
Identification, Evolution and Expression Analysis of GRF Family Reveals Their Involvement in Shoot Growth and Abiotic Stress Response in Moso Bamboo



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
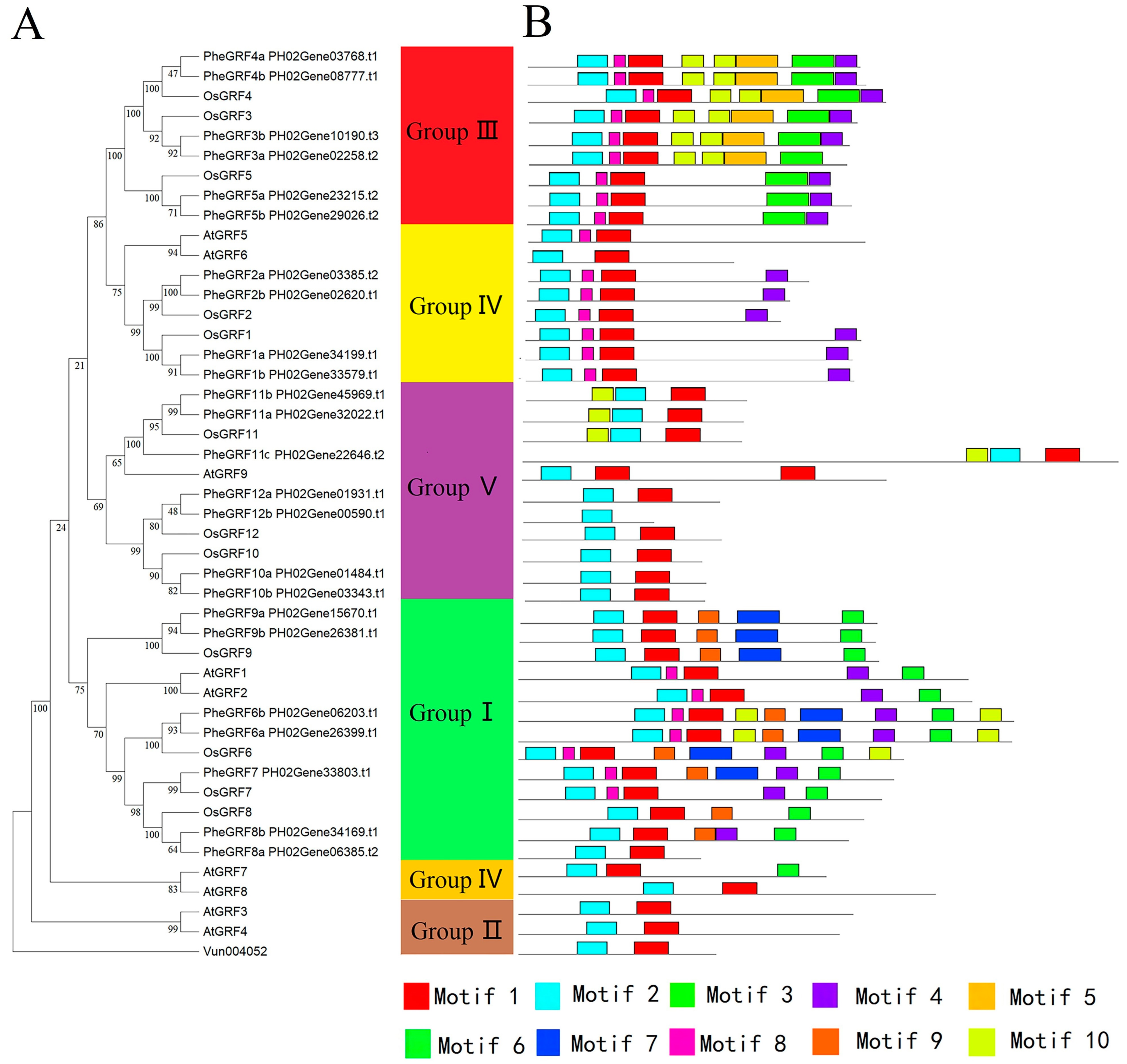
2.1. Identification and Classification of the PheGRF Gene Family
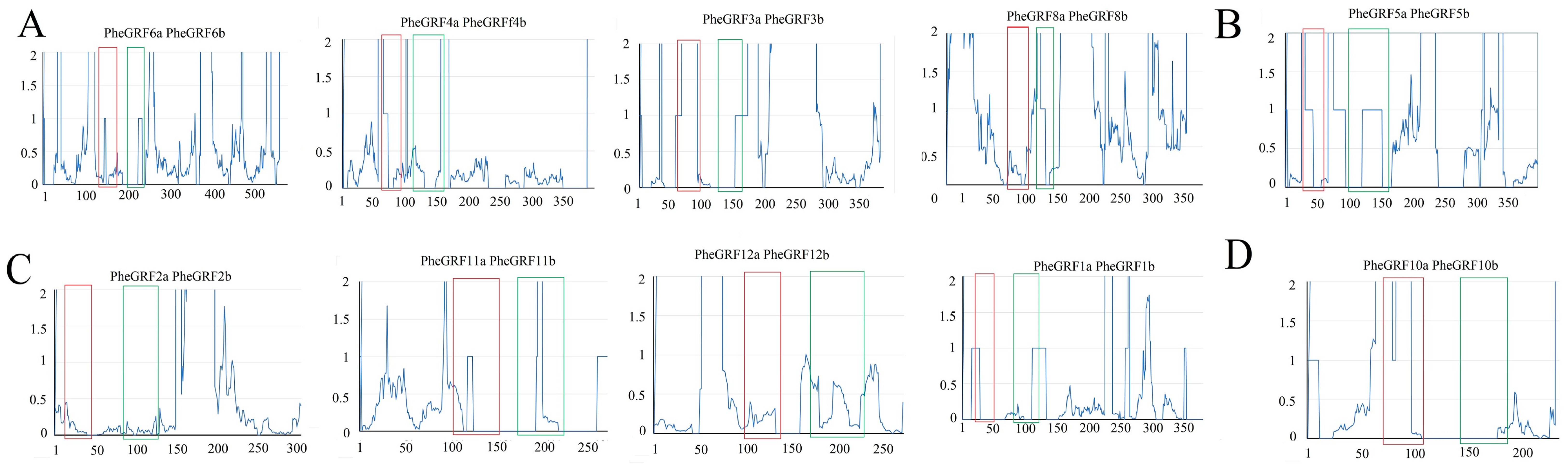
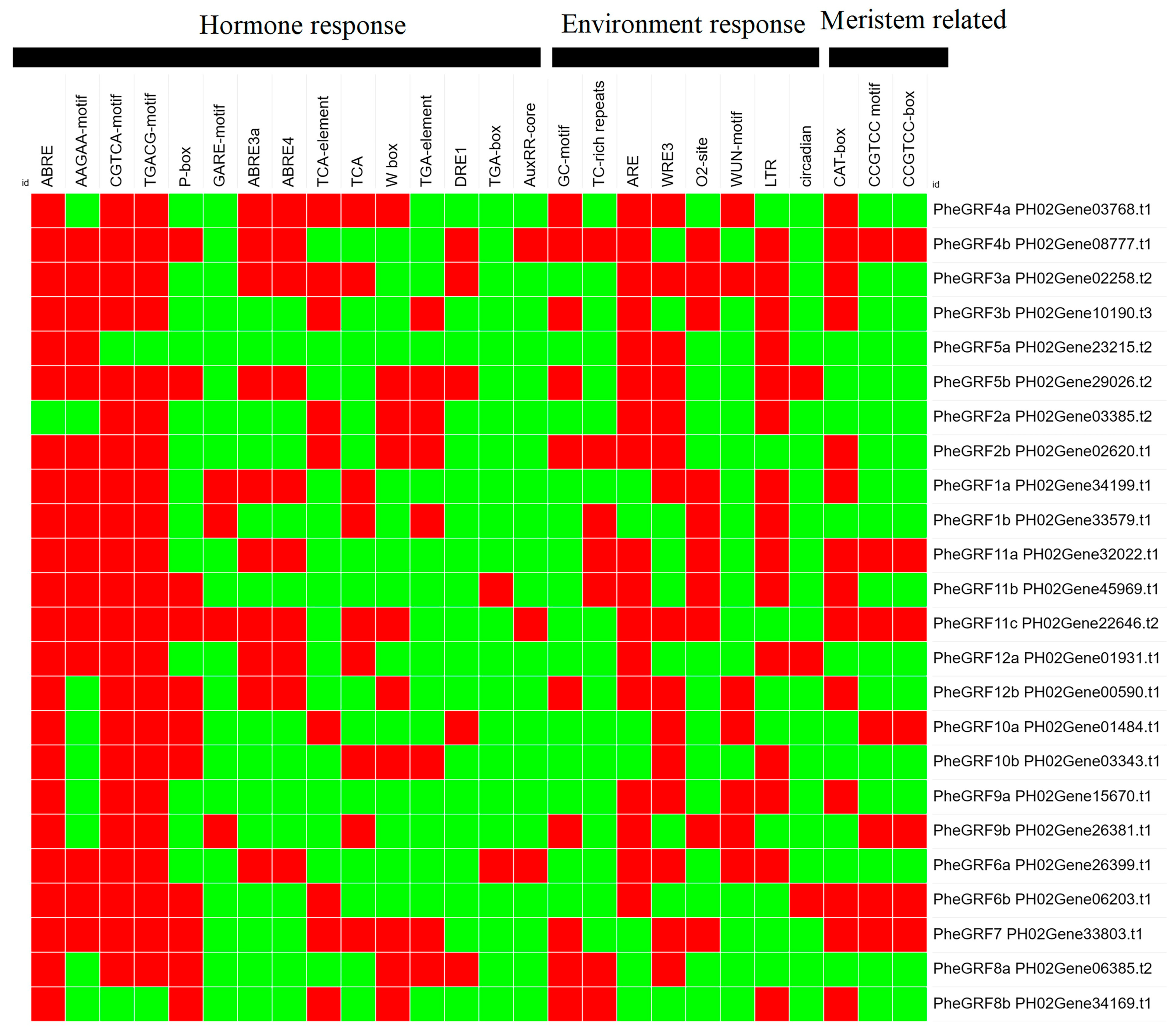
2.2. Characterization of the PheGRF Gene Family
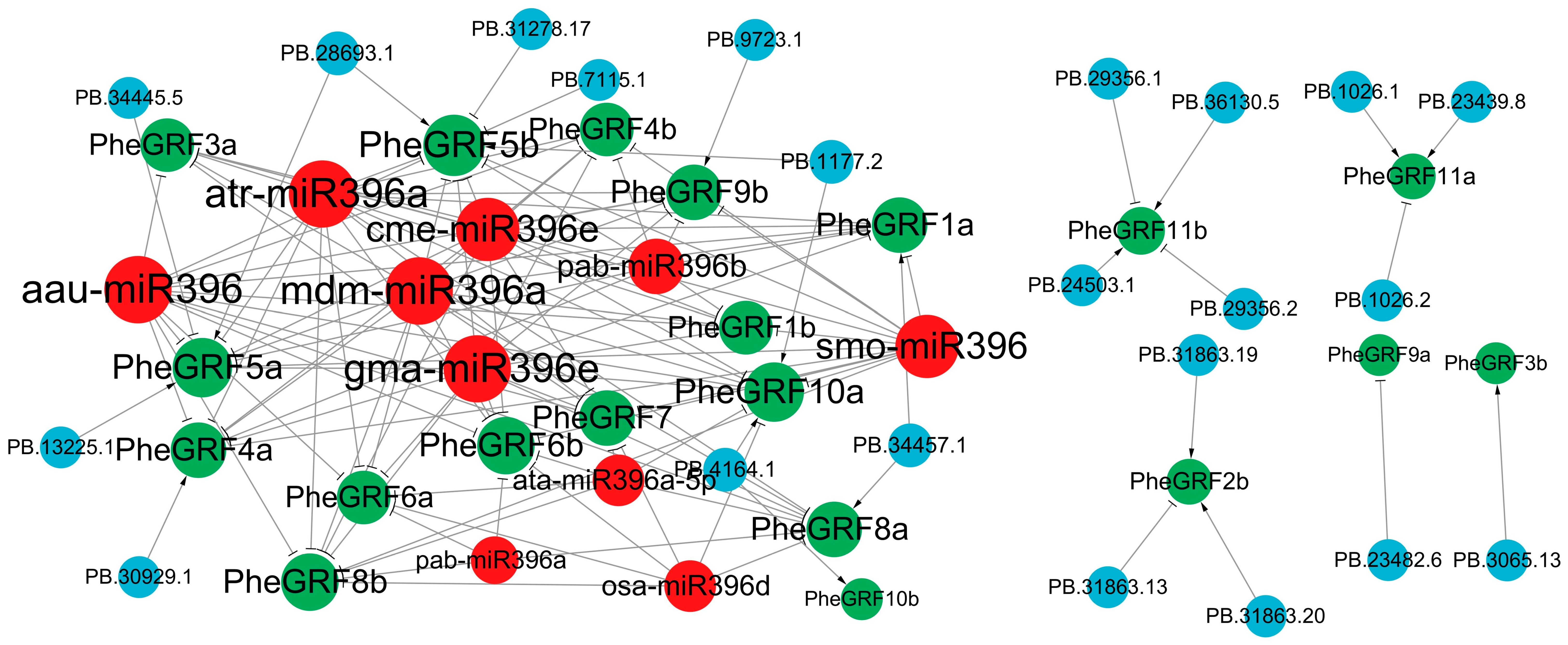
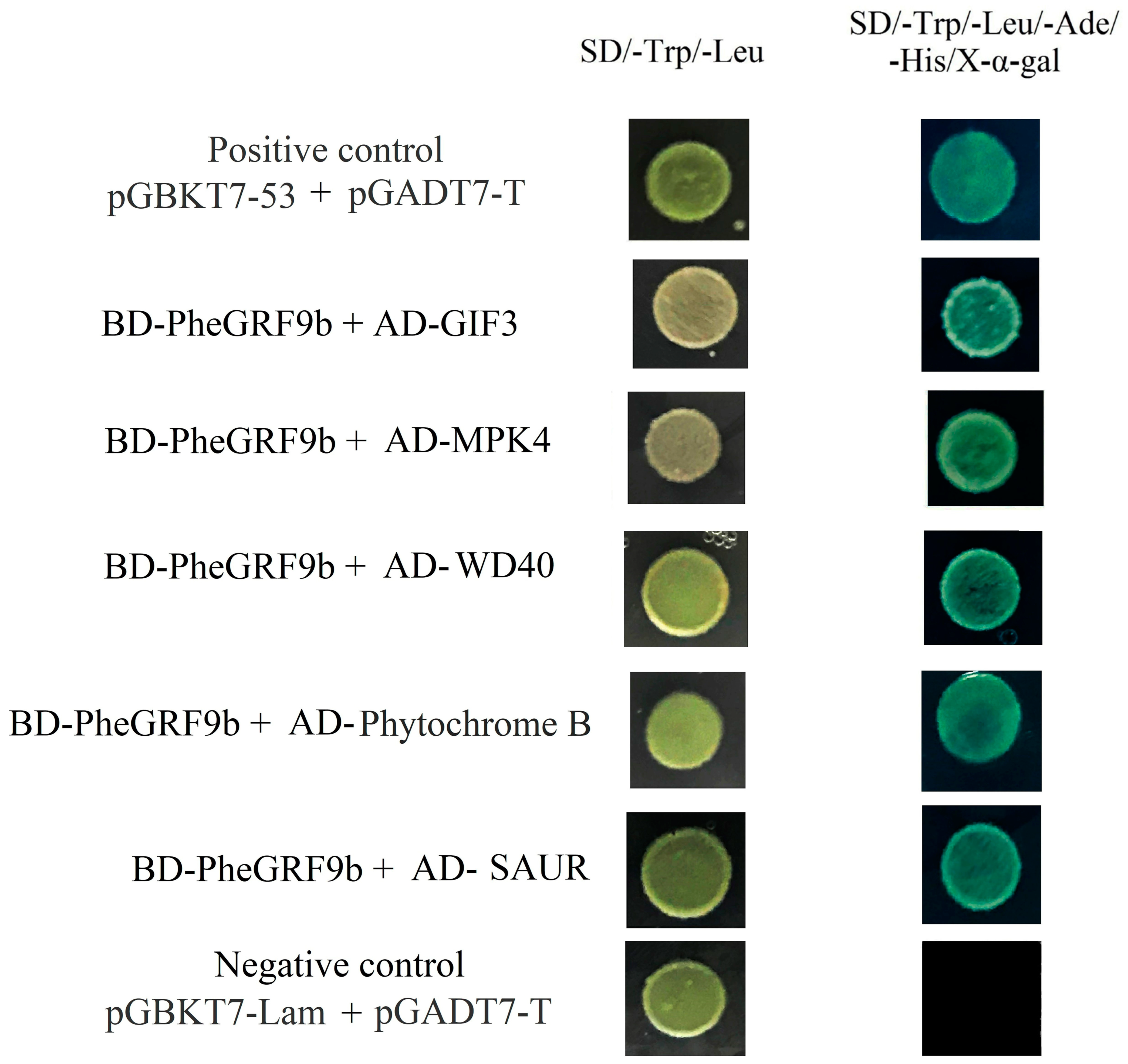
2.3. Regulatory Network of PheGRF in Moso Bamboo
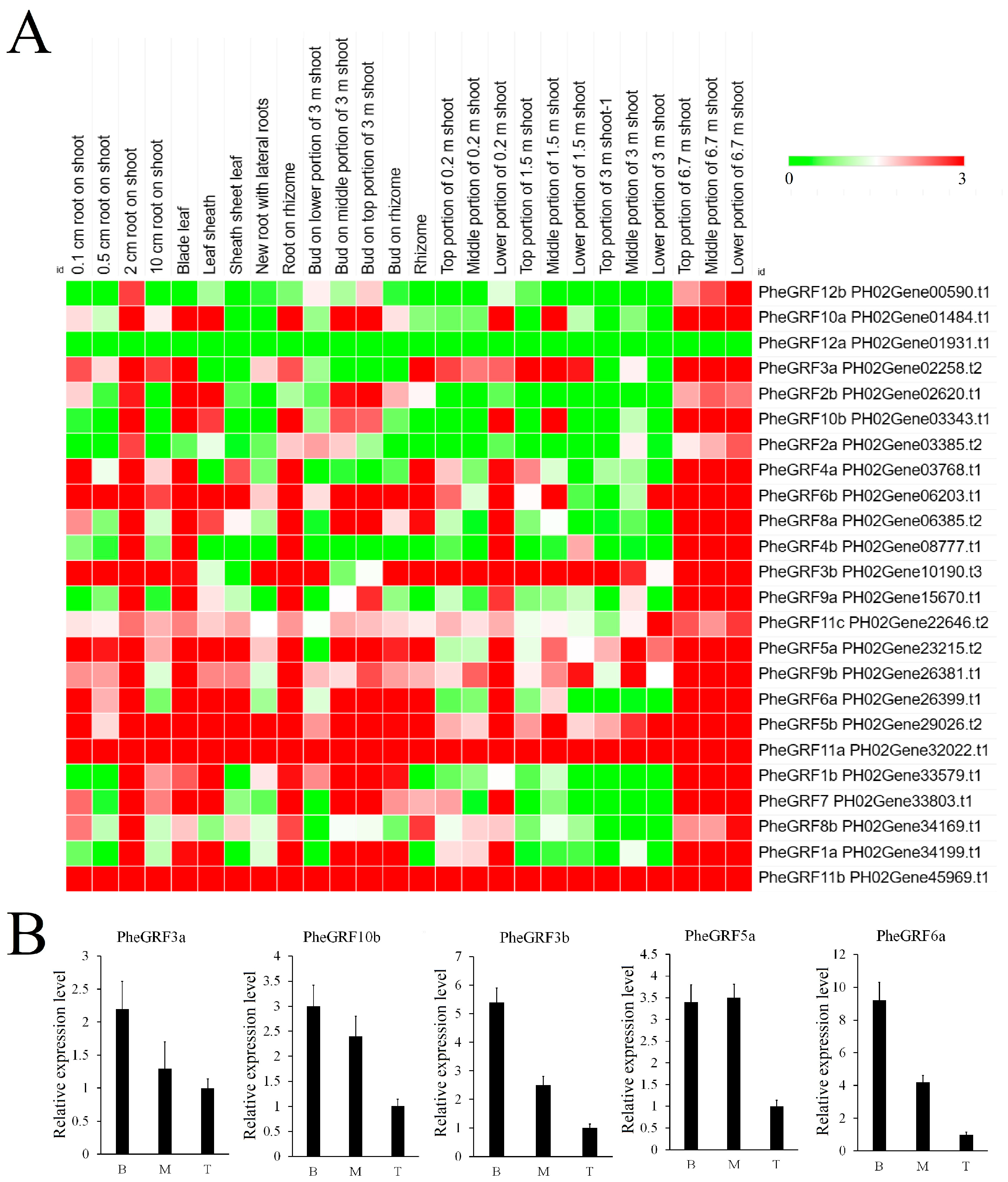
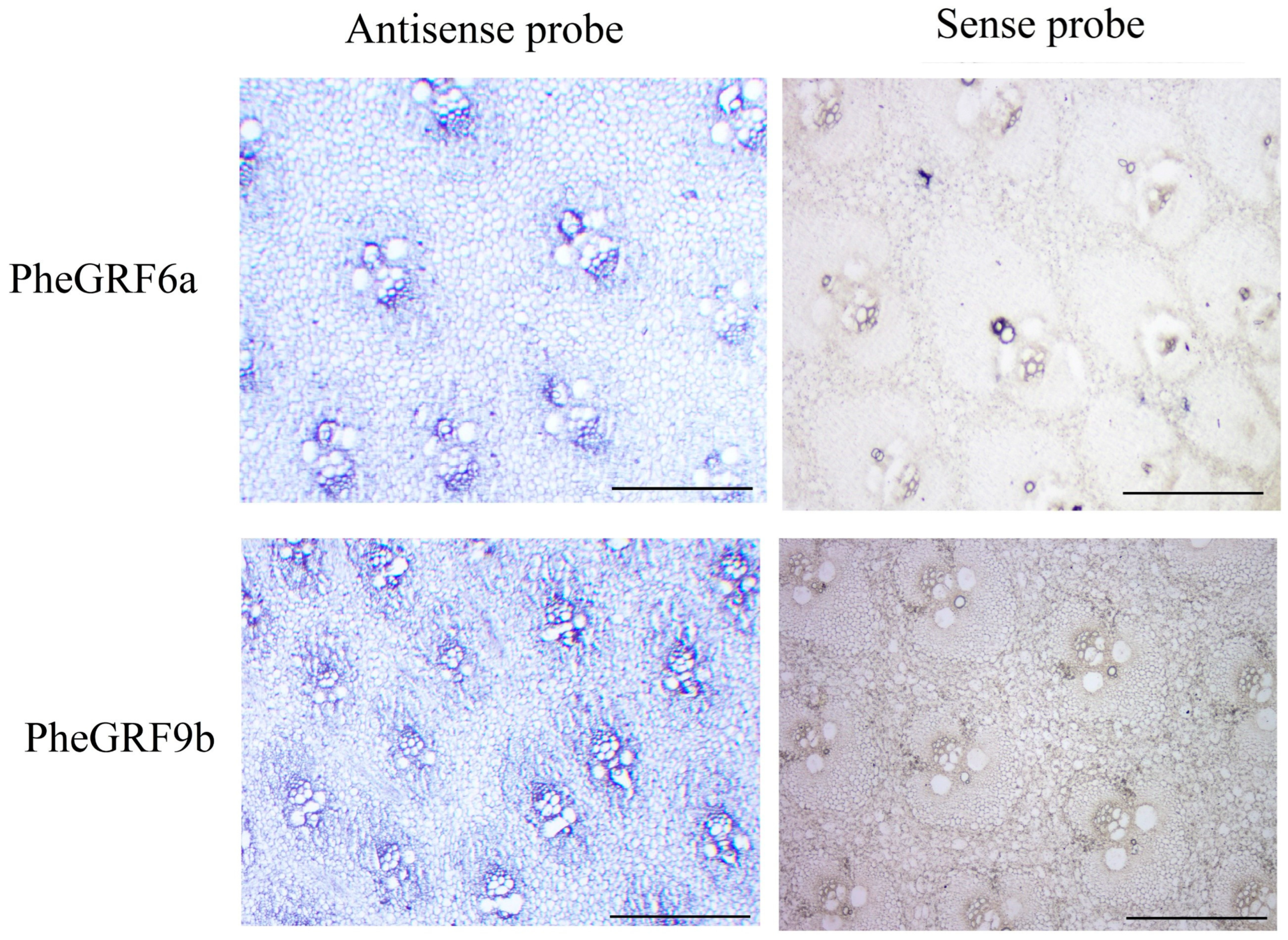
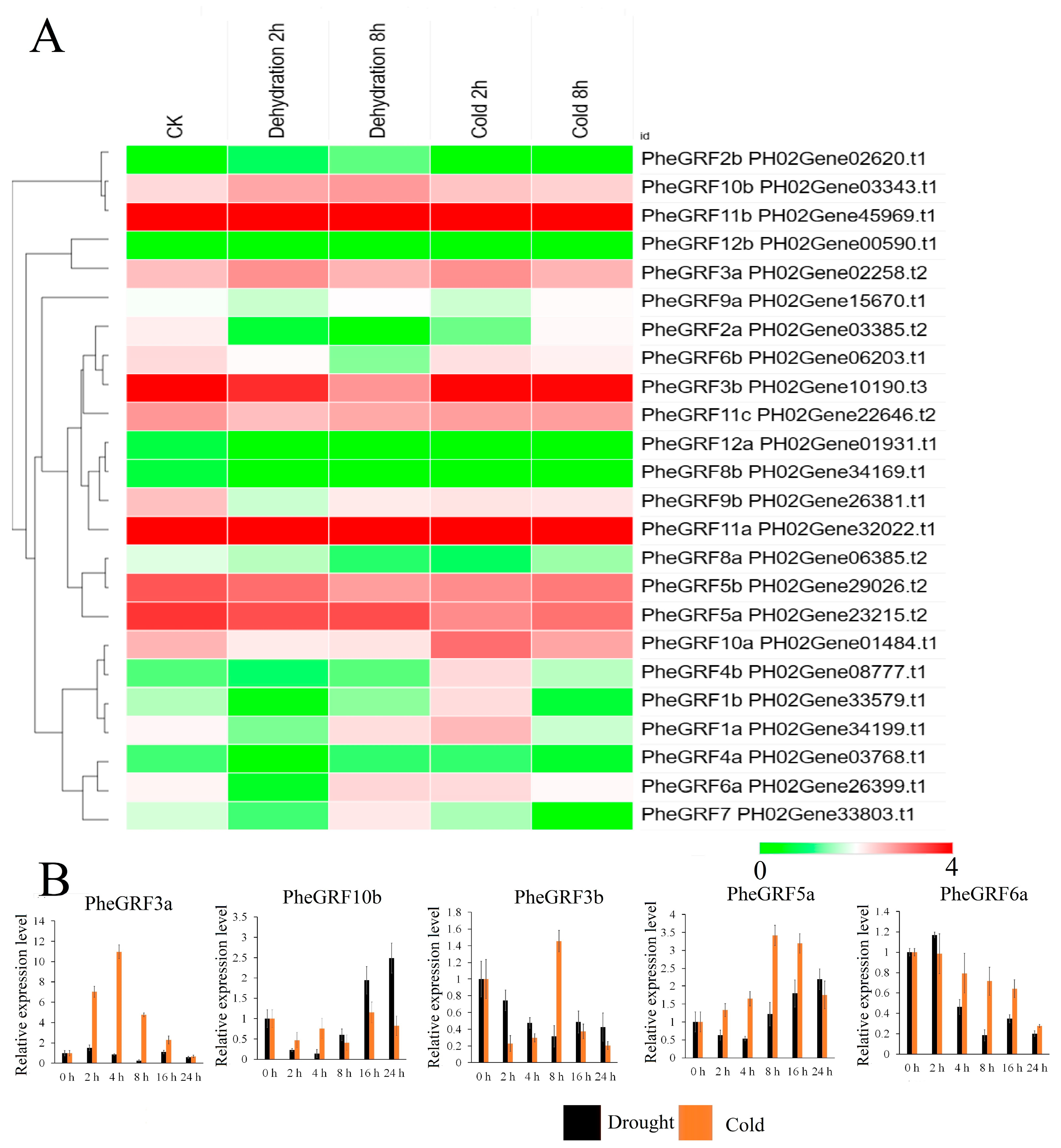
2.4. Expression Profile of PheGRF Genes
3. Discussion
4. Methods
4.1. Identification of GRF Genes in Moso Bamboo Reference Genome
4.2. Multiple Sequence Alignment and Phylogenetic Analyses
4.3. Gene Structure, Motif, and Promoter Analysis
4.4. Collinearity Analysis
4.5. Estimation of Ka, Ks, and Divergence Time in Paralogous Pairs
4.6. In Situ Hybridization
4.7. Yeast Two-Hybrid Assays
4.8. Transcriptome Data Analysis
4.9. qRT-PCR Analysis
4.10. lncRNA Identification
4.11. Degradome Sequencing and Target Gene of miRNA Identification
4.12. Regulation Network Construction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lobovikov, M.; Paudel, S.; Piazza, M.; Ren, H.; Wu, J. World Bamboo Resources: A Thematic Study Prepared in the Framework of the Global Forest Resources Assessment 2005; Non-Wood Forest Products (FAO): Rome, Italy, 2007. [Google Scholar]
- Li, L.; Cheng, Z.C.; Ma, Y.J.; Bai, Q.S.; Li, X.Y.; Cao, Z.H.; Wu, Z.N.; Gao, J. The association of hormone signaling, transcription and anatomy during shoot growth in Moso bamboo. Plant Biotechnol. J. 2018, 16, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, P.; Wang, J.; Xu, Z.Y. Growth-regulating factors: Conserved and divergent roles in plant growth and development and potential value for crop improvement. Plant J. 2023, 113, 1122–1145. [Google Scholar] [CrossRef] [PubMed]
- Fonini, L.S.; Lazzarotto, F.; Barros, P.M.; Cabreira-Cagliari, C.; Martins, M.A.; Saibo, N.J.; Turchetto-Zolet, A.C.; Margis-Pinheiro, M. Molecular evolution and diversification of the GRF transcription factor family. Genet Mol. Biol. 2020, 43, 20200080. [Google Scholar] [CrossRef] [PubMed]
- Bull, T.; Debernardi, J.; Reeves, M.; Hill, T.; Bertier, L.; Van Deynze, A.; Michelmore, R. GRF-GIF chimeric proteins enhance in vitro regeneration and Agrobacterium-mediated transformation efficiencies of lettuce (Lactuca spp.). Plant Cell Rep. 2023, 42, 629–643. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, E.; Kim, J.H.; Kende, H. A novel gibberellin-induced gene from rice and its potential regulatory role in stem growth. Plant Physiol. 2000, 122, 695–704. [Google Scholar] [CrossRef]
- Vercruysse, J.; Baekelandt, A.; Gonzalez, N.; Inzé, D. Molecular networks regulating cell division during Arabidopsis leaf growth. J. Exp. Bot. 2020, 71, 2365–2378. [Google Scholar] [CrossRef]
- Wu, W.; Li, J.; Wang, Q.; Lv, K.; Du, K.; Zhang, W.; Li, Q.; Kang, X.; Wei, H. Growth-regulating factor 5 (GRF5)-mediated gene regulatory network promotes leaf growth and expansion in poplar. New Phytol. 2021, 230, 612–628. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, T.; Yi, F.; Yu, J. SimiR396d targets SiGRF1 to regulate drought tolerance and root growth in foxtail millet. Plant Sci. 2023, 326, 111492. [Google Scholar] [CrossRef]
- Wang, P.; Xiao, Y.; Yan, M.; Yan, Y.; Lei, X.; Di, P.; Wang, Y. Whole-genome identification and expression profiling of growth-regulating factor (GRF) and GRF-interacting factor (GIF) gene families in Panax ginseng. BMC Genom. 2023, 24, 334. [Google Scholar] [CrossRef]
- Shahan, R. The Cold Never Bothered Me Anyway: DELLA-Interacting GROWTH REGULATING FACTORS Mediate Plant Growth in Cold Stress. Plant C. 2020, 32, 797–798. [Google Scholar] [CrossRef]
- Hu, Q.; Jiang, B.; Wang, L.; Song, Y.; Tang, X.; Zhao, Y.; Fan, X.; Gu, Y.; Zheng, Q.; Cheng, J.; et al. Genome-wide analysis of growth-regulating factor genes in grape (Vitis vinifera L.): Identification, characterization and their responsive expression to osmotic stress. Plant Cell Rep. 2023, 42, 107–121. [Google Scholar] [CrossRef]
- Li, A.L.; Wen, Z.; Yang, K.; Wen, X.P. Conserved miR396b-GRF regulation is involved in abiotic stress responses in Pitaya (Hylocereus polyrhizus). J. Mol. Sci. 2019, 20, 2501. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, R.; Rodriguez, R.E.; Debernardi, J.M.; Palatnik, J.F.; Casati, P. Repression of Growth Regulating Factors by the MicroRNA396 inhibits cell proliferation by UV-B radiation in Arabidopsis leaves. Plant C. 2013, 25, 3570–3583. [Google Scholar] [CrossRef] [PubMed]
- Piya, S.; Liu, J.; Burch-Smith, T.; Baum, T.J.; Hewezi, T. A role for Arabidopsis growth-regulating factors 1 and 3 in growth-stress antagonism. J. Exp. Bot. 2020, 71, 1402–1417. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Mizoi, J.; Kidokoro, S.; Maruyama, K.; Nakajima, J.; Nakashima, K.; Mitsuda, N.; Takiguchi, Y.; Ohme-Takagi, M.; Kondou, Y.; et al. Arabidopsis growth-regulating factor7 functions as a transcriptional repressor of abscisic acid- and osmotic stress-responsive genes, including DREB2A. Plant C. 2012, 24, 3393–3405. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Choi, D.; Kende, H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 2010, 36, 94–104. [Google Scholar] [CrossRef]
- Choi, D.; Kim, J.H.; Kende, H. (Whole genome analysis of the OsGRF gene family encoding plant-specific putative transcription activators in rice (Oryza sativa L.). Plant Cell Physiol. 2004, 45, 897–904. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, X.; Fu, D.; Zeng, Q.; Gao, X.; Zhang, N.; Wu, J. Genome-wide characterization and expression analysis of the growth-regulating factor family in Saccharum. BMC Plant Biol. 2022, 22, 510. [Google Scholar] [CrossRef]
- Li, G.; Chen, Y.; Zhao, X.; Yang, J.; Wang, X.; Li, X.; Hu, S.; Hou, H. Genome-wide analysis of the Growth-Regulating Factor (GRF) family in aquatic plants and their Rroles in the ABA-induced turion formation of Spirodela polyrhiza. Int. J. Mol. Sci. 2022, 23, 10485. [Google Scholar] [CrossRef]
- Zan, T.; Zhang, L.; Xie, T.; Li, L. Genome-wide identification and analysis of the Growth-Regulating Factor (GRF) gene family and GRF-interacting factor family in Triticum aestivum L. Biochem. Genet. 2020, 58, 705–724. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, J.; Li, J.; Li, Y.; Zeng, X. Genome-wide identification and analysis of the Growth-Regulating Factor family in Zanthoxylum armatum DC and functional analysis of ZaGRF6 in leaf size and longevity regulation. Int. J. Mol. Sci. 2022, 23, 9043. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tong, Y.; Luo, K.; Zhai, Z.; Liu, X.; Shi, Z.; Zhang, D.; Li, D. Identification of GROWTH-REGULATING FACTOR transcription factors in lettuce (Lactuca sativa) genome and functional analysis of LsaGRF5 in leaf size regulation. BMC Plant Biology. 2022, 21, 485. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qiu, T.T.; Zhou, Z.S.; Kang, L.Q.; Chen, R.R.; Zeng, L.M.; Yu, H.Y.; Wang, Y.H.; Song, J.B. Genome-wide analysis of the Growth-Regulating Factor family in Medicago truncatula. J. Plant Growth Regul. 2022, 23, 6905. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, H.; Gao, Y.; Wang, Y.; Wu, M.; Xiang, Y. Genome-wide identification of growth-regulating factors in moso bamboo (Phyllostachys edulis): In silico and experimental analyses. PeerJ. 2019, 12, e7510. [Google Scholar] [CrossRef]
- Peng, Z.H.; Lu, Y.; Li, L.B.; Zhao, Q.; Feng, Q. The draft genome of the fast-growing non-timber forest species Moso bamboo (Phyllostachys heterocycla). Nat. Gene 2013, 45, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Gao, Z.; Wang, L.; Wang, J.; Wang, S.; Fei, B.; Chen, C.; Shi, C.; Liu, X.; Zhang, H.; et al. Chromosome-level reference genome and alternative splicing atlas of moso bamboo (Phyllostachys edulis). Gigascience 2018, 7, giy115. [Google Scholar] [CrossRef]
- Li, L.; Shi, Q.; Jia, Y.; Deng, P.; Gao, J. Transcriptome and anatomical comparisons reveal the specific characteristics and genes involved in distinct types of growing culms. Ind. Crops Prod. 2021, 171, 113865. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, D.; Zhang, S.; Lou, Y.; An, X.; Jiang, Z.; Gao, Z. Transcriptome and miRNAome analysis reveals components regulating tissue differentiation of bamboo shoots. Plant Physiol. 2022, 188, 2182–2198. [Google Scholar] [CrossRef]
- Qin, L.; Chen, H.; Wu, Q.; Wang, X. Identification and exploration of the GRF and GIF families in maize and foxtail millet. Physiol. Mol. Biol. Plants. 2022, 28, 1717–1735. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, M.; Wei, X.; Wang, R.; Fan, F.; Wang, Z.; Tian, Z.; Li, S.; Yuan, H. Comprehensive evolutionary analysis of growth-regulating factor gene family revealing the potential molecular basis under multiple hormonal stress in Gramineae crops. Front. Plant Sci. 2023, 14, 1174955. [Google Scholar] [CrossRef]
- Duvick, J.; Fu, A.; Muppirala, U.; Sabharval, M.; Wilkerson, M.D.; Lawrence, C.J.; Lushbough, C.; Brendel, V. PlantGDB: A resource for comparative plant genomics. Nucl. Acids Res. 2008, 36, D959. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing Consortium. A chromosomebased draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef] [PubMed]
- Burr, B. Mapping and sequencing the rice genome. Plant C. 2002, 14, 521–523. [Google Scholar] [CrossRef]
- The International Brachypodium Initiative. Genome sequence analysis of the model grass Brachypodium distachyon: Insights into grass genome evolution. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010, 20, 45–58. [Google Scholar] [CrossRef]
- Mallik, S.; Tawfik, D.S.; Levy, E.D. How gene duplication diversifies the landscape of protein oligomeric state and function. Curr. Opin. Genet. Dev. 2022, 76, 101966. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef]
- Guo, Z.H.; Ma, P.F.; Yang, G.Q.; Hu, J.Y.; Liu, Y.L.; Xia, E.H.; Zhong, M.C.; Zhao, L.; Sun, G.L.; Xu, Y.X. Genome Sequences Provide Insights into the Reticulate Origin and Unique Traits of Woody Bamboos. Mol. Plant 2019, 12, 1353–1365. [Google Scholar] [CrossRef]
- Zhao, Y.; Su, X.; Wang, X.; Wang, M.; Cai, M. Comparative genomic analysis of TCP genes in six Rosaceae species and expression pattern analysis in Pyrus bretschneideri. Front. Genet. 2021, 12, 669959. [Google Scholar] [CrossRef]
- Liebsch, D.; Palatnik, J.F. MicroRNA miR396, GRF transcription factors and GIF co-regulators: A conserved plant growth regulatory module with potential for breeding and biotechnology. Curr. Opin. Plant Biol. 2020, 53, 31–42. [Google Scholar] [CrossRef]
- Cai, M.; Cheng, W.; Bai, Y.; Mu, C.; Zheng, H.; Cheng, Z.; Gao, J. PheGRF4e initiated auxin signaling during moso bamboo shoot development. Mol. Biol. Rep. 2022, 49, 8815–8825. [Google Scholar] [CrossRef] [PubMed]
- Lantzouni, O.; Alkofer, A.; Falter-Braun, P.; Schwechheimer, C. GROWTH-REGULATING FACTORS Interact with DELLAs and Regulate Growth in Cold Stress. Plant C. 2020, 32, 1018–1034. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Guo, L.; Jiao, C.; Fei, Z.; Chen, M.; Cao, J.; Ding, Y.; Yuan, Q. Characterization of the developmental dynamics of the elongation of a bamboo internode during the fast growth stage. Tree Physiol. 2019, 39, 1201–1214. [Google Scholar] [CrossRef]
- Chen, M.; Guo, L.; Ramakrishnan, M.; Fei, Z.; Vinod, K.K.; Ding, Y.; Jiao, C.; Gao, Z.; Zha, R.; Wang, C.; et al. Rapid growth of Moso bamboo (Phyllostachys edulis): Cellular roadmaps, transcriptome dynamics, and environmental factors. Plant C. 2022, 34, 3577–3610. [Google Scholar] [CrossRef] [PubMed]
- Chandran, V.; Wang, H.; Gao, F.; Cao, X.L.; Chen, Y.P.; Li, G.B.; Zhu, Y.; Yang, X.M.; Zhang, L.L.; Zhao, Z.X.; et al. miR396-OsGRFs module balances growth and rice blast disease-resistance. Front. Plant Sci. 2019, 9, 1999. [Google Scholar] [CrossRef]
- Chen, Y.; Dan, Z.; Gao, F.; Chen, P.; Fan, F.; Li, S. Rice GROWTH-REGULATING FACTOR7 Modulates Plant Architecture through Regulating GA and Indole-3-Acetic Acid Metabolism. Plant Physiol. 2020, 184, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Xia, H.; Yang, L.; Zhai, X.; Shen, Y.; Li, H. The Roles of microRNA-Long Non-coding RNA-mRNA networks in the regulation of leaf and flower development in Liriodendron chinense. Front. Plant Sci. 2022, 13, 816875. [Google Scholar] [CrossRef]
- Moison, M.; Pacheco, J.M.; Lucero, L.; Fonouni-Farde, C.; Rodríguez-Melo, J.; Mansilla, N.; Christ, A.; Bazin, J.; Benhamed, M.; Ibañez, F.; et al. The lncRNA APOLO interacts with the transcription factor WRKY42 to trigger root hair cell expansion in response to cold. Mol. Plant. 2021, 14, 937–948. [Google Scholar] [CrossRef]
- Lu, Y.; Zeng, J.; Liu, Q. The Rice miR396-GRF-GIF-SWI/SNF Module: A Player in GA Signaling. Front Plant Sci. 2022, 12, 786641. [Google Scholar] [CrossRef]
- Park, E.; Kim, Y.; Choi, G. Phytochrome B Requires PIF Degradation and Sequestration to Induce Light Responses across a Wide Range of Light Conditions. Plant C. 2018, 30, 1277–1292. [Google Scholar] [CrossRef]
- Yan, C.; Yang, T.; Wang, B.; Yang, H.; Wang, J.; Yu, Q. Genome-Wide Identification of the WD40 Gene Family in Tomato (Solanum lycopersicum L.). Genes 2023, 15, 1273. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Campbell, K.; Gifford, R.J.; Singer, J.; Hill, V.; O’Toole, A.; Rambaut, A.; Hampson, K.; Brunker, K. Making genomic surveillance deliver: A lineage classification and nomenclature system to inform rabies elimination. PLoS Pathog. 2022, 2, e1010023. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Timothy, L.; Bailey James, J.; Charles, E.G.; William, S.N. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Liang, H.; Zhou, W.; Landweber, L.F. SWAKK: A web server for detecting positive selection in proteins using a sliding window substitution rate analysis. Nucleic Acids Res. 2016, 34, W382–W384. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Huang, J.L.; Yang, Y.P.; Hu, X.Y. Analyses of the oligopeptide transporter gene family in poplar and grape. BMC Genom. 2011, 12, 465. [Google Scholar] [CrossRef]
- Huang, Z.; Zhong, X.J.; He, J.; Jin, S.H.; Guo, H.D.; Yu, X.F.; Zhou, Y.J.; Li, X.; Ma, M.D.; Chen, Q.B.; et al. Genome-wide identification, characterization, and stress-responsive expression profiling of genes encoding LEA (Late Embryogenesis Abundant) proteins in Moso bamboo (Phyllostachys edulis). PLoS ONE 2016, 11, e0165953. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Zhang, Y. miRU: An automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33, W701–W704. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, B.; Long, C.; Yao, W.; Lin, S.; Li, L. Identification, Evolution and Expression Analysis of GRF Family Reveals Their Involvement in Shoot Growth and Abiotic Stress Response in Moso Bamboo. Forests 2023, 14, 2044. https://doi.org/10.3390/f14102044
Zhou B, Long C, Yao W, Lin S, Li L. Identification, Evolution and Expression Analysis of GRF Family Reveals Their Involvement in Shoot Growth and Abiotic Stress Response in Moso Bamboo. Forests. 2023; 14(10):2044. https://doi.org/10.3390/f14102044
Chicago/Turabian StyleZhou, Binao, Cheng Long, Wenjing Yao, Shuyan Lin, and Long Li. 2023. "Identification, Evolution and Expression Analysis of GRF Family Reveals Their Involvement in Shoot Growth and Abiotic Stress Response in Moso Bamboo" Forests 14, no. 10: 2044. https://doi.org/10.3390/f14102044
APA StyleZhou, B., Long, C., Yao, W., Lin, S., & Li, L. (2023). Identification, Evolution and Expression Analysis of GRF Family Reveals Their Involvement in Shoot Growth and Abiotic Stress Response in Moso Bamboo. Forests, 14(10), 2044. https://doi.org/10.3390/f14102044