
Genetic Diversity in Peripheral Pedunculate Oak (Quercus robur L.) Provenances—Potential Climate Change Mitigators in the Center of Distribution despite Challenges in Natural Populations
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
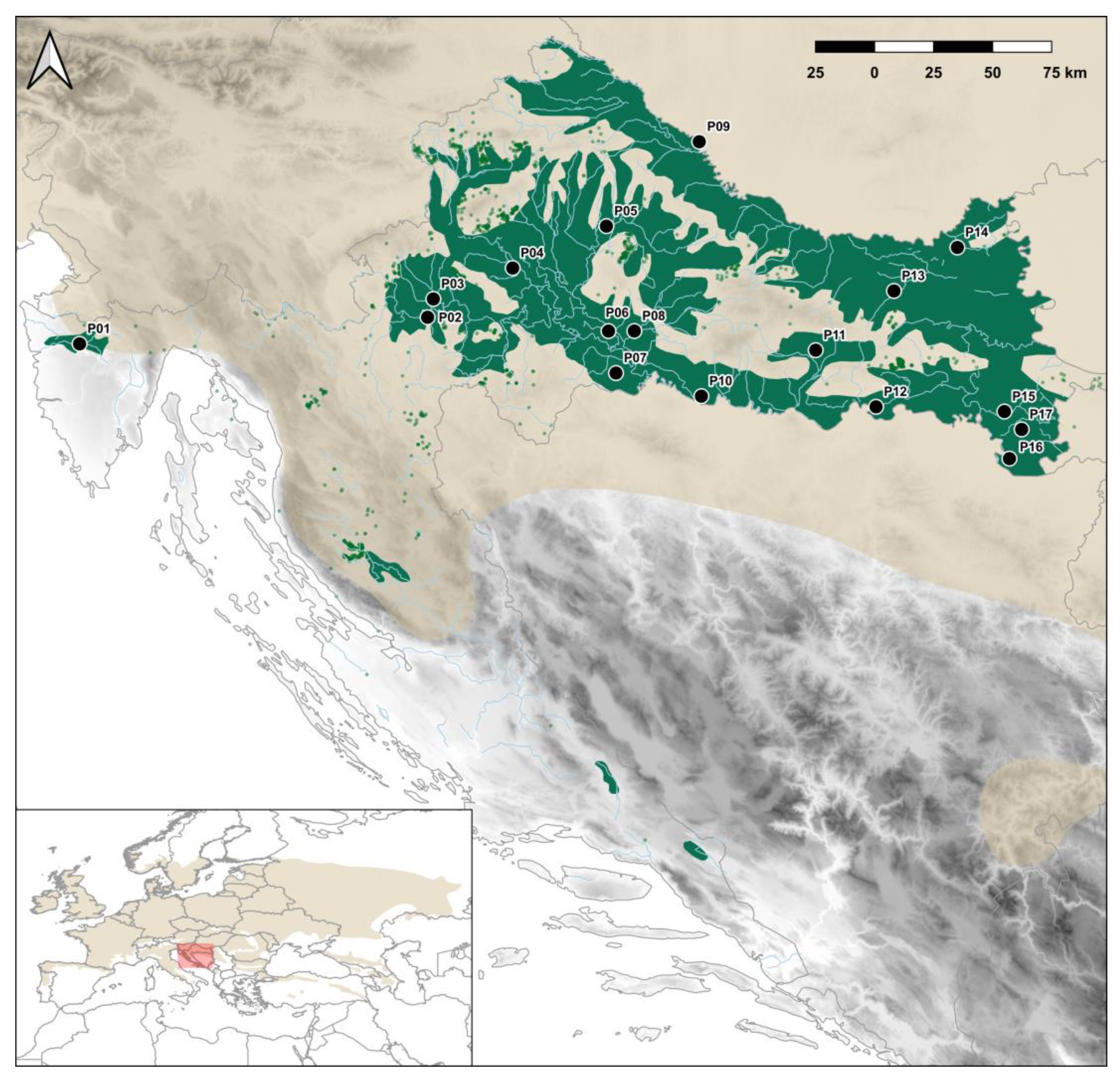
2.1. Plant Material
2.2. DNA Isolation and PCR
2.3. Statistical Analysis
2.3.1. Chloroplast Microsatellites (cpSSR)
Haplotype Diversity
Genetic Differentiation
Network/Maternal Lineages
2.3.2. Nuclear Microsatellites (nSSR)
Microsatellite Diversity
Within-Population Diversity
Population Differentiation and Structure
Spatial Genetics
Correlation with SNP and Quantitative Traits Data
3. Results
3.1. Chloroplast Microsatellites (cpSSR)
3.1.1. Haplotype Diversity
3.1.2. Genetic Differentiation
3.1.3. Network/Maternal Lineages
3.2. Nuclear Microsatellites (nSSR)
3.2.1. Microsatellite Diversity
3.2.2. Within-Population Diversity
3.2.3. Population Differentiation and Structure
3.2.4. Spatial Genetics
3.2.5. Correlation with SNP and Quantitative Traits Data
4. Discussion
4.1. Chloroplast Microsatellites
4.2. Nuclear Microsatellites
4.3. Current State and Recommendations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atkins, K.E.; Travis, J.M.J. Local Adaptation and the Evolution of Species’ Ranges under Climate Change. J. Theor. Biol. 2010, 266, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Valladares, F.; Matesanz, S.; Guilhaumon, F.; Araújo, M.B.; Balaguer, L.; Benito-Garzón, M.; Cornwell, W.; Gianoli, E.; van Kleunen, M.; Naya, D.E.; et al. The Effects of Phenotypic Plasticity and Local Adaptation on Forecasts of Species Range Shifts under Climate Change. Ecol. Lett. 2014, 17, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Croatian Forests Ltd. Forests in Croatia. Available online: https://www.hrsume.hr/sume/sume-u-hrvatskoj/ (accessed on 11 September 2023).
- Rauš, Đ. Forest Associatio of Pedunculate Oak. In Pedunculate Oak (Quercus robur L.) in Croatia; Klepac, D., Ed.; Croatian Academy of Sciences and Arts, Croatian Forests: Zagreb, Croatia, 1996; pp. 27–54. [Google Scholar]
- White, T.L.; Adams, W.T.; Neale, D.B. Forest Genetics; CABI: Wallingford, UK, 2007. [Google Scholar]
- Vukelić, J.; Mikac, S.; Baričević, D.; Bakšić, D.; Rosavec, R. Šumska Staništa i Šumske Zajednice u Hrvatskoj Nacionalna Ekološka Mreža; State Department for Nature Protection: Zagreb, Croatia, 2008; ISBN 978-953-7169-42-8.
- Burger, K.; Gailing, O. Genetic Variability of Indigenous (Quercus robur L.) and Late Flushing Oak (Quercus robur L. Subsp. Slavonica (Gáyer) Mátyás) in Adult Stands Compared with Their Natural Regeneration. Eur. J. For. Res. 2022, 141, 1073–1088. [Google Scholar] [CrossRef]
- Burger, K.; Müller, M.; Rogge, M.; Gailing, O. Genetic Differentiation of Indigenous (Quercus robur L.) and Late Flushing Oak Stands (Q. Robur L. Subsp. Slavonica (Gáyer) Mátyás) in Western Germany (North Rhine-Westphalia). Eur. J. For. Res. 2021, 140, 1179–1194. [Google Scholar] [CrossRef]
- Kesić, L.; Cseke, K.; Orlović, S.; Stojanović, D.B.; Kostić, S.; Benke, A.; Borovics, A.; Stojnić, S.; Avramidou, E.V. Genetic Diversity and Differentiation of Pedunculate Oak (Quercus robur L.) Populations at the Southern Margin of Its Distribution Range—Implications for Conservation. Divers 2021, 13, 371. [Google Scholar] [CrossRef]
- Temunović, M.; Garnier-Géré, P.; Morić, M.; Franjić, J.; Ivanković, M.; Bogdan, S.; Hampe, A. Candidate Gene SNP Variation in Floodplain Populations of Pedunculate Oak (Quercus robur L.) near the Species’ Southern Range Margin: Weak Differentiation yet Distinct Associations with Water Availability. Mol. Ecol. 2020, 29, 2359–2378. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Maroschek, M.; Netherer, S.; Kremer, A.; Barbati, A.; Garcia-Gonzalo, J.; Seidl, R.; Delzon, S.; Corona, P.; Kolström, M.; et al. Climate Change Impacts, Adaptive Capacity, and Vulnerability of European Forest Ecosystems. For. Ecol. Manag. 2010, 259, 698–709. [Google Scholar] [CrossRef]
- Lindner, M.; Fitzgerald, J.B.; Zimmermann, N.E.; Reyer, C.; Delzon, S.; van der Maaten, E.; Schelhaas, M.J.; Lasch, P.; Eggers, J.; van der Maaten-Theunissen, M.; et al. Climate Change and European Forests: What Do We Know, What Are the Uncertainties, and What Are the Implications for Forest Management? J. Environ. Manag. 2014, 146, 69–83. [Google Scholar] [CrossRef]
- Kleinschmit, J. Intraspecific Variation of Growth and Adaptive Traits in European Oak Species. Ann. Sci. For. 1993, 50, 166s–185s. [Google Scholar] [CrossRef]
- Popović, M.; Ivanković, M.; Bogdan, S. Variability of Height Growth and Survival of Progenies from Pedunculate Oak (Quercus robur L.) Seed Stands at the Field Trial ‘Jastrebarski Lugovi’–First Results. Šumarski List. Znan. Staleško Glas. Hrvat. Šumarskog Društva 2014, 138, 155–165. [Google Scholar]
- Morić, M. Genetska Raznolikost Hrasta Lužnjaka (Quercus robur L.) u Pokusnim Nasadima s Potomstvom Iz Odabranih Sjemenskih Sastojina. Ph.D. Thesis, University of Zagreb, Faculty of Forestry and Wood Technology, Zagreb, Croatia, 2016. [Google Scholar]
- Morić, M.; Bogdan, S.; Ivanković, M. Kvantitativna Genetska Diferencijacija Populacija Hrasta Lužnjaka (Quercus robur L.) u Pokusnom Nasadu »Jastrebarski Lugovi. Nov. Meh. Šumarstva Časopis Teor. Praksu Šumarskoga Inženjerstva 2018, 39, 35–45. [Google Scholar]
- Gradečki-Poštenjak, M.; Novak Agbaba, S.; Licht, R.; Posarić, D. Dynamics OfAcorn Production and Quality of English OakAcorn (Quercus robur L.) in Disrupted Ecological Conditions. Šumarski List 2011, 135, 169–180. [Google Scholar]
- Deguilloux, M.F.; Pemonge, M.H.; Petit, R.J. Use of Chloroplast Microsatellites to Differentiate Oak Populations. Ann. For. Sci. 2004, 61, 825–830. [Google Scholar] [CrossRef]
- Weising, K.; Gardner, R.C. A Set of Conserved PCR Primers for the Analysis of Simple Sequence Repeat Polymorphisms in Chloroplast Genomes of Dicotyledonous Angiosperms. Genome 1999, 42, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Kampfer, S.; Lexer, C.; Glössl, J.; Steinkellner, H. Characterization of (GA)n Microsatellite Loci from Quercus robur. Hereditas 1998, 129, 183–186. [Google Scholar] [CrossRef]
- Steinkellner, H.; Fluch, S.; Turetschek, E.; Lexer, C.; Streiff, R.; Kremer, A.; Burg, K.; Glössl, J. Identification and Characterization of (GA/CT)n-Microsatellite Loci from Quercus Petraea. Plant Mol. Biol. 1997, 33, 1093–1096. [Google Scholar] [CrossRef]
- Dow, B.D.; Ashley, M.V. Microsatellite Analysis of Seed Dispersal and Parentage of Saplings in Bur Oak, Quercus Macrocarpa. Mol. Ecol. 1996, 5, 615–627. [Google Scholar] [CrossRef]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current Trends in Microsatellite Genotyping. Mol. Ecol. Resour. 2011, 11, 591–611. [Google Scholar] [CrossRef]
- Eliades, N.-G.H.; Eliades, D.G. HAPLOTYPE ANALYSIS: Software for Analysis of Haplotype Data; Georg-August University Goettingen: Goettingen, Germany, 2009. [Google Scholar]
- Pons, O.; Petit, R.J. Measuring and Testing Genetic Differentiation with Ordered versus Unordered Alleles. Genetics 1996, 144, 1237. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987; ISBN 0231063210. [Google Scholar]
- Slatkin, M. A Measure of Population Subdivision Based on Microsatellite Allele Frequencies. Genetics 1995, 139, 457–462. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. Popart: Full-Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bowcock, A.M.; Ruiz-Linares, A.; Tomfohrde, J.; Minch, E.; Kidd, J.R.; Cavalli-Sforza, L.L. High Resolution of Human Evolutionary Trees with Polymorphic Microsatellites. Nature 1994, 368, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Minch, E.; Ruiz-Linares, A.; Goldstein, D.; Feldman, M.; Cavalli-Sforza, L.L. MICROSAT. A Computer Program for Calculating Various Statistics on Microsatellite Allele Data; Stanford University: Palo Alto, CA, USA, 1997. [Google Scholar]
- Felsenstein, J. PHYLIP-Phylogeny Inference Package, Version 3.69; University of Washington: Seattle, WA, USA, 2009. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising How the Computer Program CERVUS Accommodates Genotyping Error Increases Success in Paternity Assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipely, P. Micro-Checker: Software for Identifying and Correcting Genotyping Errors in Microsatellite Data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Chapuis, M.P.; Estoup, A. Microsatellite Null Alleles and Estimation of Population Differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A Complete Re-Implementation of the Genepop Software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- SAS Institute Inc. The SAS System for Windows, Release 9.4; Statistical Analysis Systems Institute: Cary, NC, USA, 2013; 556p. [Google Scholar]
- Luikart, G.; Allendorf, F.W.; Cornuet, J.M.; Sherwin, W.B. Distortion of Allele Frequency Distributions Provides a Test for Recent Population Bottlenecks. J. Hered. 1998, 89, 238–247. [Google Scholar] [CrossRef]
- Peery, Z.M.; Kirby, R.; Reid, B.N.; Stoelting, R.; Doucet-Bëer, E.; Robinson, S.; Vásquez-Carrillo, C.; Pauli, J.N.; Palsboll, P.J. Reliability of Genetic Bottleneck Tests for Detecting Recent Population Declines. Mol. Ecol. 2012, 21, 3403–3418. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.M.; Luikart, G. Description and Power Analysis of Two Tests for Detecting Recent Population Bottlenecks from Allele Frequency Data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Piry, S.; Luikart, G.; Cornuet, J.-M. BOTTLENECK: A Computer Program for Detecting Recent Effective Population Size Reductions from Allele Data Frequencies. J. Hered. 1999, 89, 502–503. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (Version 1.2): A Computer Program to Calculate F-Statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of Molecular Variance Inferred from Metric Distances among DNA Haplotypes-Application to Human Mitochondrial-DNA Restriction Data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring Weak Population Structure with the Assistance of Sample Group Information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software STRUCTURE: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A Website and Program for Visualizing STRUCTURE Output and Implementing the Evanno Method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A Program for Identifying Clustering Modes and Packaging Population Structure Inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Rousset, F. Genetic Differentiation and Estimation of Gene Flow from F-Statistics under Isolation by Distance. Genetics 1997, 145, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Rohlf, F.J. NTSYS-Pc: Numerical Taxonomy and Multivariate Analysis System; Applied Biostatistics, Inc.: Setauket, NY, USA, 2009. [Google Scholar]
- Petit, R.J.; Pineau, E.; Demesure, B.; Bacilieri, R.; Ducousso, A.; Kremer, A. Chloroplast DNA Footprints of Postglacial Recolonization by Oaks. Proc. Natl. Acad. Sci. USA 1997, 94, 9996–10001. [Google Scholar] [CrossRef] [PubMed]
- Katičić Bogdan, I.; Kajba, D.; Šatović, Z.; Schüler, S.; Bogdan, S. Genetic Diversity of Pedunculate Oak (Quercus robur L.) in Clonal Seed Orchards in Croatia, Assessed by Nuclear and Chloroplast Microsatellites. South-East Eur. For. 2018, 9, 29–46. [Google Scholar] [CrossRef]
- Slade, D.; Škvorc, Ž.; Ballian, D.; Gračan, J.; Papeš, D. The Chloroplast DNA Polymorphisms of White Oaks of Section Quercus in the Central Balkans. Silvae Genet. 2008, 57, 227–234. [Google Scholar] [CrossRef]
- Chmielewski, M.; Meyza, K.; Chybicki, I.J.; Dzialuk, A.; Litkowiec, M.; Burczyk, J. Chloroplast Microsatellites as a Tool for Phylogeographic Studies: The Case of White Oaks in Poland. iForest-Biogeosci. For. 2015, 8, 765. [Google Scholar] [CrossRef]
- Petit, R.J.; Brewer, S.; Bordács, S.; Burg, K.; Cheddadi, R.; Coart, E.; Cottrell, J.; Csaikl, U.M.; Van Dam, B.; Deans, J.D.; et al. Identification of Refugia and Post-Glacial Colonisation Routes of European White Oaks Based on Chloroplast DNA and Fossil Pollen Evidence. For. Ecol. Manag. 2002, 156, 49–74. [Google Scholar] [CrossRef]
- Dumolin-Lapègue, S.; Démesure, B.; Fineschi, S.; Le Corre, V.; Petit, R.J. Phylogeographic Structure of White Oaks throughout the European Continent. Genetics 1997, 146, 1475–1487. [Google Scholar] [CrossRef]
- Bordács, S.; Popescu, F.; Slade, D.; Csaikl, U.M.; Lesur, I.; Borovics, A.; Kézdy, P.; König, A.O.; Gömöry, D.; Brewer, S.; et al. Chloroplast DNA Variation of White Oaks in Northern Balkans and in the Carpathian Basin. For. Ecol. Manag. 2002, 156, 197–209. [Google Scholar] [CrossRef]
- Petit, R.J.; Csaikl, U.M.; Bordács, S.; Burg, K.; Coart, E.; Cottrell, J.; Van Dam, B.; Deans, J.D.; Dumolin-Lapègue, S.; Fineschi, S.; et al. Chloroplast DNA Variation in European White Oaks: Phylogeography and Patterns of Diversity Based on Data from over 2600 Populations. For. Ecol. Manag. 2002, 156, 5–26. [Google Scholar] [CrossRef]
- Streiff, R.; Labbe, T.; Bacilieri, R.; Steinkellner, H.; Glossl, J.; Kremer, A. Within-Population Genetic Structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. Assessed with Isozymes and Microsatellites. Mol. Ecol. 1998, 7, 317–328. [Google Scholar] [CrossRef]
- Zanetto, A.; Roussel, G.; Kremer, A. Geographic Variation of Inter-Specific Differentiation between Quercus robur L. and Quercus Petraea (Matt.) Liebl. For. Genet. 1994, 1, 111–123. [Google Scholar]
- Kremer, A.; Petit, R.J. Gene Diversity in Natural Populations of Oak Species. Ann. Sci. For. 1993, 50, 186–202. [Google Scholar] [CrossRef]
- Gregorius, H.R.; Degen, B.; König, A. Problems in the Analysis of Genetic Differentiation among Populations—A Case Study in Quercus robur. Silvae Genet. 2007, 56, 190–199. [Google Scholar] [CrossRef]
- Neophytou, C.; Aravanopoulos, F.A.; Fink, S.; Dounavi, A. Detecting Interspecific and Geographic Differentiation Patterns in Two Interfertile Oak Species (Quercus petraea (Matt.) Liebl. and Q. robur L.) Using Small Sets of Microsatellite Markers. For. Ecol. Manag. 2010, 259, 2026–2035. [Google Scholar] [CrossRef]
- Mattila, A.; Pakkanen, A.; Raisio, J.; Vakkari, P. Genetic Variation in English Oak (Quercus robur) in Finland. Silva Fenn. 1994, 28, 251–256. [Google Scholar] [CrossRef]
- Neophytou, C.; Gärtner, S.M.; Vargas-Gaete, R.; Michiels, H.G. Genetic Variation of Central European Oaks: Shaped by Evolutionary Factors and Human Intervention? Tree Genet. Genomes 2015, 11, 79. [Google Scholar] [CrossRef]
- Buche, G.; Colas, C.; Fougère, L.; Destandau, E. Oak Species Quercus robur L. and Quercus Petraea Liebl. Identification Based on UHPLC-HRMS/MS Molecular Networks. Metabolites 2021, 11, 684. [Google Scholar] [CrossRef]
- Degen, B.; Yanbaev, Y.; Ianbaev, R.; Bakhtina, S.; Tagirova, A. Genetic Diversity and Differentiation among Populations of the Pedunculate Oak (Quercus robur) at the Eastern Margin of Its Range Based on a New Set of 95 SNP Loci. J. For. Res. 2021, 32, 2237–2243. [Google Scholar] [CrossRef]
- Degen, B.; Streiff, R.; Ziegenhagen, B. Comparative Study of Genetic Variation and Differentiation of Two Pedunculate Oak (Quercus robur) Stands Using Microsatellite and Allozyme Loci. Hered 1999, 83, 597–603. [Google Scholar] [CrossRef]
- Lexer, C.; Heinze, B.; Gerber, S.; Macalka-Kampfer, S.; Steinkellner, H.; Kremer, A.; Glössl, J. Microsatellite Analysis of Maternal Half-Sib Families of Quercus robur, Pedunculate Oak: II. Inferring the Number of Pollen Donors from the Offspring. Theor. Appl. Genet. 2000, 100, 858–865. [Google Scholar] [CrossRef]
- Gömöry, D.; Yakovlev, I.; Zhelev, P.; Jedináková, J.; Paule, L. Genetic Differentiation of Oak Populations within the Quercus robur /Quercus Petraea Complex in Central and Eastern Europe. Hered 2001, 86, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Kremer, A.; Kleinschmit, J.; Cottrell, J.; Cundall, E.P.; Deans, J.D.; Ducousso, A.; König, A.O.; Lowe, A.J.; Munro, R.C.; Petit, R.J.; et al. Is There a Correlation between Chloroplastic and Nuclear Divergence, or What Are the Roles of History and Selection on Genetic Diversity in European Oaks? For. Ecol. Manag. 2002, 156, 75–87. [Google Scholar] [CrossRef]
- Zoldoš, V.; Littvay, T.; Besendorfer, V.; Jelenić, S.; Lorković, Z.; Papeš, D. Primjena Citogenetskih i Biokemijskih Analiza u Utvrđivanju Stupnja Oštećenja Šuma Hrasta Lužnjaka; Rad. Šumarskog Instituta Jastrebarsko: Jastrebarsko, Croatia, 1994; Volume 29, pp. 151–160. [Google Scholar]
- Zoldoš, V.; Besendorfer, V.; Littvay, T.; Papeš, D. The Common Oak (Quercus robur L.) as a Potential Test Plant for Cytotoxicity Monitoring. Period. Biol. 1995, 96, 490–492. [Google Scholar]
- Besendorfer, V.; Zoldos, V.; Peskan, T.; Krsnik-Rasol, M.; Littvay, T.; Papes, D. Identification of Potential Cytogenetical and Biochemical Markers in Bioindication of Common Oak Forests. Phyton 1996, 36, 139–146. [Google Scholar]
- Slade, D. Phylogenetic Origin and Distribution of Pedunculate Oak (Quercus robur L.), Sessile Oak (Q. petraea Liebl.), Pubescent Oak (Q. pubescens Thuill.) and Hungarian Oak (Q. frainetto L.) in Croatia; Rad. Hrvatski Šumarski Institut: Jastrebarsko, Croatia, 1999; Volume 34, pp. 121–131. [Google Scholar]
- Slade, D. Distribucija Haplotipova Hrasta Lužnjaka (Quercus robur L.) u Hrvatskoj; University of Zagreb: Zagreb, Croatia, 2001. [Google Scholar]
- Abdul-Muneer, P.M. Application of Microsatellite Markers in Conservation Genetics and Fisheries Management: Recent Advances in Population Structure Analysis and Conservation Strategies. Genet. Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Marwal, A.; Gaur, R.K. Molecular Markers: Tool for Genetic Analysis. Anim. Biotechnol. Model. Discov. Transl. 2020, 353–372. [Google Scholar] [CrossRef]
- Vranckx, G.; Jacquemyn, H.; Mergeay, J.; Cox, K.; Kint, V.; Muys, B.; Honnay, O. Transmission of Genetic Variation from the Adult Generation to Naturally Established Seedling Cohorts in Small Forest Stands of Pedunculate Oak (Quercus robur L.). For. Ecol. Manag. 2014, 312, 19–27. [Google Scholar] [CrossRef]
- Bakker, E.G.; Van Dam, B.C.; Van Eeuwijk, F.A.; Jacobsen, E. Population Genetics of Indigenous Quercus robur L. Populations and of Derived Half-Sib Families Has Implications for the Reproductive Management of the Species. Plant Biol. 2003, 5, 393–399. [Google Scholar] [CrossRef]
- Ballian, D.; Belletti, P.; Ferrazzini, D.; Bogunić, F.; Kajba, D. Genetic Variability of Pedunculate Oak (Quercus robur L.) in Bosnia and Herzegovina. Period. Biol. 2010, 112, 353–362. [Google Scholar]
- Craciunesc, I.; Ciocirlan, E.; Sofletea, N.; Curtu, A.L. Genetic Diversity of Pedunculate Oak (Quercus robur L.) in Prejmer Natural Reserve. Bull. Transilv. Univ. Brasov. Ser. II For. Wood Ind. Agric. Food Eng. 2011, 4, 15–20. [Google Scholar]
- Dyer, R.J. Population Graphs and Landscape Genetics. Annu. Rev. Ecol. Evol. Syst. 2015, 46, 327–342. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.; Lv, Y.; Zhang, X.; Xia, C.; Zhao, H.; Wen, C. Genetic Diversity Analysis and Variety Identification Using SSR and SNP Markers in Melon. BMC Plant Biol. 2023, 23, 39. [Google Scholar] [CrossRef] [PubMed]
- Teslak, K.; Čavlović, J.; Božić, M.; Beljan, K. Pedunculate Oak (Quercus robur L.) Trees Qualitative Structure as a Criterion of the Stand Regeneration Planning. Šumarski List 2013, 137, 367–377. [Google Scholar]
- Mikac, S.; Žmegač, A.; Trlin, D.; Paulić, V.; Oršanić, M.; Anić, I. Drought-Induced Shift in Tree Response to Climate in Floodplain Forests of Southeastern Europe. Sci. Rep. 2018, 8, 16495. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, J.E.; Munro, R.C.; Tabbener, H.E.; Milner, A.D.; Forrest, G.I.; Lowe, A.J. Comparison of Fine-Scale Genetic Structure Using Nuclear Microsatellites within Two British Oakwoods Differing in Population History. For. Ecol. Manag. 2003, 176, 287–303. [Google Scholar] [CrossRef]
- Vranckx, G. Genetic Diversity, Gene Flow and Inbreeding in Pedunculate Oak (Quercus robur L.) in Fragmented Forest Stands. Ph.D. Thesis, K.U.Leuven, Faculty of Science, Leuven, Belgium, 2014. [Google Scholar]
- Markić, A.G.; Bogdan, S.; Poštenjak, M.G.; Lanšćak, M.; Vujnović, Z.; Bogunović, S.; Ivanković, M. Acorn Yields and Seed Viability of Pedunculate Oak in a 10-Year Period in Forest Seed Objects across Croatia. South-East Eur. For. 2022, 13, 27–36. [Google Scholar] [CrossRef]
- Bogdan, S.; Ivanković, M.; Temunović, M.; Morić, M.; Franjić, J.; Katičić Bogdan, I. Adaptive Genetic Variability and Differentiation of Croatian and Austrian Quercus robur L. Populations at a Drought Prone Field Trial. Ann. For. Res. 2017, 60, 33–46. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Population | Forestry | n | nh | nh/n | nph | nE | nhr | HE |
|---|---|---|---|---|---|---|---|---|
| P01 | Buzet | 18 | 7 | 0.389 | 2 | 2.893 | 5.294 | 0.693 |
| P02 | Karlovac 1 | 19 | 4 | 0.211 | 2 | 2.560 | 2.754 | 0.643 |
| P03 | Karlovac 2 | 20 | 9 | 0.450 | 5 | 6.667 | 6.921 | 0.895 |
| P04 | Velika Gorica | 20 | 3 | 0.150 | 0 | 1.227 | 1.500 | 0.195 |
| P05 | Vrbovec | 19 | 5 | 0.263 | 1 | 2.843 | 3.575 | 0.684 |
| P06 | Kutina | 20 | 11 | 0.550 | 2 | 5.714 | 7.939 | 0.868 |
| P07 | Sunja | 18 | 12 | 0.667 | 4 | 9.000 | 9.496 | 0.941 |
| P08 | Lipovljani | 15 | 11 | 0.733 | 2 | 9.783 | 10.000 | 0.962 |
| P09 | Repaš | 19 | 9 | 0.474 | 1 | 5.085 | 6.873 | 0.848 |
| P10 | Stara Gradiška | 20 | 5 | 0.250 | 1 | 2.469 | 3.636 | 0.626 |
| P11 | Požega | 20 | 9 | 0.450 | 2 | 6.061 | 6.877 | 0.879 |
| P12 | Trnjani | 18 | 11 | 0.611 | 7 | 6.231 | 8.627 | 0.889 |
| P13 | Koška | 20 | 7 | 0.350 | 3 | 3.774 | 4.947 | 0.774 |
| P14 | Darda | 20 | 9 | 0.450 | 3 | 4.651 | 6.636 | 0.826 |
| P15 | Otok 1 | 19 | 11 | 0.579 | 3 | 5.554 | 8.246 | 0.865 |
| P16 | Gunja | 20 | 4 | 0.200 | 2 | 2.469 | 2.697 | 0.626 |
| P17 | Otok 2 | 20 | 8 | 0.400 | 1 | 2.985 | 5.645 | 0.700 |
| Population | Forestry | n | Nav | Npa | HO | HE | HE(null) | FIS | P(FIS) | PBottleneck |
|---|---|---|---|---|---|---|---|---|---|---|
| P01 | Buzet | 20 | 11.90 | 10 | 0.782 | 0.839 | 0.840 | 0.067 | *** | 0.988 |
| P02 | Karlovac 1 | 20 | 10.30 | 2 | 0.790 | 0.825 | 0.831 | 0.042 | ns | 0.813 |
| P03 | Karlovac 2 | 20 | 12.10 | 6 | 0.768 | 0.846 | 0.854 | 0.092 | *** | 0.999 |
| P04 | Velika Gorica | 20 | 10.70 | 0 | 0.785 | 0.809 | 0.817 | 0.029 | ns | 0.813 |
| P05 | Vrbovec | 20 | 11.50 | 3 | 0.785 | 0.812 | 0.816 | 0.033 | ns | 0.652 |
| P06 | Kutina | 20 | 12.20 | 1 | 0.785 | 0.817 | 0.822 | 0.040 | ns | 0.348 |
| P07 | Sunja | 20 | 11.80 | 0 | 0.790 | 0.827 | 0.833 | 0.045 | ns | 0.615 |
| P08 | Lipovljani | 20 | 11.80 | 1 | 0.770 | 0.840 | 0.847 | 0.084 | ** | 0.313 |
| P09 | Repaš | 20 | 11.60 | 3 | 0.794 | 0.816 | 0.819 | 0.027 | ns | 0.500 |
| P10 | Stara Gradiška | 20 | 13.00 | 2 | 0.765 | 0.846 | 0.849 | 0.096 | ** | 0.903 |
| P11 | Požega | 20 | 13.00 | 2 | 0.760 | 0.831 | 0.840 | 0.086 | ** | 0.993 |
| P12 | Trnjani | 20 | 11.80 | 4 | 0.814 | 0.844 | 0.826 | 0.036 | ns | 0.615 |
| P13 | Koška | 20 | 10.80 | 3 | 0.770 | 0.820 | 0.830 | 0.061 | ** | 0.652 |
| P14 | Darda | 20 | 11.90 | 0 | 0.785 | 0.839 | 0.855 | 0.064 | ns | 0.839 |
| P15 | Otok 1 | 20 | 13.70 | 4 | 0.807 | 0.836 | 0.846 | 0.035 | ns | 0.652 |
| P16 | Gunja | 20 | 13.10 | 2 | 0.760 | 0.838 | 0.849 | 0.093 | *** | 0.920 |
| P17 | Otok 2 | 20 | 12.10 | 1 | 0.785 | 0.797 | 0.801 | 0.015 | ns | 0.884 |
| Analysis | Source of Variation | df | Variance Components | % Total Variance | φST | P(φST) |
|---|---|---|---|---|---|---|
| (A) nSSR | Among populations | 16 | 0.060 | 1.47 | 0.015 | <0.0001 |
| Within populations | 663 | 4.057 | 98.53 | |||
| (B) | Among maternal lineages | 2 | 0.016 | 0.38 | 0.004 | <0.0001 |
| Within maternal lineages | 647 | 4.101 | 99.62 | |||
| (C) cpSSR | Among populations | 16 | 0.661 | 29.77 | 0.298 | <0.0001 |
| Within populations | 118 | 1.559 | 70.23 | |||
| (D) | Among maternal lineages | 2 | 1.718 | 54.23 | 0.542 | <0.0001 |
| Within maternal lineages | 63 | 1.450 | 45.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popović, M.; Katičić Bogdan, I.; Varga, F.; Šatović, Z.; Bogdan, S.; Ivanković, M. Genetic Diversity in Peripheral Pedunculate Oak (Quercus robur L.) Provenances—Potential Climate Change Mitigators in the Center of Distribution despite Challenges in Natural Populations. Forests 2023, 14, 2290. https://doi.org/10.3390/f14122290
Popović M, Katičić Bogdan I, Varga F, Šatović Z, Bogdan S, Ivanković M. Genetic Diversity in Peripheral Pedunculate Oak (Quercus robur L.) Provenances—Potential Climate Change Mitigators in the Center of Distribution despite Challenges in Natural Populations. Forests. 2023; 14(12):2290. https://doi.org/10.3390/f14122290
Chicago/Turabian StylePopović, Maja, Ida Katičić Bogdan, Filip Varga, Zlatko Šatović, Saša Bogdan, and Mladen Ivanković. 2023. "Genetic Diversity in Peripheral Pedunculate Oak (Quercus robur L.) Provenances—Potential Climate Change Mitigators in the Center of Distribution despite Challenges in Natural Populations" Forests 14, no. 12: 2290. https://doi.org/10.3390/f14122290