Signal Transduction Pathways
3.8. Identification of DEGs Involved in Starch and Sucrose Metabolism and Photosynthesis-Related Pathways
3.9. Identification of DEGs Involved in (Unsaturated) Fatty Acid Biosynthesis or Related to LT-Related Proteins
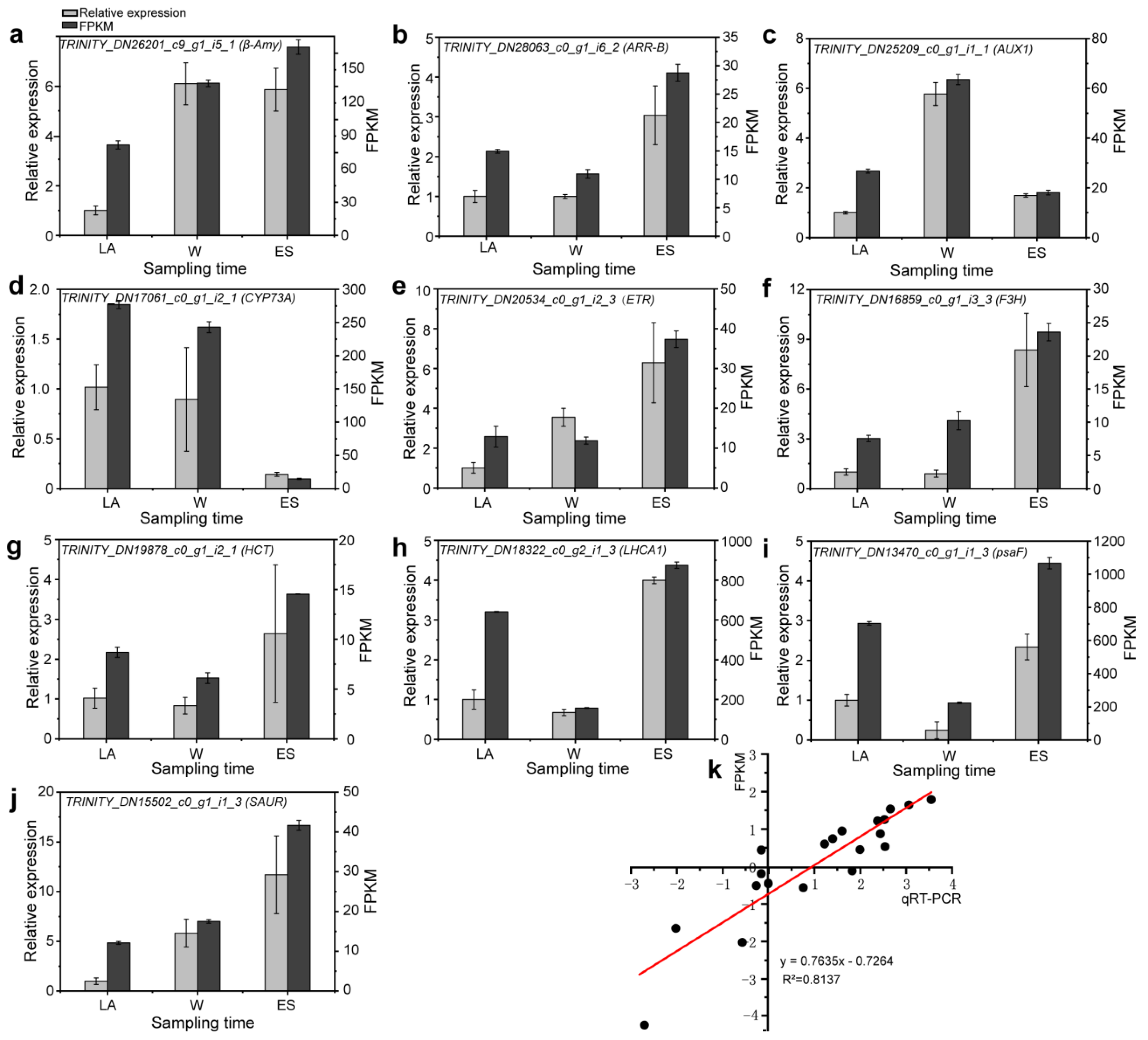
3.10. Verification of RNA-seq Results Using qRT-PCR
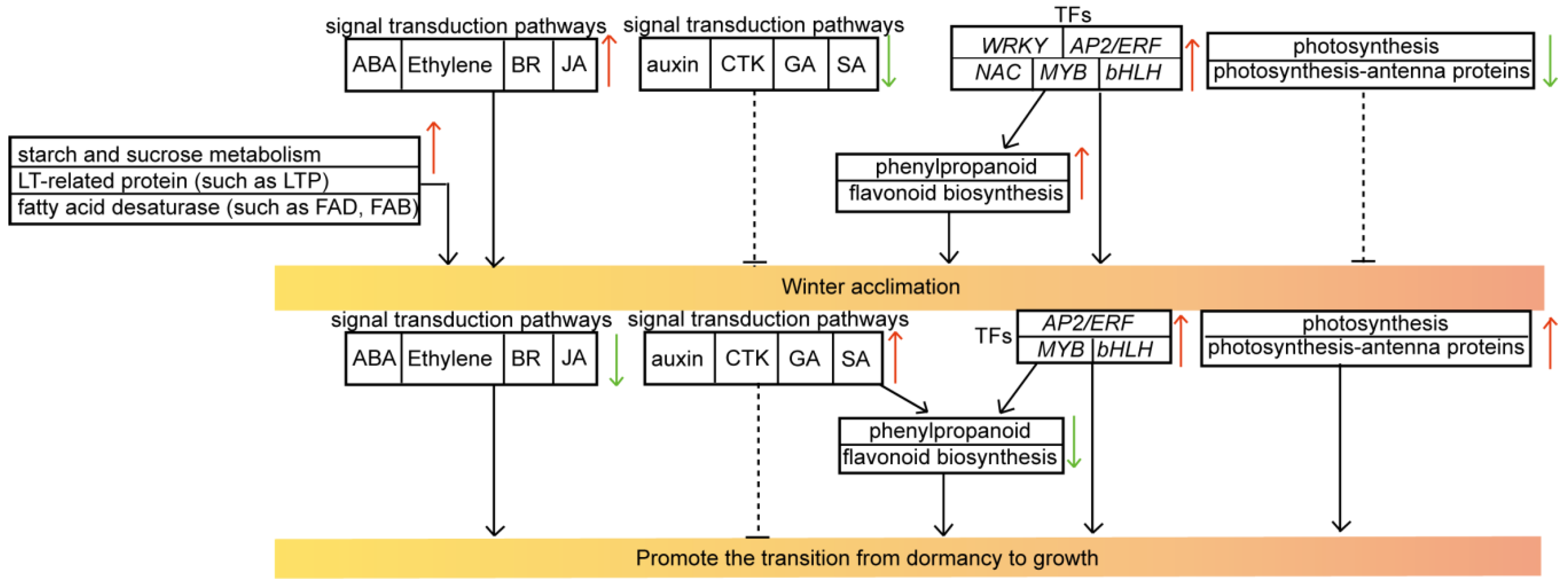
4. Discussion
4.1. Changes in Physiological Indicators in Overwintering C. fortunei
4.2. Changes in Photosynthesis in Overwintering C. fortunei
4.3. Genes Involved in the Flavonoid and Phenylpropanoid Biosynthesis Pathways Are Activated to Promote Metabolite Accumulation in Overwintering C. fortunei
4.4. Overwintering C. fortunei Plants Form a Complex Hormone Signaling Network
4.5. Fatty Acid Desaturase and LT-Related Proteins Are Activated to Promote Winter Acclimation in Overwintering C. fortunei
4.6. TFs Show Upregulated Expression in Response to Cold Winters
4.7. Comparison with Extremely and Moderately Cold-tolerant Plant Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, L.; Yu, Y.; Robert, R.M. Taxodiaceae. In Flora of China; Wu, Z., Peter, H.R., Eds.; Science Press: Beijing, China, 1999; Volume 4, pp. 54–64. [Google Scholar]
- Zhang, Y.; Zhu, Q.; Zhang, M.; Guo, Z.; Yang, J.; Mo, J.; Cui, J.; Hu, H.; Xu, J. Individual Cryptomeria fortunei hooibrenk clones show varying degrees of chilling stress resistance. Forests 2020, 11, 189. [Google Scholar] [CrossRef]
- Han, Q.; Shinohara, K.; Kakubari, Y.; Mukai, Y. Photoprotective role of rhodoxanthin during cold acclimation in Cryptomeria japonica. Plant Cell Environ. 2003, 26, 715–723. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Zhu, J.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Strimbeck, G.R.; Schaberg, P.G. Going to extremes: Low-temperature tolerance and acclimation in temperate and boreal conifers. In Plant Cold Hardiness: From the Laboratory to the Field; Gusta, L.V., Wisniewski, M., Tanino, K.K., Eds.; CABI: Oxfordshire, UK, 2009; pp. 226–239. [Google Scholar]
- Strimbeck, G.R.; Schaberg, P.G.; Fossdal, C.G.; Schroder, W.P.; Kjellsen, T.D. Extreme low temperature tolerance in woody plants. Front. Plant Sci. 2015, 6, 884. [Google Scholar] [CrossRef] [PubMed]
- Kjellsen, T.D.; Yakovlev, I.A.; Fossdal, C.G.; Strimbeck, G.R. Dehydrin accumulation and extreme low-temperature tolerance in Siberian spruce (Picea obovata). Tree Physiol. 2013, 33, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Strimbeck, G.R.; Kjellsent, T.D.; Schaberg, P.G.; Murakami, P.F. Cold in the common garden: Comparative tolerance of boreal and temperate conifer foliage. Trees 2007, 21, 557–567. [Google Scholar] [CrossRef]
- DeHayes, D.H.; Schaberg, P.G.; Strimbeck, G.R. Red spruce (Picea rubens Sarg.) cold hardiness and freezing injury susceptibility. In Conifer Cold Hardiness; Bigras, F.J., Colombo, S.J., Eds.; Springer: Dordrecht, The Netherlands, 2001; Volume 1, pp. 495–529. [Google Scholar]
- Bleecker, A.B.; Kende, H. Ethylene: A gaseous signal molecule in plants. Annu. Rev. Cell Dev. Biol. 2000, 16, 1–18. [Google Scholar] [CrossRef]
- Greer, D.; Berry, J.; Björkman, O. Photoinhibition of photosynthesis in intact bean leaves: Role of light and temperature, and requirement for chloroplast-protein synthesis during recovery. Planta 1986, 168, 253–260. [Google Scholar]
- Ishitani, M. Genetic analysis of osmotic and cold stress signal transduction in Arabidopsis: Interactions and convergence of abscisic acid-dependent and abscisic acid-independent pathways. Plant Cell 1997, 9, 1935–1949. [Google Scholar]
- Thomashow, M.F. Plant cold acclimation: Freezing tolerance genes and regulatory mechanisms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 571–599. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Hu, H.; Yang, J.; Cui, J.; Xu, J. Cloning and cold-resistance analyses of CfICE1 gene in Cryptomeria fortunei. Plant Physiol. Biochem. 2021, 162, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yu, S.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The role of WRKY transcription factors in plant abiotic stresses. BBA-Gene Regul. Mech. 2012, 1819, 120–128. [Google Scholar] [CrossRef] [PubMed]
- de Souza, R.R.; Rouhana, L.V.; Sadeque, A.; Koga, L.; Clough, S.J.; Calla, B.; de Oliveira Paiva, P.D.; Korban, S.S. Genome-wide expression of low temperature response genes in Rosa hybrida L. Plant Physiol. Biochem. 2020, 146, 238–248. [Google Scholar]
- Ruan, M.B.; Guo, X.; Wang, B.; Yang, Y.L.; Li, W.Q.; Yu, X.L.; Zhang, P.; Peng, M. Genome-wide characterization and expression analysis enables identification of abiotic stress-responsive MYB transcription factors in cassava (Manihot esculenta). J. Exp. Bot. 2017, 68, 3657–3672. [Google Scholar] [CrossRef]
- Wang, L.; Xiang, L.; Hong, J.; Xie, Z.; Li, B. Genome-wide analysis of bHLH transcription factor family reveals their involvement in biotic and abiotic stress responses in wheat (Triticum aestivum L.). 3 Biotech 2019, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- An, J.P.; Li, R.; Qu, F.J.; You, C.X.; Wang, X.F.; Hao, Y.J. An apple NAC transcription factor negatively regulates cold tolerance via CBF-dependent pathway. J. Plant Physiol. 2017, 221, 74–80. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Ding, W.; Li, H.; Shi, T.; Yang, X.; Wang, L.; Yue, Y. Genome-wide identification of Osmanthus fragrans bHLH transcription factors and their expression analysis in response to abiotic stress. Environ. Exp. Bot. 2020, 172, 103990. [Google Scholar] [CrossRef]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. BBA-Gene Regul. Mech. 2012, 1819, 86–96. [Google Scholar] [CrossRef]
- Zhou, L.; Rajesh, Y.; Jin, L.; Cao, H. Genome-wide identification and expression analysis of MYB gene family in oil palm (Elaeis guineensis Jacq.) under abiotic stress conditions. Environ. Exp. Bot. 2020, 180, 104245. [Google Scholar] [CrossRef]
- Akhtar, M.; Jaiswal, A.; Taj, G.; Jaiswal, J.P.; Qureshi, M.I.; Singh, N.K. DREB1/CBF transcription factors: Their structure, function and role in abiotic stress tolerance in plants. J. Genet. 2012, 91, 385–395. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, M.; Lee, J.H.; Lee, H.J.; Park, C.M. The unified ICE–CBF pathway provides a transcriptional feedback control of freezing tolerance during cold acclimation in Arabidopsis. Plant Mol. Biol. 2015, 89, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Thomashow, M.F. Molecular basis of plant cold acclimation: Insights gained from studying the CBF cold response pathway. Plant Physiol. 2010, 154, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K.; Jha, B. Transcription factors in plants and ABA dependent and independent abiotic stress signalling. Biol. Plant. 2010, 54, 201–212. [Google Scholar] [CrossRef]
- Huang, X.; Shi, H.; Hu, Z.; Ao, L.; Erick, A.; Chen, L.; Fu, J. ABA is involved in regulation of cold stress response in bermudagrass. Front. Plant Sci. 2017, 8, 1613. [Google Scholar] [CrossRef]
- Hormaetxe, K.; Hernandez, A.; Becerril, J.M.; García-Plazaola, J.I. Role of red carotenoids in photoprotection during winter acclimation in Buxus sempervirens leaves. Plant Biol. 2004, 6, 325–332. [Google Scholar] [CrossRef]
- Hughes, N.M. Winter leaf reddening in “evergreen” species. New Phytol. 2011, 190, 573–581. [Google Scholar] [CrossRef]
- Hormaetxe, K.; Becerril, J.M.; Hernandez, A.; Esteban, R.; García-Plazaola, J.I. Plasticity of photoprotective mechanisms of Buxus sempervirens L. leaves in response to extreme temperatures. Plant Biol. 2007, 9, 59–68. [Google Scholar] [CrossRef]
- Tanaka, A. Photosynthetic activity in winter needles of the evergreen tree Taxus cuspidata at low temperatures. Tree Physiol. 2007, 27, 641–648. [Google Scholar] [CrossRef]
- Oquist, G.; Huner, N.P. Photosynthesis of overwintering evergreen plants. Annu. Rev. Plant Biol. 2003, 54, 329–355. [Google Scholar] [CrossRef]
- Hughes, N.M.; Burkey, K.O.; Lavender-Bares, J.; Smith, W.K. Xanthophyll cycle pigment and antioxidant profiles of winter-red (anthocyanic) and winter-green (acyanic) angiosperm evergreen species. J. Exp. Bot. 2012, 63, 1895–1905. [Google Scholar] [CrossRef]
- Fischer, B.B.; Hideg, E.; Krieger-Liszkay, A. Production, detection, and signaling of singlet oxygen in photosynthetic organisms. Antioxid. Redox Signal. 2013, 18, 2145–2162. [Google Scholar] [CrossRef] [PubMed]
- Ruelland, E.; Vaultier, M.; Zachowslci, A.; Hurry, V. Cold signalling and cold acclimation in plants. Adv. Bot. Res. 2009, 49, 35–150. [Google Scholar]
- Nozzolillo, C.; Isabelle, P.; Das, G. Seasonal changes in the phenolic constituents of jack pine seedlings (Pinus banksiana) in relation to the purpling phenomenon. Can. J. Bot. 1990, 68, 2010–2017. [Google Scholar] [CrossRef]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genom. Hum. Genet. 2009, 10, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.M.; Zhou, Q.; Luo, F.; Wei, B.D.; Wang, Y.J.; Sun, H.J.; Zhao, Y.B.; Ji, S.J. Transcriptome analysis of harvested bell peppers (Capsicum annuum L.) in response to cold stress. Plant Physiol. Biochem. 2019, 139, 314–324. [Google Scholar] [CrossRef]
- Yael, K.; Adi, D.F.; Doron, H.; Ron, P. Effects of harvest time on chilling tolerance and the transcriptome of ‘Wonderful’ pomegranate fruit. Postharvest Biol. Tec. 2019, 147, 10–19. [Google Scholar]
- Wang, F.; Chen, S.; Cai, K.; Lu, Z.; Yang, Y.; Tigabu, M.; Zhao, X. Transcriptome sequencing and gene expression profiling of Pinus sibirica under different cold stresses. Breed Sci. 2021, 71, 550–563. [Google Scholar] [CrossRef]
- Wang, F.; Chen, S.; Liang, D.; Qu, G.; Chen, S.; Zhao, X. Transcriptomic analyses of Pinus koraiensis under different cold stresses. BMC Genom. 2020, 21, 10. [Google Scholar] [CrossRef]
- Vergara, A.; Haas, J.C.; Aro, T.; Stachula, P.; Street, N.R.; Hurry, V. Norway spruce deploys tissue-specific responses during acclimation to cold. Plant Cell Environ. 2022, 45, 427–445. [Google Scholar] [CrossRef]
- Angelcheva, L.; Mishra, Y.; Antti, H.; Kjellsen, T.D.; Funk, C.; Strimbeck, R.G.; Schroder, W.P. Metabolomic analysis of extreme freezing tolerance in Siberian spruce (Picea obovata). New Phytol. 2014, 204, 545–555. [Google Scholar] [CrossRef]
- Kjellsen, T.D.; Shiryaeva, L.; Schröder, W.P.; Strimbeck, G.R. Proteomics of extreme freezing tolerance in Siberian spruce (Picea obovata). J. Proteom. 2010, 73, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Nishida, I.; Murata, N. Chilling sensitivity in plants and cyanobacterta: The crucial contribution of membrane lipids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 541–568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.; Zhu, L.; Xue, J.; Hu, H.; Cui, J.; Xu, J. Identification of microRNAs and their target genes related to needle discoloration of evergreen tree Chinese cedar (Cryptomeria fortunei) in cold winters. Planta 2021, 254, 31. [Google Scholar] [CrossRef] [PubMed]
- Tsarouhas, V.; Kenney, W.A.; Zsuffa, L. Application of two electrical methods for the rapid assessment of freezing resistance in Salix eriocephala. Biomass Bioenerg. 2000, 19, 165–175. [Google Scholar] [CrossRef]
- Li, H.S.; Sun, Q.; Zhao, S.J.; Zhang, W.H. Principles and Techniques of Plant Physiology and Biochemistry Experiments, 1st ed.; Higher Education Press: Beijing, China, 2000; pp. 164–261. [Google Scholar]
- Lichtenthaler, H.K.; Wellburn, A.R. Determinations of total carotenoids and chlorophylls a and b of leaf extracts in different solvents. Analysis 1983, 11, 591–592. [Google Scholar] [CrossRef]
- Maxwell, N.; Johnson, G.N. Chlorophyll fluorescence-a practical guide. J. Exp. Bot. 2000, 51, 659–668. [Google Scholar] [CrossRef]
- Draper, H.H.; Hadley, M. Malondialdehyde determination as index of lipid peroxidation. Method. Enzymol. 1990, 186, 421–431. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Britton, C.; Maehly, A.C. Assay of catalases and peroxidases. Method. Enzymol. 1955, 2, 764–775. [Google Scholar]
- Giannopolites, C.N.; Ries, S.K. Superoxide dismutase occurrence in higher plants. Plant Physiol. 1977, 59, 309–314. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Amit, I. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2013, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Roberts, A.; Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 2013, 10, 71–73. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level-the DESeq Package. Available online: http://mirrors.ustc.edu.cn/bioc/3.4/bioc/vignettes/DESeq/inst/doc/DESeq.pdf (accessed on 12 January 2016).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, L.; Xue, J.; Yang, J.; Hu, H.; Cui, J.; Xu, J. Selection and verification of appropriate reference genes for expression normalization in Cryptomeria fortunei under abiotic stress and hormone treatments. Genes 2021, 12, 791. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, J.; Hu, H.; Xue, J.; Yang, J.; Xu, J. Integrated four comparative-omics reveals the mechanism of the terpenoid biosynthesis in two different overwintering Cryptomeria fortunei phenotypes. Front. Plant Sci. 2021, 12, 740755. [Google Scholar] [CrossRef] [PubMed]
- Bajji, M.; Kinet, J.M.; Lutts. S. The use of the electrolyte leakage method for assessing cell membrane stability as a water stress tolerance test in durum wheat. Plant Growth Regul. 2002, 36, 61–70. [Google Scholar] [CrossRef]
- Hepburn, H.A.; Naylor, R.; Stokes, D.T. Electrolyte leakage from winter barley tissue as an indicator of winter-hardiness. Ann. Appl. Biol. 1986, 108, 164–165. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, J.; Shi, K.; Xia, X.J.; Zhou, Y.H.; Yu, J.Q. Hydrogen peroxide is involved in the cold acclimation-induced chilling tolerance of tomato plants. Plant Physiol. Biochem. 2012, 60, 141–149. [Google Scholar] [CrossRef]
- McWilliam, J.R.; Kramer, P.J.; Musser, R.L. Temperature-induced water stress in chilling-sensitive plants. Funct. Plant Biol. 1982, 9, 343–352. [Google Scholar] [CrossRef]
- İşeri, Ö.D.; Körpe, D.A.; Sahin, F.I.; Haberal, M. Hydrogen peroxide pretreatment of roots enhanced oxidative stress response of tomato under cold stress. Acta Physiol. Plant. 2013, 35, 1905–1913. [Google Scholar] [CrossRef]
- Lakra, N.; Nutan, K.K.; Das, P.; Anwar, K.; Singla-Pareek, S.L.; Pareek, A. A nuclear-localized histone-gene binding protein from rice (OsHBP1b) functions in salinity and drought stress tolerance by maintaining chlorophyll content and improving the antioxidant machinery. J. Plant Physiol. 2015, 176, 36–46. [Google Scholar] [CrossRef]
- Ottander, C.; Campbell, D.; Oquist, G. Seasonal changes in photosystem II organisation and pigment composition in Pinus sylvestris. Planta 1995, 197, 176–183. [Google Scholar] [CrossRef]
- Öquist, G.; Gardeström, P.; Huner, N.P.A. Metabolic changes during cold acclimation and subsequent freezing and thawing. In Conifer Cold Hardiness; Bigras, F.J., Colombo, S.J., Eds.; Springer: Dordrecht, The Netherlands, 2001; Volume 1, pp. 137–163. [Google Scholar]
- Mochizuki, N.; Brusslan, J.A.; Larkin, R.; Nagatani, A.; Chory, J. Arabidopsis genomes uncoupled 5 (GUN5) mutant reveals the involvement of Mg-chelatase H subunit. Proc. Natl. Acad. Sci. USA 2001, 98, 2053–2058. [Google Scholar] [CrossRef]
- Barber, J. Photosystem II. Biochim. Biophys. Acta 1998, 1165, 269–277. [Google Scholar] [CrossRef]
- Malone, L.A.; Qian, P.; Mayneord, G.E.; Hitchcock, A.; Farmer, D.A.; Thompson, R.F.; Swainsbury, D.J.K.; Ranson, N.A.; Hunter, C.N.; Johnson, M.P. Cryo-EM structure of the spinach cytochrome b6f complex at 3.6 A resolution. Nature 2009, 575, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Goral, T.K.; Johnson, M.P.; Duffy, C.D.; Brain, A.P.R.; Ruban, A.V.; Mullineaux, C.W. Light-harvesting antenna composition controls the macrostructure and dynamics of thylakoid membranes in Arabidopsis. Plant J. 2012, 69, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, T.; Feng, B.; Zhang, C.; Peng, S.; Zhang, X.; Fu, G.; Tao, L. Non-photochemical quenching plays a key role in light acclimation of rice plants differing in leaf color. Front. Plant Sci. 2016, 7, 1968–1985. [Google Scholar] [CrossRef]
- Dixon, R.A.; Paiva, N.L. Stress-induced phenylpropanoid metabolism. Plant Cell 1995, 7, 1085. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, W.; Liu, J.; Liu, H.; Lv, Z.; Zhang, C.; Chen, D.; Jiao, Z. Postharvest UV-C irradiation increased the flavonoids and anthocyanins accumulation, phenylpropanoid pathway gene expression, and antioxidant activity in sweet cherries (Prunus avium L.). Postharvest Biol. Technol. 2021, 175, 111490. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Z.; Zhang, Y.; Mo, J.; Cui, J.; Hu, H.; He, Y.; Xu, J. Transcriptomic profiling of Cryptomeria fortunei Hooibrenk vascular cambium identifies candidate genes involved in phenylpropanoid metabolism. Forests 2020, 11, 766. [Google Scholar] [CrossRef]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of phenylpropanoid pathway and the role of polyphenols in plants under abiotic stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Yonekura-Sakakibara, K.; Urano, K.; Suzuki, M.; Yamada, Y.; Nishizawa, T.; Matsuda, F.; Kojima, M.; Sakakibara, H.; Shinozaki, K.; et al. Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of antioxidant flavonoids. Plant J. 2014, 77, 367–379. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Biosynthesis of flavonoids and effects of stress. Curr. Opin. Plant Biol. 2002, 5, 218–223. [Google Scholar] [CrossRef]
- Liu, X.; Dong, Y.; Yao, N.; Zhang, Y.; Wang, N.; Cui, X.; Li, X.; Wang, Y.; Wang, F.; Yang, J.; et al. De novo sequencing and analysis of the safflower transcriptome to discover putative genes associated with safflor yellow in Carthamus tinctorius L. Int. J. Mol. Sci. 2015, 16, 25657–25677. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Ma, Q.; Zou, Z.; Kang, S.; Yao, Y.; Tao, J.; Kaleri, N.A.; Li, X. Molecular link between leaf coloration and gene expression of flavonoid and carotenoid biosynthesis in Camellia sinensis cultivar ‘Huangjinya’. Front. Plant Sci. 2017, 8, 803. [Google Scholar] [CrossRef] [PubMed]
- Bari, R.; Jones, J. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Gusta, L.V.; Trischuk, R.; Weiser, C.J. Plant cold acclimation: The role of abscisic acid. J. Plant Growth Regul. 2005, 24, 308–318. [Google Scholar] [CrossRef]
- Lata, C.; Prasad, M. Role of DREBs in regulation of abiotic stress responses in plants. J. Exp. Bot. 2011, 62, 4731–4748. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N. Abscisic acid and abiotic stress signaling. Plant Signal. Behav. 2007, 2, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.S.; Liu, J.H.; Chen, X.J. Overexpression of PtrABF gene, a bZIP transcription factor isolated from Poncirus trifoliata, enhances dehydration and drought tolerance in tobacco via scavenging ROS and modulating expression of stress-responsive genes. BMC Plant Biol. 2010, 10, 230. [Google Scholar] [CrossRef] [PubMed]
- Berger, S. Jasmonate-related mutants of Arabidopsis as tools for studying stress signaling. Planta 2002, 214, 497–504. [Google Scholar] [CrossRef]
- Rehman, M.; Singh, Z.; Khurshid, T. Methyl jasmonate alleviates chilling injury and regulates fruit quality in ‘Midknight’ Valencia orange. Postharvest Biol. Technol. 2018, 141, 58–62. [Google Scholar] [CrossRef]
- Yu, X.M.; Griffith, M.; Wiseman, S.B. Ethylene induces antifreeze activity in winter rye leaves. Plant Physiol. 2001, 126, 1232–1240. [Google Scholar] [CrossRef]
- Pedranzani, H.; Sierra-de-Grado, R.; Vigliocco, A.; Miersch, O.; Abdala, G. Cold and water stresses produce changes in endogenous jasmonates in two populations of Pinus pinaster Ait. Plant Growth Regul. 2007, 52, 111–116. [Google Scholar] [CrossRef]
- Stumpff, N.J. Ethylene Production by Loblolly Pine Seedlings during Cold Storage and Water Stress. Master’s Thesis, VPISU, Blacksburg, Virginia, 1984. [Google Scholar]
- Ramirez, V.E.; Poppenberger, B. Modes of brassinosteroid activity in cold stress tolerance. Front. Plant Sci. 2020, 11, 583666. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Purugganan, M.M.; Polisensky, D.H.; Antosiewicz, D.M.; Fry, S.C.; Braam, J. Arabidopsis TCH4, regulated by hormones and the environment, encodes a xyloglucan endotransglycosylase. Plant Cell 1995, 7, 1555–1567. [Google Scholar]
- Davière, J.M.; Achard, P. A pivotal role of DELLAs in regulating multiple hormone signals. Mol. Plant 2016, 9, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Richards, D.E.; Fleck, B.; Xie, D.; Burton, N.; Harberd, N.P. The Arabidopsis mutant sleepy1 gar2-1 protein promotes plant growth by increasing the affinity of the SCFSLY1 E3 ubiquitin ligase for DELLA protein substrates. Plant Cell 2004, 16, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Ueguchi-Tanaka, M.; Matsuoka, M. GID1-mediated gibberellin signaling in plants. Trends Plant Sci. 2008, 13, 192–199. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, K.M.; Thomas, S.G.; Soule, J.D.; Strader, L.C.; Zale, J.M.; Sun, T.P.; Steber, C.M. The Arabidopsis SLEEPY1 gene encodes a putative F-box subunit of an SCF E3 ubiquitin ligase. Plant Cell 2003, 15, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Itoh, H.; Gomi, K.; Ueguchi-Tanaka, M.; Ishiyama, K.; Kobayashi, M.; Jeong, D.; An, G.; Kitano, H.; Ashikari, M.; et al. Accumulation of phosphorylated repressor for gibberellin signaling in an F-box mutant. Science 2003, 299, 1896–1898. [Google Scholar] [CrossRef]
- Choi, H.; Oh, E. PIF4 integrates multiple environmental and hormonal signals for plant growth regulation in Arabidopsis. Mol. Cells 2016, 39, 587–593. [Google Scholar] [CrossRef]
- Dar, A.A.; Choudhury, A.R.; Kancharla, P.K.; Arumugam, N. The FAD2 gene in plants: Occurrence, regulation, and role. Front. Plant Sci. 2017, 8, 1789. [Google Scholar] [CrossRef]
- Chi, X.; Yang, Q.; Pan, L.; Chen, M.; He, Y.; Yang, Z.; Yu, S. Isolation and characterization of fatty acid desaturase genes from peanut (Arachis hypogaea L.). Plant Cell Rep. 2011, 30, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Murata, N.; Ishizaki-Nishizawa, O.; Higashi, S.; Hayashi, H.; Tasaka, Y.; Nishida, I. Genetically engineered alteration in the chilling sensitivity of plants. Nature 1992, 356, 710–713. [Google Scholar] [CrossRef]
- Kentaro, S.; Kirilov, C.N.; Sakae, T.; Imai, R. Identification of a novel LEA protein involved in freezing tolerance in wheat. Plant Cell Physiol. 2014, 55, 136–147. [Google Scholar]
- Wise, M.J. LEAping to conclusions: A computational reanalysis of late embryogenesis abundant proteins and their possible roles. BMC Bioinform. 2003, 4, 52–70. [Google Scholar] [CrossRef]
- Goyal, K.; Walton, L.J.; Tunnacliffe, A. LEA proteins prevent protein aggregation due to water stress. Biochem. J. 2005, 388, 151–157. [Google Scholar] [CrossRef]
- Gao, T.; Mo, Y.; Huang, H.; Yu, J.; Wang, Y.; Wang, W. Heterologous expression of Camellia sinensis late embryogenesis abundant protein gene 1 (CsLEA1) confers cold stress tolerance in Escherichia coli and yeast. Hortic. Plant J. 2021, 7, 8. [Google Scholar] [CrossRef]
- Hincha, D.K. Cryoprotectin: A plant lipid-transfer protein homologue that stabilizes membranes during freezing. Philos. Trans. R. Soc. Lond. 2002, 357, 909–916. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, W.; Fang, L.; Sun, X.; Su, L.; Liang, Z.; Wang, N.; Londo, J.P.; Li, S.; Xin, H. Genome-wide identification of WRKY family genes and their response to cold stress in Vitis vinifera. BMC Plant Biol. 2014, 14, 103. [Google Scholar] [CrossRef]
- Zhang, T.Q.; Zhao, Y.L.; Wang, Y.C.; Liu, Z.Y.; Gao, C.Q. Comprehensive analysis of MYB gene family and their expressions under abiotic stresses and hormone treatments in Tamarix hispida. Front. Plant Sci. 2018, 9, 1303. [Google Scholar] [CrossRef]
- Mondal, S.K.; Roy, S. Genome-wide sequential, evolutionary, organizational and expression analyses of phenylpropanoid biosynthesis associated MYB domain transcription factors in Arabidopsis. J. Biomol. Struct. Dyn. 2017, 36, 1577–1601. [Google Scholar] [CrossRef]
- Moyano, E.; Martínez-Garcia, J.F.; Martin, C. Apparent redundancy in MYB gene function provides gearing for the control of flavonoid biosynthesis in antirrhinum flowers. Plant Cell 1996, 8, 1519–1532. [Google Scholar] [PubMed]
- Yogendra, K.N.; Sarkar, K.; Kage, U.; Kushalappa, A.C. Potato NAC43 and MYB8 mediated transcriptional regulation of secondary cell wall biosynthesis to contain Phytophthora infestans infection. Plant Mol. Biol. Rep. 2017, 35, 519–533. [Google Scholar] [CrossRef]
- Wang, N.; Liu, W.; Zhang, T.; Jiang, S.; Xu, H.; Wang, Y.; Zhang, Z.; Wang, C.; Chen, X. Transcriptomic analysis of red-fleshed apples reveals the novel role of MdWRKY11 in flavonoid and anthocyanin biosynthesis. J. Agric. Food Chem. 2018, 66, 7076–7086. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, X.; Gong, Q.; Cao, J.; Shen, W.; Yin, X.; Grierson, D.; Zhang, B.; Xu, C.; Li, X.; et al. Three AP2/ERF family members modulate flavonoid synthesis by regulating type IV chalcone isomerase in citrus. Plant Biotechnol. J. 2020, 19, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, P.; Deswal, R. A novel class I chitinase from Hippophae rhamnoides: Indications for participating in ICE-CBF cold stress signaling pathway. Plant Sci. 2017, 259, 62–70. [Google Scholar] [CrossRef]
- Chen, L.; Zhong, H.; Feng, R.; Guo, Q.Q.; Hu, X.P.; Li, X.B. A novel cold-regulated gene, COR25, of Brassica napus is involved in plant response and tolerance to cold stress. Plant Cell Rep. 2011, 30, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Wu, L.H.; Wei, D.H.; Chen, H.; Zhou, M.Q.; Yao, X.H.; Lin, J. Promoter analysis of cold-responsive (COR) gene from Capsella bursa-pastoris and expression character in response to low temperature. Int. J. Agric. Biol. 2016, 18, 346–352. [Google Scholar] [CrossRef]
- Yi, Y.; Yu, X.; Ping, W. Comparison and evolution analysis of two rice subspecies LATERAL ORGAN BOUNDARIES domain gene family and their evolutionary characterization from Arabidopsis. Mol. Phylogenet. Evol. 2006, 39, 248–262. [Google Scholar]
- Chinnusamy, V.; Zhu, J.; Zhu, J.K. Gene regulation during cold acclimation in plants. Physiol. Plantarum 2010, 126, 52–61. [Google Scholar] [CrossRef]
- Li, T.; Yang, S.; Kang, X.; Lei, W.; Qiao, K.; Zhang, D.; Lin, H. The bHLH transcription factor gene AtUPB1 regulates growth by mediating cell cycle progression in Arabidopsis. Biochem. Biophys. Res. Commun. 2019, 518, 565–572. [Google Scholar] [CrossRef]
- Zhu, N.; Cheng, S.F.; Liu, X.; Du, H.; Dai, M.; Zhou, D.; Yang, W.; Zhao, Y. The R2R3-type MYB gene OsMYB91 has a function in coordinating plant growth and salt stress tolerance in rice. Plant Sci. 2015, 236, 146–156. [Google Scholar] [CrossRef]
- Qi, W.; Fan, S.; Wang, Q.; Chen, M.; Huang, Y.; Feng, Y.; Luo, X.; Yang, J. Rice ethylene-response AP2/ERF factor OsEATB restricts internode elongation by down-regulating a gibberellin biosynthetic gene. Plant Physiol. 2011, 157, 216–228. [Google Scholar] [CrossRef]
- Gocal, G.F.; Sheldon, C.C.; Gubler, F.; Moritz, T.; Bagnall, D.J.; MacMillan, C.P.; Li, S.F.; Parish, R.W.; Dennis, E.S.; Weigel, D.; et al. GAMYB-like genes, flowering, and gibberellin signaling in Arabidopsis. Plant Physiol. 2001, 127, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Varaud, E.; Brioudes, F.; Szécsi, J.; Leroux, J.; Brown, S.; Perrot-Rechenmann, C.; Bendahmanea, M. AUXIN RESPONSE FACTOR8 regulates Arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell 2011, 23, 973–983. [Google Scholar] [CrossRef]
- Van den Broeck, L.; Dubois, M.; Vermeersch, M.; Storme, V.; Matsui, M.; Inzé, D. From network to phenotype: The dynamic wiring of an Arabidopsis transcriptional network induced by osmotic stress. Mol. Syst. Biol. 2017, 13, 961. [Google Scholar] [CrossRef]
- Colebrook, E.H.; Thomas, S.G.; Phillips, A.L.; Hedden, P. The role of gibberellin signaling in plant responses to abiotic stress. J. Exp. Biol. 2014, 217, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kazan, K. Diverse roles of jasmonates and ethylene in abiotic stress tolerance. Trends Plant Sci. 2015, 20, 219–229. [Google Scholar] [CrossRef]
- Nolan, T.M.; Brennan, B.; Yang, M.R.; Chen, J.N.; Zhang, M.C.; Li, Z.H.; Wang, X.L.; Bassham, D.C.; Walley, J.; Yin, Y.H. Selective autophagy of BES1 mediated by DSK2 balances plant growth and survival. Dev. Cell 2017, 41, 33–46. [Google Scholar] [CrossRef]
- Wan, Y.; Schwaninger, H.; Li, D.; Simon, C.; Wang, Y.; He, P. The eco-geographic distribution of wild grape germplasm in China. VITIS 2008, 47, 77. [Google Scholar]
- Xu, W.; Li, R.; Zhang, N.; Ma, F.; Jiao, Y.; Wang, Z. Transcriptome profiling of Vitis amurensis, an extremely cold-tolerant Chinese wild Vitis species, reveals candidate genes and events that potentially connected to cold stress. Plant Mol. Biol. 2014, 86, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.G.; Hayashi, T.; Yazawa, S.; Katoh, T.; Yasuda, Y. Acute morphological changes of palisade cells of Saintpaulia leaves induced by a rapid temperature drop. J. Plant Res. 1996, 109, 339–342. [Google Scholar] [CrossRef]
- Kratsch, H.; Wise, R. The ultrastructure of chilling stress. Plant Cell Environ. 2000, 23, 337–350. [Google Scholar] [CrossRef]
- Zhuang, Q.; Chen, S.; Jua, Z.; Yao, Y. Joint transcriptomic and metabolomic analysis reveals the mechanism of low temperature tolerance in Hosta ventricosa. PLoS ONE 2021, 16, e0259455. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, T.; Rao, P.; Gao, K.; Yang, X.; Chen, Z.; An, X. Transcriptome profiling of Populus tomentosa under cold stress. Ind. Crop. Prod. 2019, 135, 283–293. [Google Scholar] [CrossRef]
- Lu, C.; Yin, L.; Li, K. Proteome expression patterns in the stress tolerant evergreen Ammopiptanthus nanus under conditions of extreme cold. Plant Growth Regul. 2010, 62, 65–70. [Google Scholar] [CrossRef]
- Kaplan, F.; Kopka, J.; Haskell, D.W.; Zhao, W.; Schiller, K.C.; Gatzke, N.; Sung, D.Y.; Guy, C.L. Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiol. 2004, 136, 4159–4168. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, H.; Zhang, J.; Liu, P.; Chen, X.; Li, Z.; Xu, Y.; Lu, P.; Cao, P. Integrated transcriptomics and metabolomics analysis to characterize cold stress responses in Nicotiana tabacum. BMC Genom. 2017, 18, 496. [Google Scholar] [CrossRef]
- Guy, C.; Kaplan, F.; Kopka, J.; Selbig, J.; Hincha, D.K. Metabolomics of temperature stress. Physiol. Plant. 2008, 132, 220–235. [Google Scholar] [CrossRef]
- Peng, Y.; Lin, W.L.; Wei, H.; Krebs, S.L.; Arora, R. Phylogenetic analysis and seasonal cold acclimation-associated expression of early light-induced protein genes of Rhododendron catawbiense. Physiol. Plant. 2008, 132, 44–52. [Google Scholar] [CrossRef]
- Gage, K.; Jens, R.; Wilson, R.C.; Kopka, J.; Erban, A.; Winge, P.; Bones, A.M.; Davik, J.; Alsheikh, M.K.; Randall, S.K. Integrative “omic” analysis reveals distinctive cold responses in leaves and roots of strawberry, Fragaria × ananassa ‘Korona’. Fron. Plant Sci. 2015, 6, 826. [Google Scholar]
- Maruyama, K.; Urano, K.; Yoshiwara, K.; Morishita, Y.; Sakurai, N.; Suzuki, H.; Kojima, M.; Sakakibara, H.; Shibata, D.; Saito, K.; et al. Integrated analysis of the effects of cold and dehydration on rice metabolites, phytohormones, and gene transcripts. Plant Physiol. 2014, 164, 1759–1771. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Time | Sample 1 | Raw Reads (M) | Clean reads (M) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|
| Late autumn (November 28) | LA_1 | 53.01 | 52.70 | 95.99 | 44.38 |
| LA_2 | 54.49 | 54.16 | 96.07 | 44.28 | |
| LA_3 | 54.12 | 53.79 | 96.06 | 44.27 | |
| Winter (December 28) | W_1 | 52.23 | 51.90 | 95.69 | 44.74 |
| W_2 | 47.81 | 47.50 | 95.36 | 44.20 | |
| W_3 | 48.82 | 48.49 | 95.60 | 44.26 | |
| Early spring (February 15) | ES_1 | 50.88 | 50.53 | 95.60 | 44.19 |
| ES_2 | 49.66 | 49.30 | 95.81 | 44.33 | |
| ES_3 | 49.25 | 48.92 | 96.02 | 44.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Hu, H.; Yang, J.; Xue, J.; Xu, J. Physiological, Transcriptomic and Metabolomic Analyses of Overwintering Cryptomeria fortunei Needles. Forests 2022, 13, 1249. https://doi.org/10.3390/f13081249
Zhang Y, Hu H, Yang J, Xue J, Xu J. Physiological, Transcriptomic and Metabolomic Analyses of Overwintering Cryptomeria fortunei Needles. Forests. 2022; 13(8):1249. https://doi.org/10.3390/f13081249
Chicago/Turabian StyleZhang, Yingting, Hailiang Hu, Junjie Yang, Jinyu Xue, and Jin Xu. 2022. "Physiological, Transcriptomic and Metabolomic Analyses of Overwintering Cryptomeria fortunei Needles" Forests 13, no. 8: 1249. https://doi.org/10.3390/f13081249
APA StyleZhang, Y., Hu, H., Yang, J., Xue, J., & Xu, J. (2022). Physiological, Transcriptomic and Metabolomic Analyses of Overwintering Cryptomeria fortunei Needles. Forests, 13(8), 1249. https://doi.org/10.3390/f13081249