
Comprehensive Analysis of Chloroplast Genome of Hibiscus sinosyriacus: Evolutionary Studies in Related Species and Genera

Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction, Sequencing, Assembly, and Annotation
2.2. Comparative Analyses of cp Genome Sequences
2.3. Simple Sequence Repeats (SSRs) Analysis
2.4. Detection of Variants and Statistical Analyses
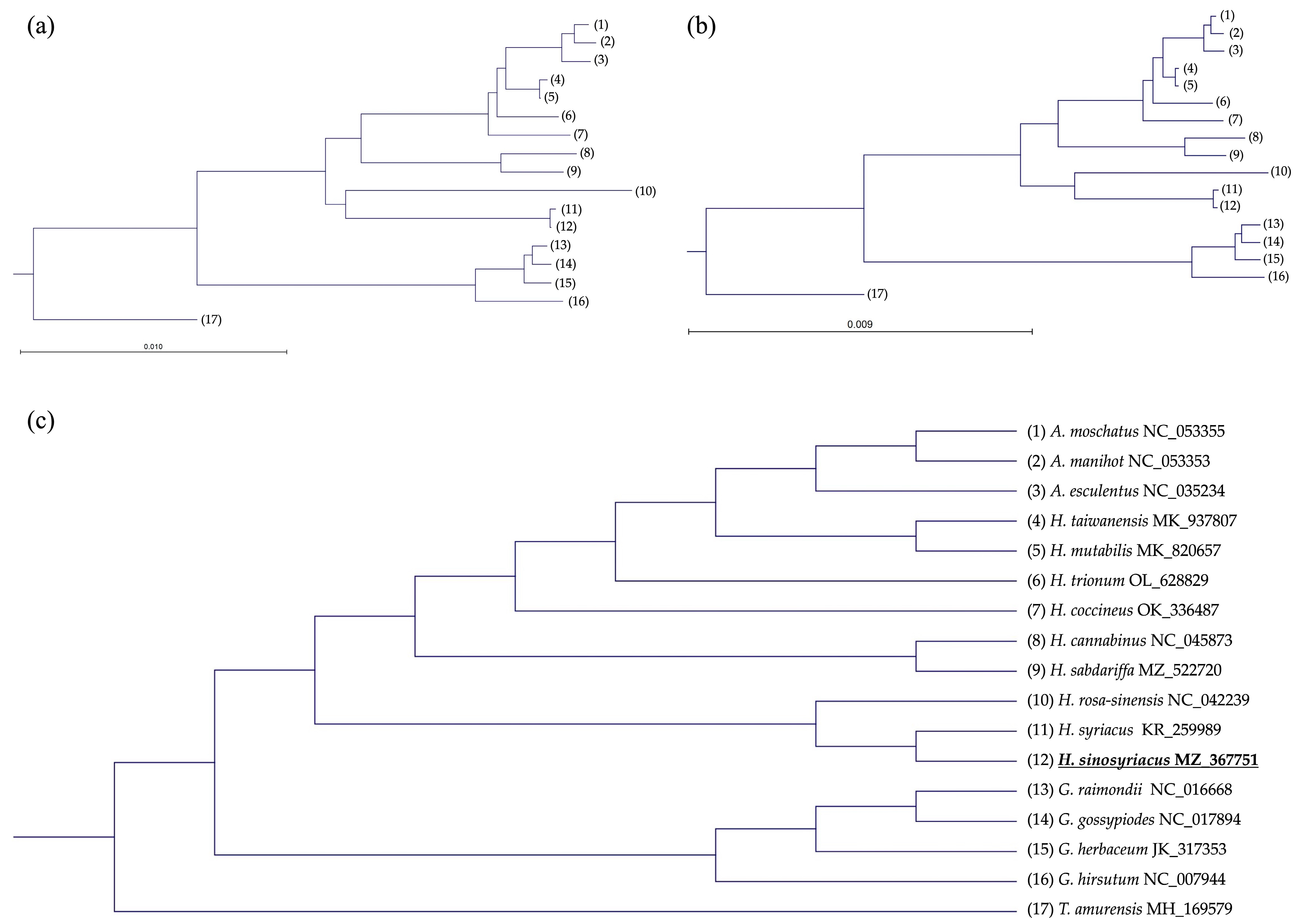
2.5. Phylogenetic Tree Analysis
3. Results
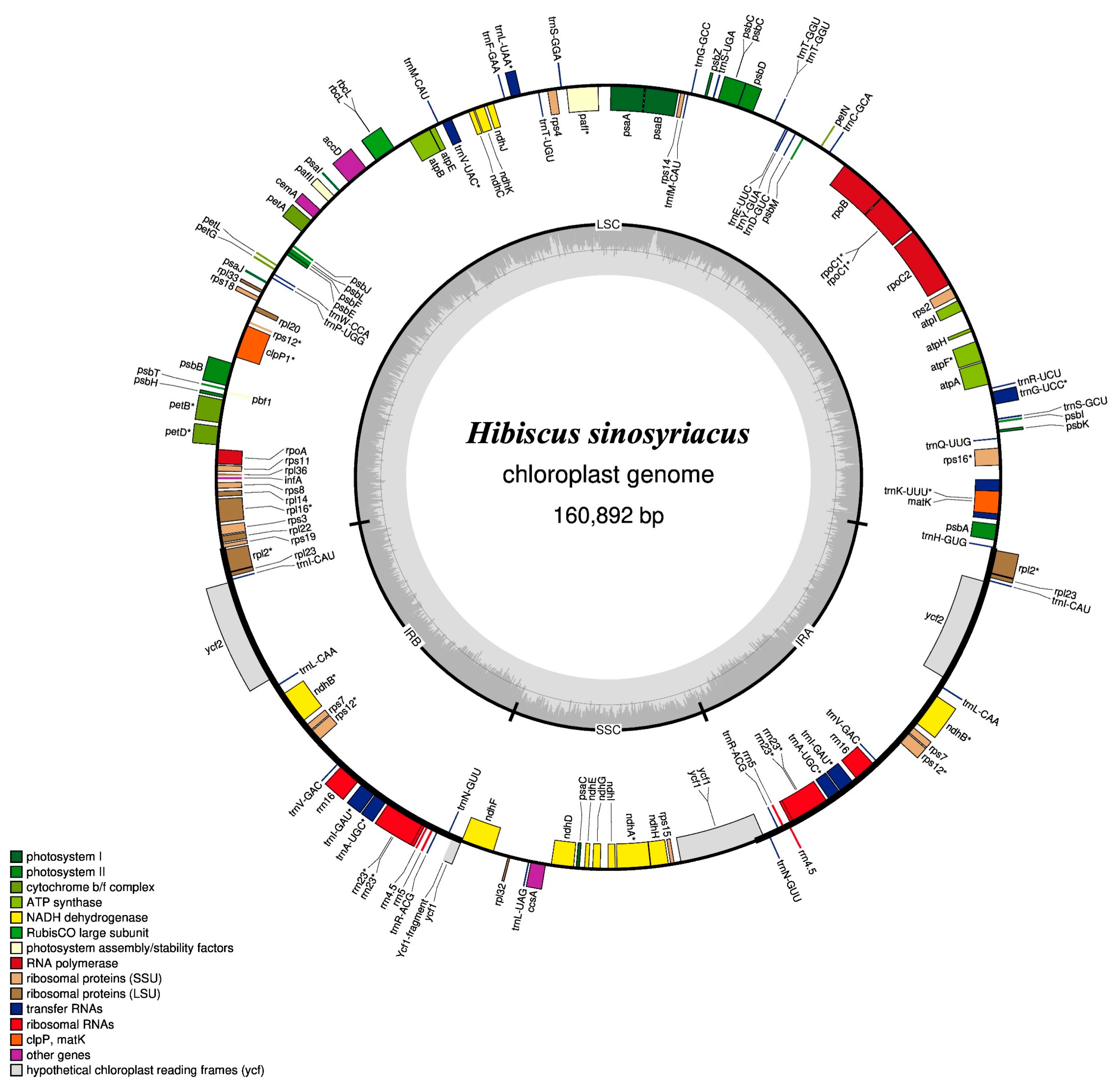
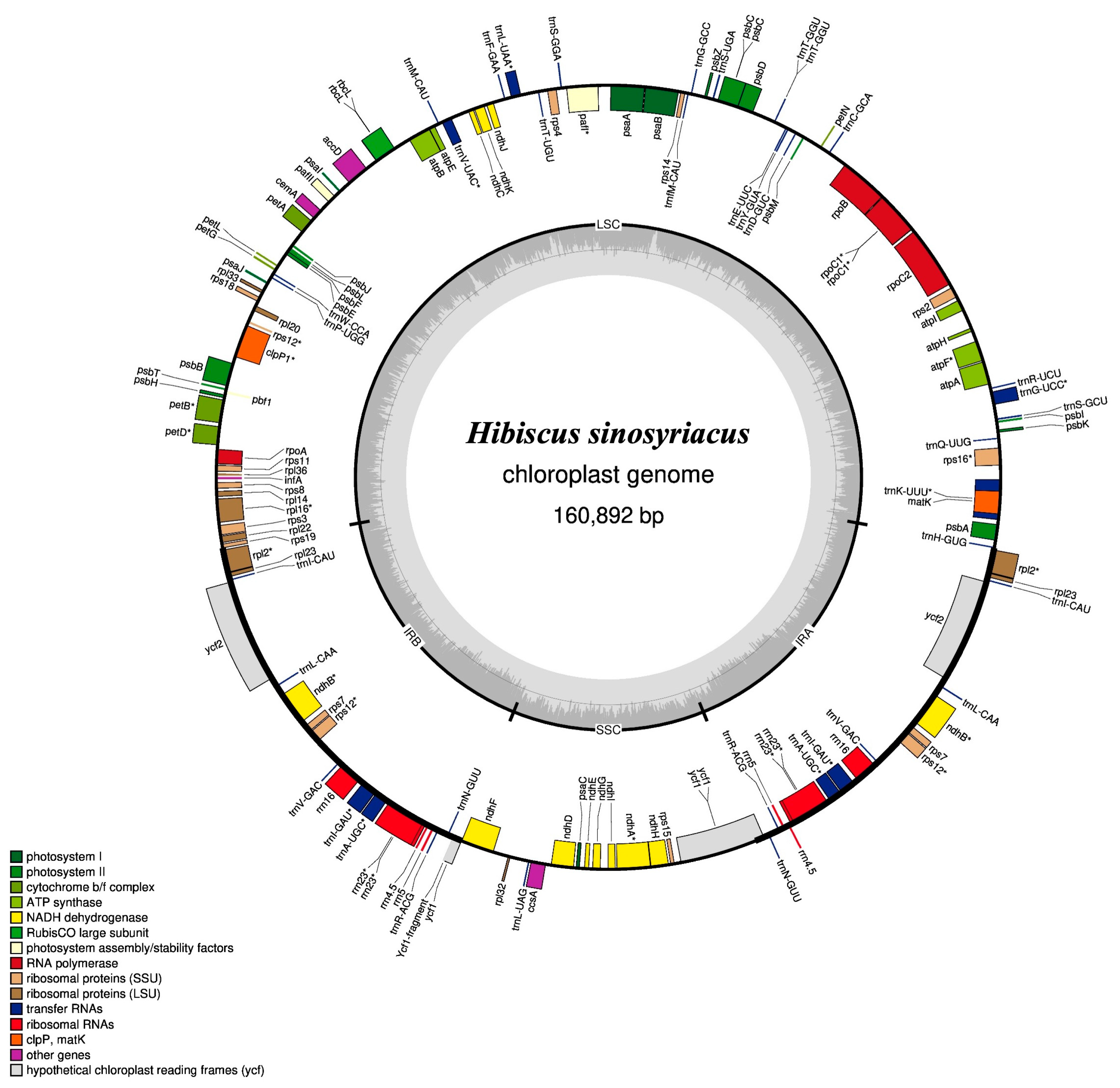
3.1. Cp Genome Assembly and Annotation of H. sinosyriacus Genes
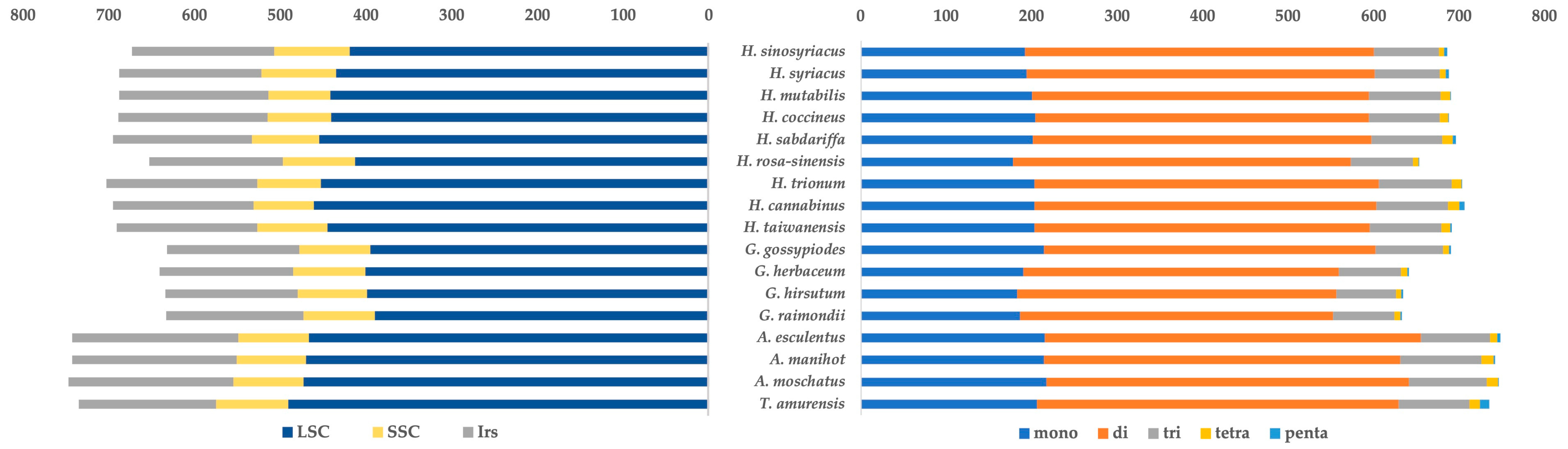
3.2. Comparative Structural Analysis
3.3. SSRs Analysis
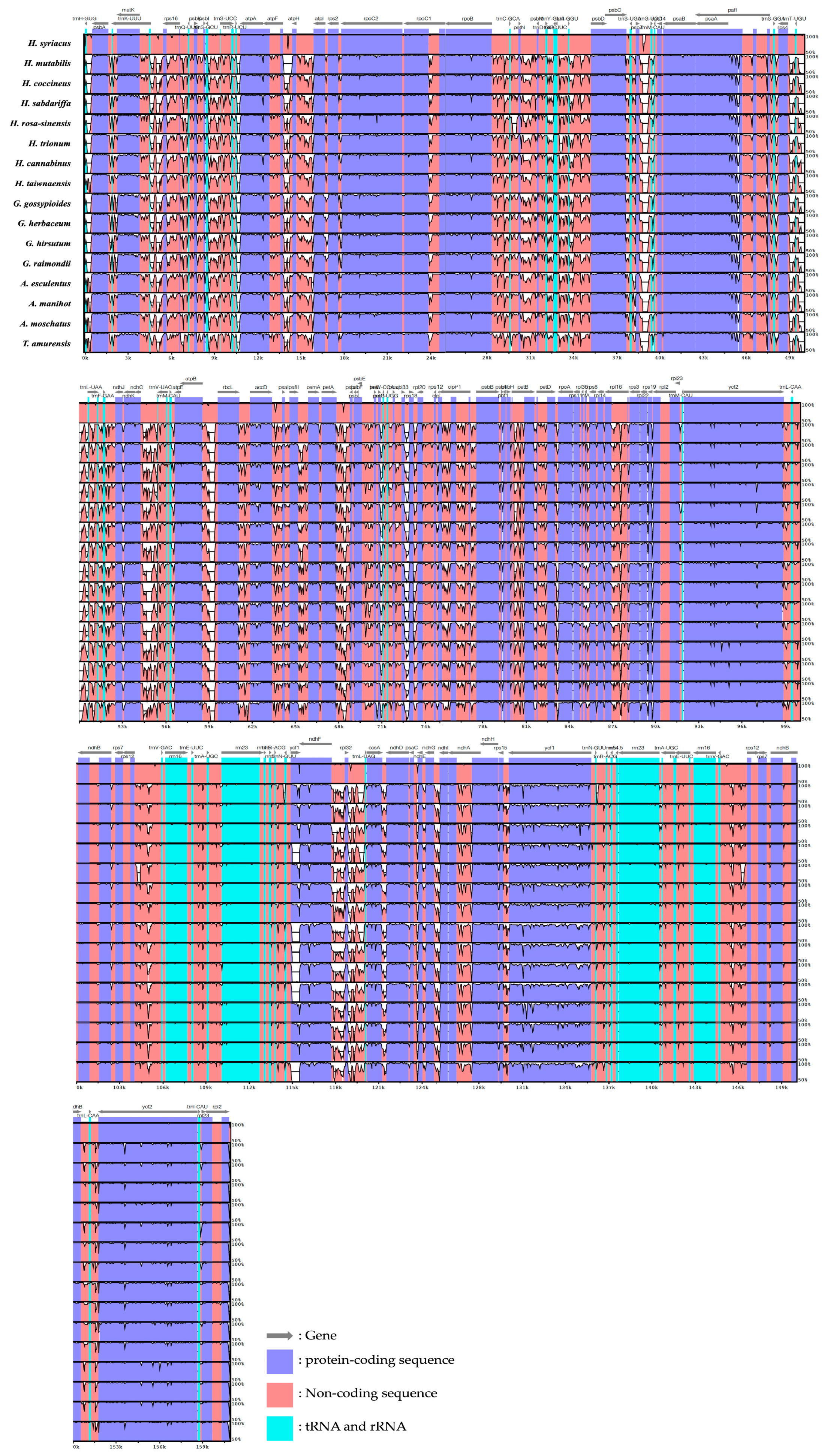
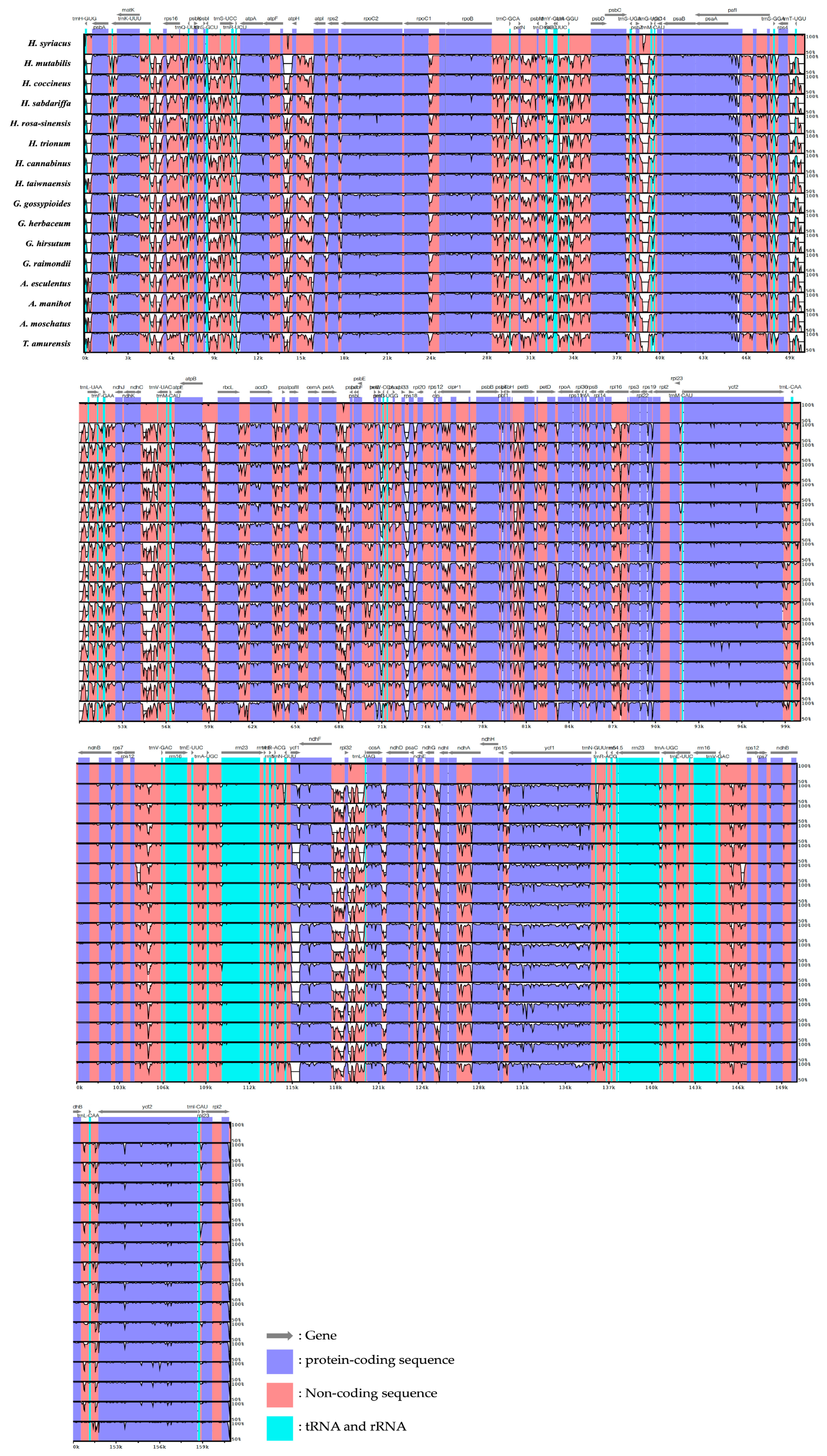
3.4. Comparative Sequence Identification Analysis via Visualization
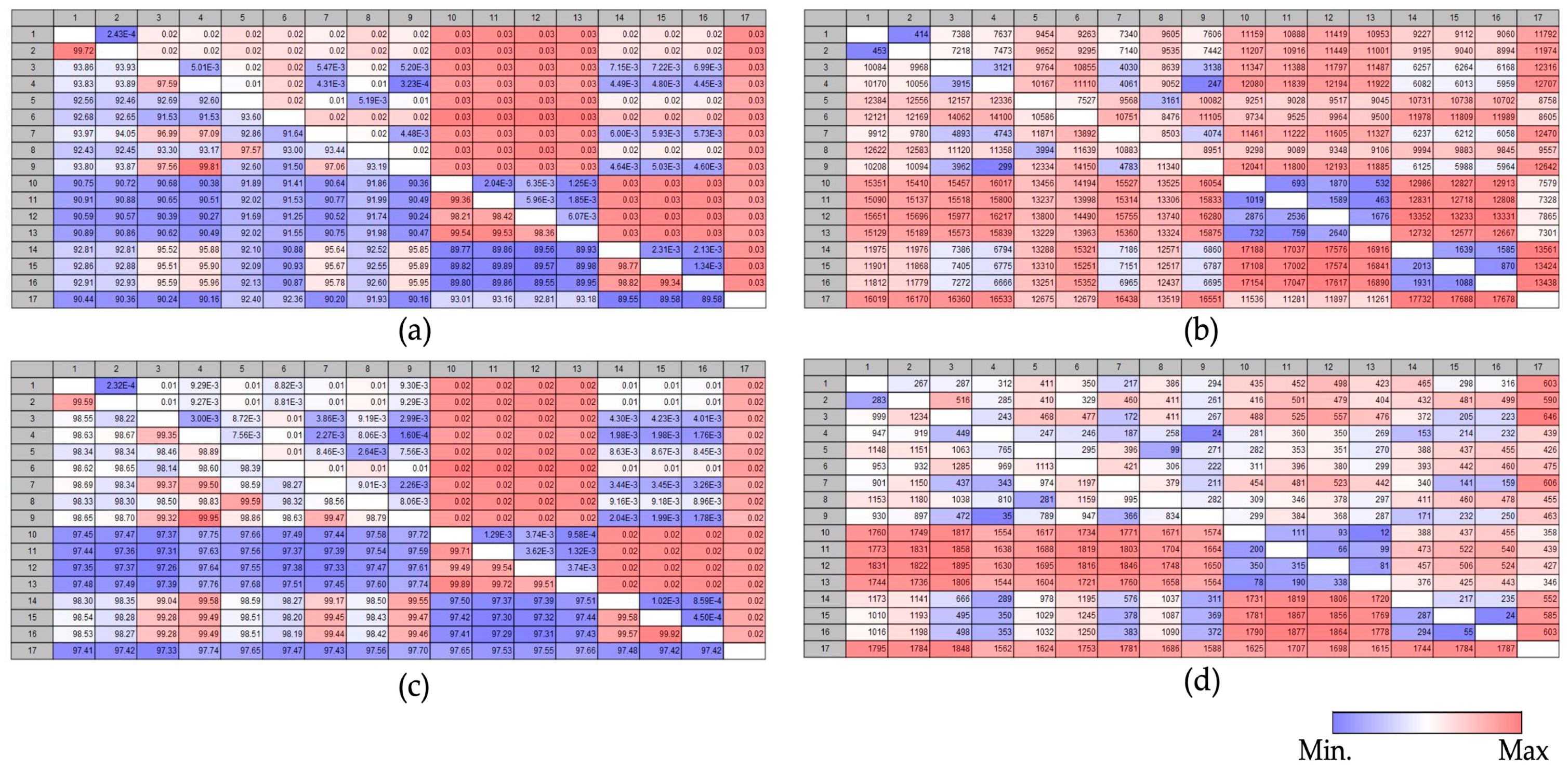
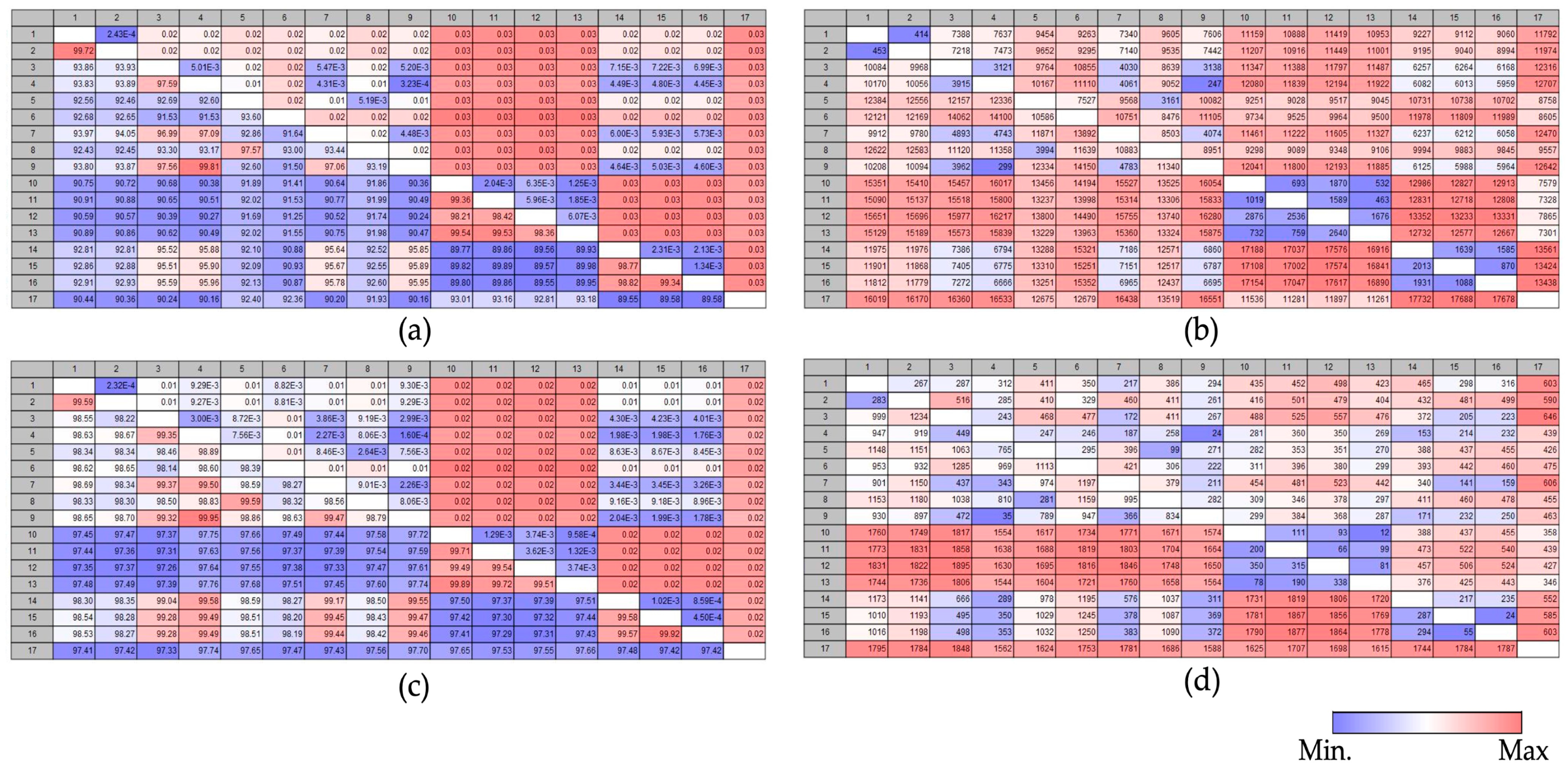
3.5. Comparison Analysis of Pairwise Heatmap
3.6. Exploration of Variants in the CDS of Hibiscus spp.
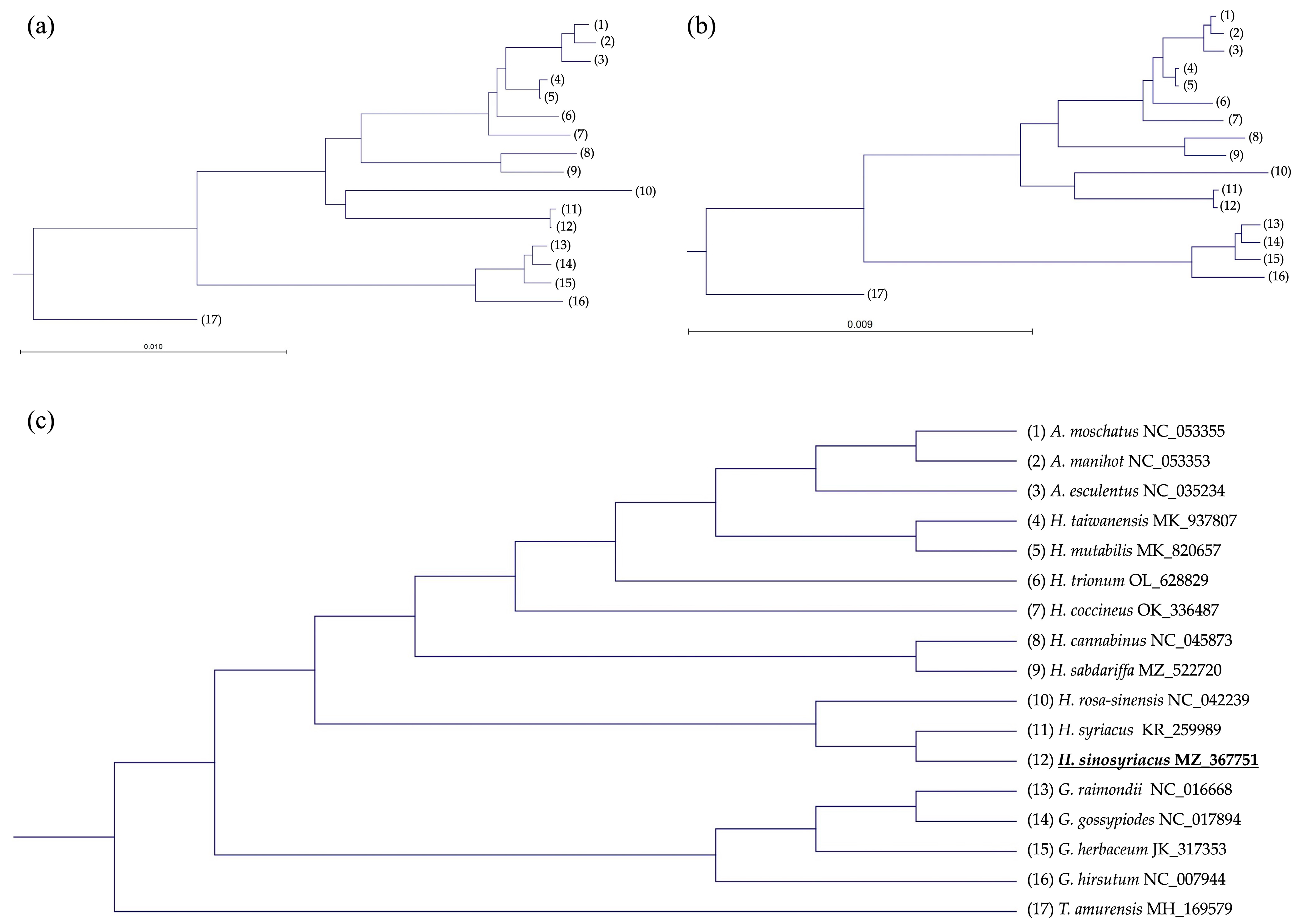
3.7. Compararive Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yamazaki, Y.; Kajita, T.; Takayama, K. Spatiotemporal process of long-distance seed dispersal in a pantropically distributed sea hibiscus group. Mol. Ecol. 2023, 32, 1726–1738. [Google Scholar] [CrossRef]
- Contreras, R.; Lattier, J.D. Targeted breeding of rose-of-sharon to improve a garden classic. Digger 2014, 215227, 47–50. [Google Scholar]
- Holser, R.A.; King, J.W.; Bost, G. Extraction of Lipid Components from Hibiscus Seeds by Supercritical Carbon Dioxide and Ethanol Mixtures; Los Alamos National Laboratory: Los Alamos, NM, USA, 2002; Available online: https://digital.library.unt.edu/ark:/67531/metadc928040/ (accessed on 7 November 2023).
- Mandaji, C.M.; Pena, R.d.S.; Chisté, R.C. Encapsulation of bioactive compounds extracted from plants of genus Hibiscus: A review of selected techniques and applications. Food Res. Int. 2022, 151, 110820. [Google Scholar] [CrossRef] [PubMed]
- Hsu, R.J.; Hsu, Y.C.; Chen, S.P.; Fu, C.L.; Yu, J.C.; Chang, F.W.; Chen, Y.H.; Liu, J.M.; Ho, J.Y.; Yu, C.P. The triterpenoids of Hibiscus syriacus induce apoptosis and inhibit cell migration in breast cancer cells. BMC Complement. Altern. Med. 2015, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Maganha, E.G.; da Costa Halmenschlager, R.; Rosa, R.M.; Henriques, J.A.; de Paula Ramos, A.L.; Saffi, J. Pharmacological evidences for the extracts and secondary metabolites from plants of the genus Hibiscus. Food Chem. 2010, 118, 1–10. [Google Scholar] [CrossRef]
- Yang, J.E.; Ngo, H.T.T.; Hwang, E.; Seo, S.A.; Park, S.W.; Yi, T.H. Dietary enzyme-treated Hibiscus syriacus L. protects skin against chronic UVB-induced photoaging via enhancement of skin hydration and collagen synthesis. Arch. Biochem. Biophys. 2019, 662, 190–200. [Google Scholar] [CrossRef]
- Molagoda, I.M.N.; Lee, K.T.; Choi, Y.H.; Jayasingha, J.A.C.C.; Kim, G.Y. Anthocyanins from Hibiscus syriacus L. inhibit NLRP3 inflammasome in BV2 microglia cells by alleviating NF-κB and ER stress-induced Ca2+ accumulation and mitochondrial ROS production. Oxid. Med. Cell. Longev. 2021, 2021, 1246491. [Google Scholar] [CrossRef]
- Beers, L.; Howie, J. Growing Hibiscus; Kangaroo Press: Kenthurst, UK, 1992; p. 94. [Google Scholar]
- Van Laere, K.; Van Huylenbroeck, J.M.; Van Bockstaele, E. Interspecific hybridisation between Hibiscus syriacus, Hibiscus sinosyriacus and Hibiscus paramutabilis. Euphytica 2007, 155, 271–283. [Google Scholar] [CrossRef]
- Yu, T.Y.; Yeam, D.Y. A survey on flower types and colors in Hibiscus syriacus L. J. Korean Soc. Hortic. Sci. 1972, 11, 55–61. [Google Scholar]
- Kim, J.H.; Lee, K.C. Studies on the flower color variation in Hibiscus syriacus L., 1; Spectral properties of fresh petals and flower color classification. J. Korean Soc. Hortic. Sci. 1991, 32, 102–110. [Google Scholar]
- Ha, Y.-M.; Shim, K.-K.; Kang, H.-C.; Lim, K.-B. A new cultivar ‘Tohagol Red’ with unique flower shape and color through interspecific hybridization of Hibiscus species. Flower Res. J. 2014, 22, 278–282. [Google Scholar] [CrossRef]
- Kretschmer, M.; Damoo, D.; Djamei, A.; Kronstad, J. Chloroplasts and plant immunity: Where are the fungal effectors? Pathogens 2020, 9, 19. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, L.; Qi, J.; Zhang, L. Complete chloroplast genome sequence of Hibiscus cannabinus and comparative analysis of the Malvaceae family. Front. Genet. 2020, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Jin, S.; Zhu, X.G.; Gitzendanner, M.A.; Soltis, D.E.; Soltis, P.S. Green giant—A tiny chloroplast genome with mighty power to produce high-value proteins: History and phylogeny. Plant Biotechnol. J. 2021, 19, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Jheng, C.F.; Chen, T.C.; Lin, J.Y.; Chen, T.C.; Wu, W.L.; Chang, C.C. The comparative chloroplast genomic analysis of photosynthetic orchids and developing DNA markers to distinguish Phalaenopsis orchids. Plant Sci. 2012, 190, 62–73. [Google Scholar] [CrossRef]
- Takayama, K.; Kajita, T.; Murata, J.; Tateishi, Y. Phylogeography and genetic structure of Hibiscus tiliaceus—Speciation of a pantropical plant with sea-drifted seeds. Mol. Ecol. 2006, 15, 2871–2881. [Google Scholar] [CrossRef]
- Nguyen, V.; Park, H.; Lee, S.; Lee, J.; Park, J.; Yang, T. Authentication markers for five major Panax species developed via comparative analysis of complete chloroplast genome sequences. J. Agric. Food Chem. 2017, 65, 6298–6306. [Google Scholar] [CrossRef]
- Padolina, J.; Linder, C.R.; Simpson, B.B. A phylogeny of Phalaenopsis using multiple chloroplast markers. Selbyana 2005, 26, 23–27. [Google Scholar]
- Olsson, S.; Grivet, D.; Cid-Vian, J. Species-diagnostic markers in the genus Pinus: Evaluation of the chloroplast regions matK and ycf1. For. Syst. 2018, 27, e016. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Houston, R. Evaluation of the trnK-matK-trnK, ycf3, and accD-psal chloroplast regions to differentiate crop type and biogeographical origin of Cannabis sativa. Int. J. Legal Med. 2021, 135, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Susanna, A.; Garcia-Jacas, N.; Hidalgo, O.; Vilatersana, R.; Garnatje, T. The Cardueae (Compositae) revisited: Insights from ITS, trnL-trnF, and matK nuclear and chloroplast DNA analysis. Ann. Mo. Bot. Gard. 2006, 93, 150–171. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, E18. [Google Scholar] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Ostertagová, E.; Ostertag, O.; Kováč, J. Methodology and application of the Kruskal-Wallis test. Appl. Mech. Mater. 2014, 611, 115–120. [Google Scholar] [CrossRef]
- Dinno, A.; Dinno, M.A. Package ‘dunn. test’. CRAN Repos 2017, 10, 1–7. [Google Scholar]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Matvienko, M. CLC Genomics Workbench. In Proceedings of the Plant and Animal Genome Conference, San Diego, CA, USA, 10–14 January 2015; CLC Bio: Aarhus, Denmark, 2015. [Google Scholar]
- Lu, G.; Moriyama, E.N. Vector NTI, a balanced all-in-one sequence analysis suite. Brief. Bioinform. 2004, 5, 378–388. [Google Scholar] [CrossRef]
- Reginato, M.; Neubig, K.M.; Majure, L.C.; Michelangeli, F.A. The first complete plastid genomes of Melastomataceae are highly structurally conserved. PeerJ 2016, 4, e2715. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, T.; Bai, G.; Zhao, Y. Complete chloroplast genome sequence of Fagopyrum dibotrys: Genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 2018, 8, 12379–12388. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046. [Google Scholar] [CrossRef] [PubMed]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Yong, B.; Jiang, H.; An, Z.; Wang, Y.; Li, B.; Yang, C.; Zhu, W.; Chen, Q.; He, C. A loss-of-function mutant allele of a glycosyl hydrolase gene has been co-opted for seed weight control during soybean domestication. J. Integr. Plant Biol. 2023. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Size (bp) | LSC | IRB | SSC | IRA | Number of Genes | Protein Coding Genes | tRNA | rRNA | GC Contents (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| H. sinosyriacus | 160,892 | 89,747 | 25,742 | 19,661 | 25,742 | 130 | 85 | 37 | 8 | 36.85 |
| H. syriacus | 161,022 | 89,701 | 25,745 | 19,831 | 25,745 | 130 | 85 | 37 | 8 | 36.83 |
| H. mutabilis | 160,879 | 89,353 | 26,300 | 18,926 | 26,300 | 130 | 85 | 37 | 8 | 36.92 |
| H. coccineus | 160,280 | 89,121 | 26,243 | 18,673 | 26,243 | 130 | 85 | 37 | 8 | 36.92 |
| H. sabdariffa | 162,428 | 90,327 | 26,100 | 19,901 | 26,100 | 130 | 85 | 37 | 8 | 36.74 |
| H. rosa-sinensis | 160,951 | 89,511 | 25,597 | 20,246 | 25,597 | 130 | 85 | 37 | 8 | 36.99 |
| H. trionum | 160,530 | 89,272 | 26,152 | 18,954 | 26,152 | 130 | 85 | 37 | 8 | 36.90 |
| H. cannabinus | 162,903 | 90,351 | 26,533 | 19,486 | 26,533 | 130 | 85 | 37 | 8 | 36.65 |
| H. taiwanensis | 161,056 | 89,538 | 25,419 | 20,680 | 25,419 | 130 | 85 | 37 | 8 | 36.89 |
| G. gossypiodes | 159,959 | 88,779 | 25,588 | 20,004 | 25,588 | 129 | 84 | 37 | 8 | 37.31 |
| G. herbaceum | 160,140 | 88,711 | 25,604 | 20,221 | 25,604 | 129 | 84 | 37 | 8 | 37.31 |
| G. hirsutum | 160,301 | 88,817 | 25,602 | 20,280 | 25,602 | 129 | 84 | 37 | 8 | 37.24 |
| G. raimondii | 160,161 | 88,654 | 25,651 | 20,205 | 25,651 | 129 | 84 | 37 | 8 | 37.31 |
| A. esculentus | 163,121 | 88,091 | 27,999 | 19,032 | 27,999 | 133 | 88 | 37 | 8 | 36.74 |
| A. manihot | 163,428 | 88,214 | 28,140 | 18,934 | 28,140 | 133 | 88 | 37 | 8 | 36.70 |
| A. moschatus | 163,430 | 88,263 | 28,118 | 18,931 | 28,118 | 133 | 88 | 37 | 8 | 36.71 |
| T. amurensis | 162,564 | 91,100 | 25,493 | 20,478 | 25,493 | 129 | 84 | 37 | 8 | 36.51 |
| Role | Group of Gene | Name of Gene | No. |
|---|---|---|---|
| Photosynthesis | Photosystem I | psaA, psaB, pasC, psaI, psaJ | 5 |
| Photosystem II | psbA, psbK, psbI, psbM, psbD, psbF, psbC, psbH, psbJ, psbL, psbE, psbN, psbB | 13 | |
| Cytochrome b/f complex | psbT, psbZ, petN, petA, petL, petG, petD 1, petB 1 | 8 | |
| ATP synthase | atpI, atpH, atpA, atpF 1, atpE, atpB | 6 | |
| Cytochrome c-type synthesis | ccsA | 1 | |
| Assembly/stability of photosystem I | ycf3(pafI) 3, ycf4(pafII) | 2 | |
| NADPH dehydrogenase | ndhB *1, ndhH, ndhA 1, ndhI, ndhG, ndhJ, ndhE, ndhF, ndhC, ndhK, ndhD | 12 | |
| Rubisco | rbcL | 1 | |
| Transcription and translation | Small subunit of ribosome | rpoA, rpoC2, rpoC1 1, rpoB, rps16 1, rps2, rps14, rps4, rps18, rps12 ***1, rps11, rps8, rps3, rps19, rps7 *, rps15 | 18 |
| Large subunit of ribosome | rpl33, rpl20, rpl36, rpl14, rpl16 1, rpl22, rpl2 *1, rpl23 *, rpl32 | 11 | |
| Translational initiation factor | infA | 1 | |
| Ribosomal RNA | rrn16 *, rrn4.5 *, rrn5 *, rrn23 * | 8 | |
| Transfer RNA | trnH-GUG, trnK-UUU 1, trnQ-UUG, trnS-GCU, trnS-UCC 1, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC 1**, trnI-GGU, trnS-UGA, trnG-UCC, trnfM-CAU **, trnS-GGA, trnT-UGU, trnL-UAA 1, trnF-GAA, trnV-UAC 1, trnW-CCA, trnP-GGU, trnL-CAA *, trnV-GAC *, trnA-UGC 1*, trnR-ACG *, trnN-GUU *, trnL-UAG, trnI-CAU | 37 | |
| Other | RNA processing | matK | 1 |
| Carbon metabolism | cemA | 1 | |
| Fatty acid synthesis | accD | 1 | |
| Proteolysis | clpP1 2 | 1 | |
| Component of TIC complex | ycf1 | 1 | |
| Hypothetical proteins | ycf2 * | 2 | |
| Total number of genes | 130 | ||
| Variants Type | Kruskal–Wallis Test | Post hoc Analysis | ||
|---|---|---|---|---|
| p-Value | H-G | G-A | A-H | |
| SSRs | 7.39 × 10−3 ** | 1.32 × 10−1 | 2.60 × 10−3 ** | 6.04 × 10−2 |
| Identities | 4.64 × 10−8 *** | 9.95 × 10−7 *** | 1.00 | 6.64 × 10−4 *** |
| Differences | 8.52 × 10−3 ** | 3.56 × 10−2 * | 4.42 × 10−3 ** | 3.24 × 10−1 |
| Gaps | 1.39 × 10−2 * | 2.30 × 10−2 * | 1.17 × 10−2 * | 6.87 × 10−1 |
| Gaps and differences | 1.21 × 10−2 * | 2.67 × 10−2 * | 8.55 × 10−3 ** | 5.46 × 10−1 |
| Name | SNP | Indels | Variants Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Species-Specific SNP | Common | Total SNP | Species-Specific Insert | Species-Specific Deletion | Common Insert | Common Deletion | Total Indel | ||
| H. sinosyriacus | 4 | – | 4 | – | – | – | – | – | 4 |
| H. syriacus | 4 | 6 | 10 | 3 | – | – | – | 3 | 13 |
| H. coccineus | 136 | 608 | 744 | 6 | 18 | 99 | 75 | 198 | 942 |
| H. mutabilis | 4 | 655 | 659 | – | – | 87 | 69 | 156 | 815 |
| H. sabdariffa | 76 | 708 | 784 | 19 | 15 | 156 | 109 | 299 | 1083 |
| H. rosa-sinensis | 326 | 311 | 637 | 102 | 15 | 51 | 72 | 240 | 877 |
| H. trionum | 89 | 624 | 713 | 26 | 8 | 105 | 75 | 214 | 927 |
| H. cannabinus | 101 | 705 | 806 | 6 | 2 | 144 | 109 | 261 | 1067 |
| H. taiwanensis | 4 | 655 | 659 | – | – | 87 | 75 | 162 | 821 |
| Gene Name | H. sinosyriacus | H. syriacus | H. coccineus | H. mutabilis | H. sabdariffa | H. rosa-sinensis | H. trionum | H. cannabinus | H. taiwanensis |
|---|---|---|---|---|---|---|---|---|---|
| atpB | TGA | TGA | TGA | TGA | TAG | TGA | TGA | TAG | TGA |
| accD | TAG | TAG | TAG | TAG | TAG | TAG | TAA | TAG | TAG |
| petA | TAA | TAA | TAG | TAG | TAG | TAG | TAG | TAG | TAG |
| rpl16 | TAG | TAG | TAA | TAG | TAG | TAG | TAG | TAG | TAG |
| ccsA | TGA | TGA | TGA | TGA | TGA | TAA | TGA | TGA | TGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, S.-H.; Kwon, H.-Y.; Choi, Y.-I.; Shin, H. Comprehensive Analysis of Chloroplast Genome of Hibiscus sinosyriacus: Evolutionary Studies in Related Species and Genera. Forests 2023, 14, 2221. https://doi.org/10.3390/f14112221
Kwon S-H, Kwon H-Y, Choi Y-I, Shin H. Comprehensive Analysis of Chloroplast Genome of Hibiscus sinosyriacus: Evolutionary Studies in Related Species and Genera. Forests. 2023; 14(11):2221. https://doi.org/10.3390/f14112221
Chicago/Turabian StyleKwon, Soon-Ho, Hae-Yun Kwon, Young-Im Choi, and Hanna Shin. 2023. "Comprehensive Analysis of Chloroplast Genome of Hibiscus sinosyriacus: Evolutionary Studies in Related Species and Genera" Forests 14, no. 11: 2221. https://doi.org/10.3390/f14112221
APA StyleKwon, S.-H., Kwon, H.-Y., Choi, Y.-I., & Shin, H. (2023). Comprehensive Analysis of Chloroplast Genome of Hibiscus sinosyriacus: Evolutionary Studies in Related Species and Genera. Forests, 14(11), 2221. https://doi.org/10.3390/f14112221