Abstract
Masson pine (Pinus massoniana Lamb.) is an important tree species in China, but its genomic research has been hindered due to a large genome size. Genotyping-by-sequencing (GBS) has been a powerful approach to revolutionize the field of genomic research by facilitating the discovery of thousands of single nucleotide polymorphisms (SNPs) and genotyping in non-model organisms, at relatively low cost. Here, we performed de novo SNP discovery and genotyping in 299 trees via the genotyping-by-sequencing (GBS) approach. The effort produced 9.33 × 109 sequence reads, 265,525 SNP-associated contigs, and 6,739,240 raw SNPs. Further filtering and validation of the SNP-associated contigs for reliable SNPs were performed using blasting against the Pinus tabuliformis reference genome, functional annotation, technical replicates, and custom parameter settings for the optimization. The 159,372 SNP-associated contigs were aligned and validated for SNP prediction, in which 60,038 contigs were searched with hits in the NCBI nr database. We further improved the SNP discovery and genotyping with multiple technical replicates and custom parameter settings filtering. It was found that the use of blasting, annotation, technical replicates, and specific parameter settings removed many unreliable SNPs and identified 20,055 more precise and reliable SNPs from the 10,712 filtered contigs. We further demonstrated the informativeness of the identified SNPs in the inference of some genetic diversity and structure. These findings should be useful to stimulate genomic research and genomics-assisted breeding of Masson pine.
1. Introduction
Masson pine (Pinus massoniana Lamb., 2n = 24), a diploid conifer of the family Pinaceae, is one of the most commercially important timber tree species in China, providing timber, fiber pulp, rosin, and pollen pini for industrial, chemical, and medical use [1]. Genomics has the potential to revolutionize conventional forest tree breeding with promising genotype-based genomic selection and increased genetic gain [2,3]. However, affordable, reliable, and sufficient genome-wide markers are lacking for Masson pine, due to its large and complex genome, which is similar to that of most gymnosperms [4,5,6], such as the sequenced genomes of 23.2 Gbp in Pinus taeda [5] and 25.4 Gbp in Pinus tabuliformis [6]. This has hindered the adoption of genomic selection in breeding programs. Efforts have been made to obtain single nucleotide polymorphisms (SNPs) using RNA-seq [7] and a set of genome-wide genetic variation using SLAF-seq [8]. Those efforts have facilitated the development of high-throughput SNPs for Masson pine, but it remains expensive to genotype large numbers of samples with sufficient read depths through higher degrees of multiplexing, as forest tree breeding programs traditionally operate on a large number of individuals [3]. Fixed array platforms have been regarded as the gold standard for robust and reliable high-throughput genotyping [9]. However, the development of a species-specific SNP array for species like Masson pine without sufficient genetic resources requires a much more competitive price. Therefore, developing cost-effective genotyping platforms is essential for incorporating genomics into Masson pine breeding schemes.
Genotyping-by-sequencing (GBS), also known as reduced representation sequencing (RRS), is a widely used approach to reduce the complexity of large genomes to identify high-throughput SNP markers [10,11,12,13]. This approach enables subset diverse but identical enzyme-target and recognized genomic regions for sequencing multiple samples. For species without a reference genome, a pseudo-reference or homogenous reference genome is used for the identification of SNPs [14]. GBS can provide rapid, cost-effective, and comprehensive reduction of complex genomes, and thus is suitable to develop genome-wide molecular markers in non-model species, especially in tree species, as most forest trees are non-model species with large and complex genomes [15]. The compromise of sequence read depth and the number of sequenced individuals makes GBS a cost-effective molecular marker development platform [16]. It has been applied to some tree species with large genomes [17,18], such as Pinus sylvestris [18], and has discovered large volumes of SNPs in studied species in the absence of species-specific genomic resources. However, concerns about the precision and reliability of the genetic data resulting from the GBS approach are increasing [19,20]. As raw SNP datasets resulting from all genotyping experiments are typically inaccurate and incomplete [21,22], it is technically difficult to identify reliable SNPs for such a large-genome species without a sequenced genome. Thus, quality control (QC) procedures are important steps to identify more precise and reliable SNPs via GBS.
QC involves many aspects from the initial data preparation to core bioinformatics analysis [20], including filtering out poor-quality or suspected artifactual SNP loci; filtering out individuals related to missing data, anomalous genotype call, and genetic synonymies; and characterizing the identified SNPs [22]. Generally, the custom per-marker QC of GBS data consists of at least three steps: (i) filtering out SNPs with an excessive missing genotype, (ii) the removal of all markers with very low minor allele frequency (MAF) or minor allele count (MAC), and (iii) the removal of all individuals with large missing data [23]. For a non-model species without a reference genome, such as in forest tree species, extra approaches have also been used to ensure more accurate reads or contigs used for SNP prediction, such as blasting reads against the sequenced reference genome for filtering reads, performing SNP-associated contigs annotation, and using technical replicates to mitigate the genotyping errors [24,25,26]. For example, a significantly higher proportion of good loci have been obtained using paired replicates in GBS for Fagus sylvatica and Quercus robur L. [26]. However, little is known about the characteristics of identified SNPs resulting from the GBS approach in Masson pine with its un-sequenced, complex, and massive genome.
The specific objectives of this study were to (1) employ the GBS approach to generate a set of reliable SNPs in 299 Masson pine samples; (2) validate the de novo assembled contigs for SNP prediction by blasting against the well-assembled Pinus taeda and Pinus tabuliformis reference genomes and functional annotation; (3) illustrate the characteristics of SNPs identified through GBS application by the means of technical replicates; and (4) validate the informativeness of optimized SNPs with population genetic diversity analysis. These efforts aimed to generate a set of highly reliable SNP markers for Masson pine via GBS and reveal the characteristics of identified SNPs, as well as to provide more understanding of high-throughput molecular marker development using GBS in non-model tree species with large genomes.
2. Materials and Methods
2.1. Plant Material and DNA Extraction
The study material comprised 299 samples, including 293 open pollinated progenies from 65 selected families and an additional six parents with two replicates each from the advanced Masson pine (Pinus massoniana Lamb.) seed orchard in the Shanghang Baisha State-Owned Forest Farm in Fujian province, China. Information about the 299 sample collection is displayed in Table S1. There were 293 progenies from 65 families in 15 subpopulations from four local locations with a range of 1–25 individuals per family and the extra six parents. A set of 305 samples of young needles including six replicates were collected on 20–28 July 2019, sealed in a bag with full silica gel, and delivered to the lab in 4 °C containers, and then stored at −25 °C prior to DNA extraction. Genomic DNA was extracted from the dry needles using a modified cetyl trimethyl ammonium bromide (CTAB) method [27]. The quality and quantity of DNA were measured using the NanoDrop 1000 spectrophotometer (ThermoFisher Scientific, Wilmington, DE 19810, USA). The final concentration of DNA was adjusted to 50 ng/μL for GBS library construction.
2.2. GBS Library Preparation and Sequencing
A GBS library of each sample was prepared according to the protocol described in the Illumina® TruSeq® Nano DNA LT Library Prep kits (FC-121-4001). The 200 ng of genomic DNA from each sample was restriction-digested in a total volume of 50 µL, containing 5 units each of EcoRV and ScaI-HF, as well as 5 µL NEB 10 × CutSmart Buffer (New England Biolabs, Ipswich, MA, USA). The reaction was incubated at 37 °C for 16 h for digestion and then heat-inactivated at 80 °C for 10 min. The enzyme digestion products were purified and recovered with magnetic beads in TruSeq® Library Building Kit. Barcoded and common adapters were designed as described in Illumina® TruSeq® Nano DNA LT Library Preparation kits to complement the restriction overhangs created by EcoRV and ScaI-HF, respectively, and the overhangs resulting from fragmentation were converted into blunt ends using End Repair Mix 2. Following end repair, the appropriate library size of about 400–550 bp was selected using different ratios of the SPB (Sample Purification Beads). After adenylating 3ʹ ends, each restriction-digested sample was then ligated to a unique 5′ barcoded adapter and a common 3′ adapter. The process of ligating adapters was performed to ligate multiple indexing adapters to the ends of the DNA fragments for sequencing use, and then clean up the ligated fragments. To enrich the ligated DNA and perform size selection, PCR amplification was performed using 10 µL of the ligated products’ DNA, 25 µL of KAPA HiFi HotStart Ready Mix PCR Kit, and the common PCR1 and indexed PCR2 primers, respectively. The PCR primers used were specific to each adapter and comprised an Illumina index sequence and flow cell annealing complementary sequences. The ligated products were amplified with PCR by the following conditions: initial denaturation at 95 °C for 3 min; 8 cycles of denaturation at 98 °C for 15 s, annealing at 60 °C for 15 s, and extension at 72 °C for 30 s; and then final extension at 72 °C for 5 min. After electrophoresis, the PCR products of the library with the length of the inserted fragment in the target interval (500–550 bp) were cut and purified for subsequent sequencing according to the preset scheme in TIANGEN (DP209-02).
The quality of individual libraries and median fragment size were assessed on the Agilent Technologies 2100 Bioanalyzer using Qubit™ 1X dsDNA HS Assay Kits. Indexed DNA libraries were normalized to 10 nM in the DCT plate and then pooled in equal volumes in the PDP plate. The pooled library underwent a final 0.8X Ampure XP bead cleanup to remove any remaining residual fragments shorter than 500 bp. The concentration of the final bead-cleaned library was determined in preparation for sequencing. Sequencing was performed using the Illumina NovaSeq 6000 SP Reagent Kit v1.5 (500 cycles) for 150 bp paired-end sequencing (Illumina, 20028402) in LC-BIO Co., Ltd. (Hangzhou, China).
2.3. Sequence Quality Analysis and Filtering
Sequence reads were de-multiplexed by using the outer dual index barcode information and assigned to sequenced samples. Reads containing the correct restriction sites in R1 and R2 were obtained by searching restriction site sequences in the raw reads. Quality filtration was carried out as follows: the adapters were removed, the low quality reads with lengths less than 40 bp were removed, and the unstable bases in the read within the first 10 bp and the last 8 bp were trimmed using Trimmomatic v0.39 [28] and Fastp v0.23.1 pipelines [29].
2.4. De Novo Assembly, Read Alignment, and SNP Calling
A set of 24 samples was used for de novo assembly using MEGAHIT v1.2.9 [30] with kmer-size ranging from 29 to 141bp and the optional de novo contigs generated with kmer-size of 141 were selected. The obtained de novo assembly was first blasted against the Lobolly pine (v2.0) sequenced reference genome in the local database constructed from NCBI (https://www.ncbi.nlm.nih.gov/assembly/GCA_000404065.3, accessed on 2 March 2022) [31] using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 2 March 2022) [32]. The aligned assembly (contigs with identity ≧ 95% and length ≧ 141 bp) was then split into 40 subsets, each containing ~20,000 contigs. Pre-individual variants were called using a set of custom shell scripts named “parallelGeneratingBamFiles.sh” implemented with Bowtie2 v2.4.2 [33], Samtools v1.11 [34], and BCFtools [35] for parallel runs for subset BAM files generating separately on the 40 genomic subsets before a joint genotype call over all 305 samples using ANGSD [36]. In general, the bowtie2-build command was applied to index the reference contigs first, then the samtools faidx was used to deal with the indexed reference; then, the custom program cycle with the application of Bowtie2 v2.4.2 and SAMtools v1.11 pipeline was run in parallel to speed up the multiple sample-specific SAM file construction process. Based on the SAM files, SAMtools v1.11 was used to create, sort, and index BAM files for SNP calling. Based on allele frequencies that were estimated with genotype likelihoods from the sorted.bam files, the SNP calling based on certain subset reference contigs using ANGSD was carried out by using the following commands: angsd -bam bam.filelist -GL 2 -out gatk_outfile -doMaf 2 -doMajorMinor 1 -SNP_pval 1e-6 -doGeno 5 -doPost 1 -postCutoff 0.95.
2.5. SNP Filtering
A total of 305 samples with an extra six replicates for six samples were sequenced. The six replicates were processed with the 299 individuals at the same time. Initial analyses were conducted based on the filtering of missing data using Further_deletion.sh in npGeno pipeline [37] to filter out the singleton, duplicated, and homogenous loci with missing threshold < 30%. The obtained SNPs with missing < 10% were formatted using PLINK v1.9 [38] and the minor allele frequency (MAF) was calculated. Only the loci with MAF > 0.01 were treated as the initial SNPs set, and the resulting SNPs for use were 0.67 million. By using the six sample replicates for genotyping error detection, a custom R script was developed to label each SNP with a distinct genotyping error label, including missing data (MD), locus error (LE), missing loci (ML), and missing allele (MA), as well as good loci (GL); these are described in Table 1. The repx1SNP included the SNPs in which both replicates in the paired replicates had consistent alleles but there were non-missing data among the selected pairs of replicates in certain repx filtering scenarios. For example, the rep11SNP was obtained by the removal of all inconsistent data in one pair of replicates; the rep61SNP was the selected good loci with consistent alleles between each pair of replicates across six sets of replicates. Those SNP-associated contigs were blasted against the well-assembled Chinese pine (Pinus tabuliformis) reference genome (https://db.cngb.org/search/project/CNP0001649/, accessed on 2 March 2022) [6] to remove contigs with identity < 95% and multi-location hits against the reference genome (Blasted). The remaining SNP-associated contigs were further searched for validation with functional annotation (Annotated). Another filtering was considered to delete the loci with less than four samples with a minimum number of minor genotypes per loci (minimum minor genotype count ≥ 4), and different MAF values at different missing levels ranged from 0 (M0), 5% (M5) to 10% (M10). Extra filtering was performed to exclude all SNPs within 35 bp distance of those markers. The remaining SNPs were obtained for statistics summary. As each SNP loci had a distinct genotyping error label, the SNP count according to genotyping error tags in different filtering scenarios could be determined accordingly.

Table 1.
Key indicators used to assess genotyping error types.
To simply mark the custom SNP filtering scenario, seven parameters were included as follows: missing level (M), minor allele frequency (MAF), minimum minor genotype count (mC), different paired replicates for SNP filtering (repx), blasted against Chinese pine reference genome (blast), SNP-associated contigs annotation (Annot), and 35bpFiltering (35 bp). A certain SNP filtering strategy was the combination of the above several parameters labeled as SNP filtering scenario. For example, an SNP dataset with missing < 5% (M5), minor allele frequency (MAF) > 0.05 (F05), the minimum minor genotype count ≥ 4 (C4), using six paired replicates (rep6), blasted (blast), annotation contigs (Annot), and 35 bp distance filtered (35 bp) for SNP filtering was recorded as a “rep6M5F05C4blastAnnot35bp” filtering scenario.
2.6. SNP-Associated Contig Validation
BLAST research for the SNP-associated contig in the 0.67 million SNPs was performed, taking advantage of the well-assembled 25.4 Gb chromosome-level assembly of Chinese pine downloaded from GSA (https://db.cngb.org/search/project/CNP0001649/, accessed on 2 March 2022) [6]. Those contigs with identity ≧ 95% and unique position hit were selected as the reference-located contigs. Those reference-located contigs were then used to extract the reference-located-SNP from the 0.67 million SNPs. Those reference-located-SNP-associated contigs were further used for functional annotation analysis. The reference-located-SNP-associated contigs that might putatively encode proteins were searched against the non-redundant protein database at the NCBI (National Center for Biotechnology Information) (USA) with minimum E-value of <1.0 × 10−6 as the threshold to extract the Gene Ontology (GO) terms associated with the blasted hits using the program Blast2GO [39]. The three major GO terms, cellular component (CC), biological process (BP), and molecular function (MF) were also determined with the e-value hit filter < 1.0 × 10−6. In a final step, details of the pathway annotation with the reference-located-SNP-associated contigs were produced using the KEGG (Kyoto Encyclopedia of Genes and Genomes) database accessed on 12 March 2022 [40].
2.7. Diversity Analyses
To evaluate the impact of obtained SNPs with different filtering scenarios in genetic diversity, the genetic associations of the 299 Masson pine samples were assessed using principal component analysis from the R program of AveDissR v6 [41,42], and the PCoA plots of the first three resulting principal components were made based on the obtained SNPs in different filtering scenarios. For individual comparison, the resulting PCoA plots were individually labeled with respect to the sample’s original source group. To investigate the impact of missing SNP data on genetic diversity analysis, another PCoA analysis was performed with the MAF > 0.05 at different missing levels of 0, 5%, and 10% at rep6F05C4blastAnnot35bp filtering scenarios.
3. Results
3.1. High Throughput Sequencing and Assembly
Following the workflow outlined in Figure S1, this study generated a total of 9.33 × 109 clean sequence reads in 610 FASTQ files. The average number of reads after cleaning was obtained with 1.52 × 107 reads/sample, ranging from 0.6 × 107 to 7.6 × 107 reads/sample. It was found that the replicated pairs with higher read numbers generated slightly more SNPs; however, larger differences in the numbers of reads between individuals within the pair led to a slight decrease in the quantity of data obtained per pair.
A set of 24 paired FASTQ files with sequence reads ranging between 2.05 × 107 to 7.63 × 107/sample were de novo assembled using MEGAHIT. To select a set of reliable contigs as a de novo assembly for SNP calling, the de novo assembled contigs were first blasted against the sequenced genome of Lobolly pine (Pinus taeda) and those contigs with identity < 95% and length < 141 bp were filtered out. The remaining 843,351 contigs were used as a de novo assembly for SNP discovery. In the polished de novo assembly, a majority of 98.93% of the contigs had a length ranging between 150 and 1100 bp, with an average of 329 bp.
3.2. SNP Calling and Filtering
All 305 samples (including each replicate of six samples) were used for SNP calling using custom shell scripts of ref-ANGSD pipeline based on the polished 843,351 contigs. The obtained 0.67 million raw SNP data from 265,525 contigs were filtered with minor allele frequency (MAF) > 0.01 and SNP calling rate > 90% firstly based on PLINK v1.9 formatted data. A statistical summary of SNP count under varied SNP filtering strategies is illustrated in detail in Table 2.
Table 2.
SNP filtering process and SNP count and contig numbers with and without one or six replicates filtering.
The SNP and contig counts with and without replicate filtering in all the SNP filtering processes are displayed in Table 2. It was found that different filtering steps resulted in a reduced number of SNPs and contigs, which meant that more rigorous filtering was being carried out. For example, the blasting and annotating filtering at rep0M10F01 with missing < 10% (M10), MAF > 0.01 (F01) resulted in a sharp decrease from 6,739,240 to 2,199,317 in total SNP count. With further filtering at rep0M10F05C4blastAnnot, the SNP count was decreased from 2,199,317 to 627,362. The custom parameter settings on MAF, mC at different missing levels at rep0F05C4blastAnnot resulted in remaining SNPs with the proportions of 9.31% (627,362; M10), 3.63% (244,948; M5), and 0.10% (6787; M0). Obviously, the blasting, annotating, and custom parameters filtering removed a majority of 90.69% (6,739,240 vs. 627,362) or more un-reliable SNPs from the raw SNP sets in the rep0 scenario. There was only a small proportion of 1.08% (6787) without missing data in the M10 (627,362) SNP set at rep0M10F05C4blastAnnot. The small proportion of 1.08% of SNPs with non-missing data showed that the identified 627,362 SNPs in M10 were generally harbored with missing data in the Masson pine GBS application. Along with the missing level in the identified SNPs reduced from M10 and M5 to M0 at the rep0F05C4blastAnnot scenario, the contigs were sharply decreased from 20,554 and 15,434 to 2312, respectively. A steep reduction of 13,122 (85.02%) in SNP-associated contigs was observed when the missing level decreased from M5 to M0 at rep0F05C4blastAnnot, which meant a sharp shrinking of the SNP coverage in the de novo assembly. After the exclusion of all SNPs within a 35 bp distance in each set of SNPs in M10, M5, and M0, the remaining SNPs were reduced to 9780, 14,612, and 2942 at rep0F05C4blastAnnot35bp, respectively.
The effects of technical replicates in obtained SNP counts are also displayed in Table 2. The initial 6,739,240 SNPs dataset at rep0M10F01 was used for paired replicates filtering. A notable decrease in SNP count was observed among the three datasets from 6,739,240 (100%) to 5,420,678 (80.43%) and 2,626,541 (38.97%) in rep01SNP, rep11SNP, and rep61SNP, respectively. At the same time, the contigs decreased from 265,525 (100%) and 256,986 (98.78%) to 204,433 (76.99%) in rep01SNP, rep11SNP, and rep61SNP, respectively. The combination of blasting, annotation, and six replicates filtering resulted in a proportion of 12.53% (844,311 vs. 6,739,240) of SNPs being retained and 19.01% (50,447 vs. 265,525) of contigs being retained at the rep6M10F01blastAnnot scenario. With further filtering with MAF and missing and minimum minor genotype counts in each SNP set, the SNPs decreased from 844,311 to 95,115 at the rep6M10F05C4blastAnnot scenario (Table 2). Different missing levels at rep6F05C4blastAnnot resulted in 95,115, 60,143, and 2532 SNPs in M10, M5, and M0, respectively. The removal of all SNPs within a 35 bp distance retained 26,680, 20,055, and 1641 SNPs in M10, M5, and M0, respectively, at rep6F05C4blastAnnot35bp. Compared with the identified SNPs in the two scenarios at rep0F05C4blastAnnot35bp and rep6F05C4blastAnnot35bp with different missing levels, the SNP count with missing in the rep6 scenario obtained more markers than in the rep0 scenario. For example, there were 9780 vs. 26,680 in M10 and 14,612 vs. 20,055 in M5, due to a longer distance between markers resulting from the removal of inconsistent loci between each pair of replicates in the rep6 scenario. Consequently, more SNP were retained in rep6 than in the rep0 scenario.
Statistics were conducted to illustrate the distribution of the identified 20,055 SNPs and related SNP-associated contigs on each of twelve chromosome-level linkage groups and contigs of the Chinese pine reference genome at rep6M5F05C4blastAnnot35bp (Table 3). Except for the seventh linkage group, evenly distributed SNPs with unique locations were observed across the 11 chromosome linkage groups and contigs, which indicated that the identified SNPs were detected across the whole genome. Further investigation of the SNPs in the seventh linkage group showed that there were no unique hits, but rather two or more hits, recorded for the SNP-associated contigs located in this group. Consequently, no SNP with unique location was obtained in the seventh linkage group.
Table 3.
The distribution of SNPs at rep6M5F05C4blastAnnot35bp across the Chinese pine reference genome in the twelve linkage groups and contigs.
3.3. Characterization of Identified SNPs
Based on the initial 0.67 million SNPs, the statistics of the SNP counts at different missing levels of M0, M5, and M10 within different filtering steps are illustrated in Table 4. A greatly reduced proportion of filtered SNPs was observed along with the QC steps at different missing levels. For example, the proportions of filtered SNPs decreased from the largest of 100%, 71.96%, and 53.21% to the smallest of 9.95%, 4.61%, and 2.96% in the M0, M5, and M10, respectively (Table 4). The notably reduced proportions of the SNP counts were seen in the rep6 replicates filtering process with proportions decreased more than 61.95%, 45.24%, and 24.44% in the M0, M5, and M10, respectively. Interestingly, the smallest reduction proportions were seen in the Blasted filter process based on the rep6 replicates filtered GL SNP results.
Table 4.
The SNP counts and each key indicator genotyping error count for different missing levels in QC steps.
Simultaneously, the key indicators of filtered SNPs in each filtering step in rep0, rep0blast, and rep0blastAnnot at F05C4 were counted according to the genotyping error label for each SNP locus. Generally, the proportion of GL decreased as the missing levels increased from M0 to M10 at F05C4 in different filtering steps. The missing allele (MA) and missing locus (ML) types always occupied the largest proportion of genotyping error types within each of the identified SNPs. For example, the two genotyping error types of MA and ML occupied the very large proportions of 73.77% and 82.17% in M5 and M10 at rep0F05C4, respectively. After the Blasted and Annotated filtering, the two genotyping error types of MA and ML remained at the very large proportions of 74.71% and 82.39% in M5 and M10 at rep0F05C4blastAnnot. Meanwhile, the proportion of GL within each identified SNP set remained stable around 25.47%~24.55% and 15.31%~15.16% in M5 and M10, respectively.
Specific allele distribution with respect to the minor allele frequency is illustrated in Figure 1 at M5C4 with rep0 (A), rep1 (B), rep6 (C), rep6blast (D), rep6blastAnnot (E), and rep6blastAnnot35bp (F). Interestingly, more rigorous filtering altered the distribution of minor allele frequency among the six datasets from similar patterns of U shape (A, B) to reverse L shape (C, D, E, F) in SNP count numbers. The reverse L shape patterns observed in the last four datasets indicated that a higher proportion of heterozygous loci were retained and more low-frequency minor alleles were removed in these SNP datasets.
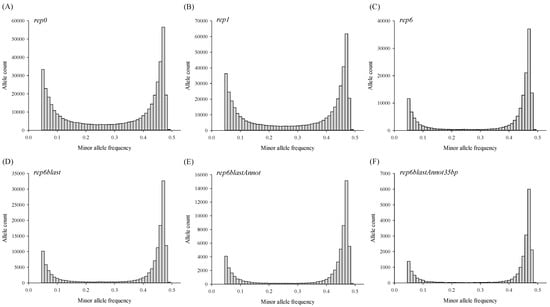
Figure 1.
The minor allele frequency distribution in different SNP datasets of rep0 (A), rep1 (B), rep6 (C), rep6blast (D), rep6blastAnnot (E), and rep6blastAnnot50bp (F) with the missing <= 5% and minimum minor genotype count ≥ 4 in 299 Masson pine samples.
3.4. Functional Analysis of SNP-Associated Contigs
A total of 265,525 SNP-Associated contigs for 6,739,240 SNPs prediction at M10F01 were blasted against the Chinese pine reference genome and there were 159,372 contigs with identity ≥ 95% and unique location in the reference genome for functional annotation. The length distributions of the searched 159,372 contigs are displayed in Figure 2A. The aligned SNP-Associated-blasted 159,372 contigs were searched against the nr (NCBI non-redundant protein sequences database) via BLAST with a minimum E-value of <1.0 × 10−6 as a similarity threshold (Table S2). There were 60,038 contigs corresponding to known protein sequences with a proportion of 37.67% annotated among the 159,372 contigs.
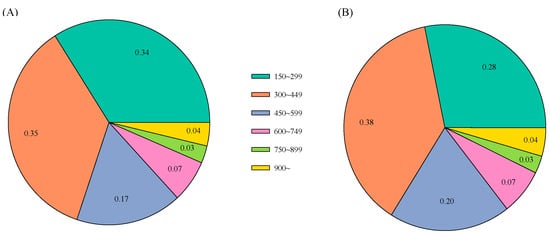
Figure 2.
The contig length distribution based on the searched 159,372 SNP-Associated-blasted contigs in the de novo assembly for reliable SNP prediction (A), the number of the 60,038 annotated SNP-Associated-blasted contigs with blasted hits (B).
A total of 17,396 contigs were searched in GO annotation with a proportion of 10.92% annotated. The functional annotations resulted in 50 GO terms (Figure S2A). These 50 GO terms were further classified into three functional categories such as cellular component (CC, 16 GO terms), molecular function (MF, 11 GO terms), and biological process (BP, 23 GO terms). Some contigs matched with more than one GO term, whereas a few matched only one GO term. The three most predominant GO subcategories in the CC category were associated with cell (category I; GO:0005623) with 3984 contigs, cell parts (category II; GO: 0044464) with 3981 contigs, and organelle (category III; GO: 0043226) with 3635 contigs. The two most predominant GO subcategories in the MF category were associated with function activity (category I, GO: 0003824) with 10,897 contigs and binding (category II, GO: 0005488) with 10,879 contigs. The two most predominant GO subcategories in the BP category were associated with metabolic process (category I; GO: 0008152) with 12,942 contigs and cellular process (category II, GO: 0009987) with 12,220 contigs.
Analysis of KEGG pathway details from annotation results showed that a total of 3694 contigs were involved in five categories and 19 subtypes (Figure S1). Based on the greatest number of contigs identified in each functional category, the largest functional category detected most often was the Metabolism category, which involved the largest number of genes and was divided into 11 subcategories. Among them, the largest two were annotated in the global and overview maps (3157 contigs) and nucleotide metabolism (1793 contigs). Another high number of genes was detected in the category of Genetic Information Processing, with the high number of 919 genes involved in transcription folding, 439 genes involved in translation folding, and so on. There were small numbers of genes detected in the other three categories of Environmental Information Processing, Cellular Processes, and Organismal Systems. Those annotated contigs would be a set of valuable genetic resources to stimulate Masson pine genomic research in the future.
3.5. Patterns of Genetic Relationship in Obtained SNP Sets
The impacts on the genetic analysis based on the obtained SNPs from different filtering scenarios were analyzed to explore the extent and influences displayed in downstream genetic analysis using different filtering scenarios. According to the original background of 299 samples from four local populations displayed in Table S1, the clustering patterns among samples were used to reveal the precise genetic background in the genetic structure analysis. The impact of missing levels in the SNPs set was evaluatedbased on the PCoA analysis using the obtained three sets of SNPs at repxblastAnnot35bp in 299 Masson pine samples with different missing levels, 0 (M0), 5% (M5), and 10% (M10). The PCoA plots resulting fromSNPs with different missing levels illustrated that the removal of all missing data from the SNPs would result in problematic problem on the relationship inference compared to the patterns inferred with missing data of 5% (M5) and 10% (M10) (Figure 3), as four more distinct clusters according to the four local sample locations were obtained in the M5 and M10 SNP sets than in the M0 SNP set. However, the cluster patterns in the M5 and M10 sets seem similar to each other.
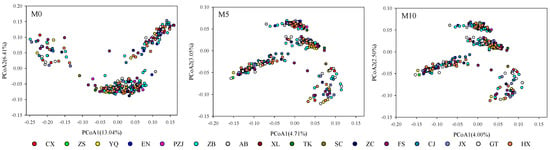
Figure 3.
PCoA plots of 299 Masson pine samples based on the SNPs with different missing levels of M0, M5, and M10. The different signals of colored circles with the related abbreviations represent the original resource of subpopulations from four local populations displayed in Table S1.
The effects of identified SNPs in downstream genetic analysis in different QC steps were illustrated in PCoA plots based on the obtained six sets of SNPs in rep0, rep1, rep6 and rep6blast, rep6blastAnnot, and rep6blastAnnot35bp at M5F05 (Figure 4A–F). The same signal of individuals in the PCoA plots represents the same original subpopulation of families which would indicate more common background retained within the original subpopulations. It was found that the open pollination of parents from four local populations in the advanced seed orchard had resulted in a heterozygous genetic background among the 293 progenies from 65 families, seen as the same color signal scattering among the four clusters in the PCoA plots. When the number of replicates increased from zero, one to six, a more reasonable four distinct gathering patterns according with the four local populations of the samples’ original sources were observed in the PCoA plots based on the obtained SNPs from the rep0 to rep6 filtering scenarios (Figure 4A–C). Four more stable clustering patterns were observed along with the QC procedure from rep0 to rep6blastAnnot35bp at M5F05. A little more overall compacted but more distinct separation among different color signals in the gathering patterns was displayed in the PCoA plots in Figure 4C–F. According to the signal clustering patterns, the PCoA plots based on SNPs in the rep6blastAnnot35bp filtering scenario displayed more useful information in revealing the heterozygous genetic background of samples, as more variations that were displayed among individuals both in the same color signals with more gathering patterns and different color signals with more distinct separation and less overlapping patterns (Figure 4F and Figure S3F). Furthermore, to display the usefulness of the QC procedure for the optimization of identified SNPs resulting from different filtering steps, the two subpopulations of YQ and ZB were taken as an example and the PCoA plot was highlighted in Figure S3 based on the same QC procedure from rep0 to rep6blastAnnot35bp at M5F05. It was found that more variations were observed with the scattering patterns within and among subpopulations than in the rep6 scenario (Figure S3C,F). The scattering trend of the highlighted samples in the YQ and ZB subpopulations developed along with the SNP sets ranged from rep6, rep6blast, rep6blastAnnot, and rep6blastAnnot35bp step by step (Figure S3C–F). Overall, those clustering patterns illustrated that it was helpful for facilitating the improvement in the reliability of SNPs to conduct the filtering strategy of blasting, annotation, and the removal of SNPs within a 35 bp distance in SNP filtering, as well as the custom filtering parameter settings.

Figure 4.
PCoA plots of 299 Masson pines based on the SNPs obtained without or with replicates in different filtering scenarios from zero to six pairs. Panels (A–F) were the PCoA plots for SNPs of rep0 (443,571) (A), rep1 (435,039) (B), rep6 (153,942) (C), rep6blast (134,611) (D), rep6blastAnnot (60,143) (E), and rep6blastAnnot35bp (20,055) (F). The different signals of colored circles with the related abbreviations represent the original resource of subpopulations from four local populations displayed in Table S1.
4. Discussion
GBS is considered as one of most cost-effective and powerful approaches to develop high-throughput SNPs for non-model tree species without a reference genome [15,16,43,44]. To date, the genetic resources for SNP discovery in Masson pine remain limited [45]. In this study, we set out to develop a set of useful genetic resources and genome-wide SNPs of Masson pine in a cost- and time-efficient manner using the GBS approach. We selected the combination of EcoRV and ScaI-HF enzymes to sample subsets of genome in Masson pine GBS to develop genetic resources and perform more accurate SNP discovery in 299 Masson pines in the absence of a reference genome. Considering the possible problem of genotyping errors in GBS, the SNP quality control tools were applied to deal with the precision and reliability of the identified SNPs by the combined QC strategies of Blasted, Annotated, technical replicates, as well as custom filtering parameter settings in SNP call rate, MAF and mC [23]. Those QC processes filtered most of the unreliable SNPs and improved the downstream genetic structure analysis illustrated with 299 individuals in PCoA analysis (Figure 4 and Figure S3). The application of GBS in 299 Masson pine samples generated 20,055 SNPs and 159,372 contigs as a set of reliable SNPs and informative genetic resources for Masson pine. The Blasted against related databases and the homologous reference genomes of Pinus taeda and Pinus tabuliformis revealed alignments with lengths of roughly 26.09Mb. The validated 60,038 functional-associated contigs can be used as informative genetic resources in Masson pine breeding. These efforts are available to stimulate more reliable, confident, and high-throughput SNP discovery in Masson pine, as well as in the tree species with large genomes, using GBS approach.
It is reported that the Pinus ssp genomes have sizes of more than 22–32 Gbp, of which more than 80% are repetitive sequences and hypermethylated [46,47]. Those repetitive sequences cause ambiguous assembly of paralogous loci and thus genotyping errors have occurred in SNP identification via GBS [19,20,21]. With the help of blasting against available genetic resources of closely relative well-assembled reference genomes [16,43,44], the precision of target contigs could be improved much to facilitate the reliability of SNP discovery in the species under study [48]. In this study, we applied two pines’ sequenced reference genomes (Pinus taeda and Pinus tabuliformis) for the correction of the obtained de novo assembly in Masson pine with identity >= 95% to ensure more accurate contigs were used for SNP discovery (Table 2). Both the two reference genomes were helpful to identify the SNPs across the genome. Our blasting with SNP-associated contigs against the sequenced Pinus tabuliformis reference genome provided more repeatability and reliability for the identification of SNPs, as well as a set of available genetic resources for Masson pine genomic research.
Peculiarities of SNPs with high missing data are a common concern in GBS application [19]. A custom choice of disregarding the missing SNPs with SNP calling rates lower than 80% across all the assayed samples was performed to deal with the missing data in GBS application. However, filtering out all the missing data from the identified SNPs dataset seemed to not be a good solution to deal with missing data for downstream analysis according to our study, as a problematic problem on the clustering pattern was observed in the M0 dataset compared to the patterns in M5 and M10 (Figure 3). The empirical data analysis in our study showed that the removal of all missing data from M5 to M0 resulted in a substantial loss of 18,414 (91.82%) reliable SNPs (20,055 vs. 1641) and a decrease of a majority proportion of 88.56% of the contigs (10,712 vs. 1225) at rep6F05C4blastAnnot35bp filtering scenario (Table 4), which indicated that the balance between the missing data and the reliable SNPs, along with the contigs genome coverage representation, should be evaluated in the SNP filtering. Nevertheless, to obtain informative SNPs, high-quality DNA must be prepared first to avoid a large proportion of missing data in tree GBS application [25].
The genotyping errors, inconsistent alleles detected with the help of paired replicates, are universally known in molecular marker development [19,20,21,24,49,50]. The use of technical replicates facilitated the detection and evaluation of genotyping errors in genetic data within different filtering steps to characterize the identified SNPs in this study. The count number based on the key indicators of the filtered SNPs in different filtering steps illustrated the detailed characteristics of identified SNPs in GBS application in Masson pine (Table 4). With the help of technical replicates, the detection and selection of GL from the identified SNPs were more feasible in GBS application. The detected genotyping errors types of MA and ML displayed high proportions of 74.14% and 82.39% in M5 and M10 at F05C4 in the rep0balstAnnot scenario, respectively (Table 4). Those genotyping errors retained in the identified SNPs would heavily bias the inference of genetic analysis [19,20]. For example, the patterns of PCoA plots based on two sets of SNPs (rep0 vs. rep6) displayed notable differences (Figure 4A,C), which prompted a great concern to address the precision and reliability of SNPs identified through the GBS approach in massive-genome tree species. Thus, great concern should be addressed to the optimization and monitoring of genotyping errors in large-genome forest tree GBS applications prior to sequencing experiment design.
Much higher genotyping error rates were observed in the obtained SNPs set in this study of Masson pine GBS application compared to GBS applications in forest tree species with small genomes [24,26]. For example, a high proportion of 47.14% genotyping errors on MA between replicates was detected at rep0M5F05C4blastAnnot in this study. However, Mastretta-Yanes et al. [24] found that only a small fluctuation between 5.9% and 8.8% of alleles were not concordantly called between replicates, based on the optional parameter settings in the de novo assembly in Berberis alpina. Another study found that a wider range of 1.96% to 22.66% genotyping error rates was observed in de novo assembly-based SNP calling with the help of replicates in Fagus sylvatica and Quercus robur L. using three reduced-genome genotyping approaches [26]. The difference between our study and these reported genotyping error rates would be due to the structure of the genomes related to genome size and genome complexity in the four forest tree species [24,26]. It is well known that the Pinus ssp have huge genomes of more than 22 GB (Chinese pine 25.4 GB vs. Loblolly pine 22.1 GB) and those genomes are filled with repetitive DNA sequences [6,47,48], while a much smaller genome compared to Pinus ssp is observed in the sequenced genome of Q. robur L. [51]. These characterizations of identified SNPs in the Masson pine GBS application provide more understanding to identify high-throughput SNPs based on GBS in non-model forest trees with a large genome.
Our analysis also focused on the reliability of identified SNPs in Masson pine and a series of quality control (QC) procedures were performed for the optimization (Table 2 and Table 4). Converting the raw GBS sequences into high-throughput SNP markers involved a number of steps, each of which contributed to the accuracy of the final genotype calls [20]. In this study, along with the QC procedures, most of the SNPs from the raw SNPs set were filtered out. For example, the count was reduced from 4,339,365 SNPs in 263,822 contigs at rep0F05 to 20,055 SNPs in 10,712 contigs at rep6F05C4blastAnnot35bp with only a small proportion of 0.46% SNPs retained (Table 2); however, more informative relationships were illustrated in the resulting small number of 20,055 SNP sets (Figure 4 and Figure S3). Obviously, the searching and filtering using different QC processes including technical replicates, Blasted and Annotated on the consistent SNPs, and reliable contigs checking had removed a majority of the unreliable SNPs and contigs from the SNPs and SNP-associated contigs; consequently, the precision and reliability of the identified SNPs in the remaining SNPs set and contigs should be much improved.
The informativeness of the resulting SNPs sets from the QC procedure was displayed in the PCoA plots according to the 299 individuals’ genetic background from four local populations. The open pollination of the sampled 65 families in the advanced seed orchard had resulted in a heterozygous genetic background, as illustrated by the scattering pattern of the same color signals in the resulting four clusters in the PCoA plots (Figure 4). More stable cluster patterns were obtained in the PCoA plots from the rep6 to rep6blastAnnot35bp filtered scenarios (Figure 4C–F). Further SNP filtering resulted in more reasonable genetic relationships of 293 individuals with more overall compacted but less overlapped patterns among different color signals of the 15 original subpopulations. The overall compacted clustering patterns along with the QC procedure displayed more similarities illustrated within each family based on the optimized SNPs. The clustering patterns in the PCoA plots with two highlighted colors of the YQ and ZB subpopulations clearly revealed more variations that were illustrated with more gathering within the same color signals and less overlapping between different color signals in the two subpopulations (Figure S3C–F). Those patterns indicated that more precise original genetic background in the sampled individuals were illustrated along with the optimization of identified SNPs.
5. Conclusions
We developed a protocol for the identification of reliable SNPs and optimized SNP-associated contigs for Masson pine via the GBS approach. The QC procedures for the precision and reliability of identified SNPs via the GBS approach were achieved by the combination of blasting the de novo assembly on available sequenced reference genomes, functional annotation, technical replicates, and 35 bp interval distance filtering, as well as the custom parameter settings on missing, minor allele frequency, and minimum minor genotype count. The use of available reference genomes and technical replicates during the generation of SNPs provided possible solutions for the mitigation of the effect of genotyping errors. The derived SNPs may have some problematic problems if no optimization was carried out on those SNPs because of the detected high genotyping error rate in Masson pine GBS in this study, which has prompted notable attention to the mitigation of genotyping errors in GBS application in large-genome forest tree species, such as the Pinus ssp. Thus, for any high-throughput sequencing data, the characteristics of raw data, assembly quality, the utilization of reference genomes, and the range of parameter values used for bioinformatics analysis should be carefully considered for precise genotyping in forest tree GBS application. However, our research is encouraging, as a continuous search for more affordable, accurate, and reliable SNP discovery in forest tree breeding for the adoption of genomic selection is possible and may yield more accurate prediction in molecular forest tree breeding. These findings facilitate Masson pine genomic research and provide more understanding of the characteristics of high-throughput SNP discovery via the GBS approach in forest tree species, especially in large-genome conifer tree species.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/f14020387/s1, Table S1: List of information about collection of the 305 samples in this study; Table S2: BLAST results of 159,372 SNP-associated contigs against the Chinese pine reference genome with identity ≥ 95% and unique location; Table S3: The identified 20,055 SNPs set; Table S4: BLAST results of SNP-associated contigs compared with the non-redundant (nr) protein database; Table S5: Gene Ontology (GO) annotation results from the 159,372 SNP-associated contigs; Table S6: The Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation results based on the 159,372 SNP-associated contigs; Figure S1. Workflow for the application of genotyping-by-sequencing in combination with technical replicates filtering and homologous reference genome blasted and validated for reliable, informative SNPs and contigs in the development of genomic resources in Masson pine genomic research; Figure S2. Gene Ontology classification of SNP-associated contigs identified from GBS in Masson pine (A), KEGG Pathway details of annotated contigs in Masson pine (B); Figure S3. Highlighting of YQ and ZB subpopulations in the PCoA plots of 299 Masson pines based on the SNPs obtained without or with replicates in different filtering scenarios from zero to six pairs. Panels (A) to (F) were the PCoA plots for SNPs of rep0 (443,571) (A), rep1 (435,039) (B), rep6 (153,942) (C), rep6blast (134,611) (D), rep6blastAnnot (60,143) (E), and rep6blastAnnot35bp (20,055) (F).
Author Contributions
Conceptualization, M.-H.Y.; methodology, M.-H.Y. and P.-L.L.; software, X.-L.J. and H.-L.D.; validation, F.-L.Z., Q.-Y.X. and W.W.; formal analysis, P.-L.L., M.-H.Y., X.-L.J. and H.X.; investigation, P.-L.L., X.-L.J., H.X. and F.-L.Z.; resources, Y.-H.H. and N.-Q.L.; data curation, M.-H.Y., Y.-H.H. and N.-Q.L.; writing—original draft preparation, P.-L.L. and M.-H.Y.; writing—review and editing, P.-L.L., M.-H.Y. and Y.-H.H.; visualization, P.-L.L., F.-L.Z., Q.-Y.X. and W.W.; supervision, M.-H.Y. and Y.-H.H.; project administration, M.-H.Y.; funding acquisition, M.-H.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by grants from the National Natural Science Foundation of China (Grant No. 32171825, 31670678) to M.-H.Y., the Postgraduate Scientific Research Innovation Project of Hunan (Grant No. CX20210888) to P.-L.L. and the Forestry Programs of Science and Technology in Fujian Province (Grant No. 201501) to Y.-H.H.
Institutional Review Board Statement
Not applicable. As our studies are not involving humans or animals.
Data Availability Statement
The GBS raw sequence data were deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in the National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA009032), and are publicly accessible at https://ngdc.cncb.ac.cn/gsa, available after 13 November 2023; https://doi.org/10.6084/m9.figshare.c.6426587.v1. The samples analyzed may be available upon request after a share transfer agreement. The datasets generated during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
The authors thank Xiao-Feng Tan, De-Yi Yuan, and Hong-Xu Long in the Key Laboratory of Cultivation and Protection for Non-Wood Forest Trees, Central South University of Forestry and Technology, for their support with the experimental apparatus. The authors also thank Bing-Zhang Zou and Jing-Quan Lin in the Shanghang Baisha State-Owned Forest Farm in Fujian province, for their support with the experimental material resources.The authors also thank the staff members of LC-BIO Co., Ltd. (Hangzhou, China) for their sequencing for our study on the Illumina sequencing platform.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ding, G.; Zhou, Z.; Wang, Z. Cultivation and Utilization of Masson Pine Pulpwood Forest; China Forestry Publishing House: Beijing, China, 2006; pp. 1–10. [Google Scholar]
- Grattapaglia, D. Twelve years into genomic selection in forest trees: Climbing the slope of enlightenment of marker assisted tree breeding. Forests 2022, 13, 1554. [Google Scholar] [CrossRef]
- Grattapaglia, D.; Silva-Junior, O.B.; Resende, R.T.; Cappa, E.P.; Müller, B.S.F.; Tan, B.; Isik, F.; Ratcliffe, B.; El-Kassaby, Y.A. Quantitative genetics and genomics converge to accelerate forest tree breeding. Front. Plant Sci. 2018, 9, 1693. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Birol, I.; Bousquet, J.; Ingvarsson, P.K.; Jansson, S.; Jones, S.J.M.; Keeling, C.I.; MacKay, J.; Nilsson, O.; Ritland, K.; et al. Insights into conifer giga-genomes. Plant Physiol. 2014, 166, 1724–1732. [Google Scholar] [CrossRef]
- Neale, D.B.; Wegrzyn, J.L.; A Stevens, K.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; E Holtz-Morris, A.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Li, J.; Bo, W.; Yang, W.; Zuccolo, A.; Giacomello, S.; Chen, X.; Han, F.; Yang, J.; Song, Y.; et al. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 2022, 185, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xie, Y.; Liu, B.; Zhou, Z.; Feng, Z.; Chen, Y. A transcriptomic variation map provides insights into the genetic basis of Pinus massoniana Lamb. evolution and the association with oleoresin yield. BMC Plant Biol. 2020, 20, 1–14. [Google Scholar] [CrossRef]
- Bai, Q.; Cai, Y.; He, B.; Liu, W.; Pan, Q.; Zhang, Q. Core set construction and association analysis of Pinus massoniana from Guangdong province in southern China using SLAF-seq. Sci. Rep. 2019, 9, 13157. [Google Scholar] [CrossRef]
- Kastally, C.; Niskanen, A.K.; Perry, A.; Kujala, S.T.; Avia, K.; Cervantes, S.; Haapanen, M.; Kesälahti, R.; Kumpula, T.A.; Mattila, T.M.; et al. Taming the massive genome of Scots pine with PiSy50k, a new genotyping array for conifer research. Plant. J. 2022, 109, 1337–1350. [Google Scholar] [CrossRef]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 123, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Jahner, J.P.; Uckele, K.A.; Galland, L.M.; Eckert, A.J. RADseq approaches and applications for forest tree genetics. Tree Genet. Genomes 2018, 14, 39. [Google Scholar] [CrossRef]
- Karam, M.J.; Lefèvre, F.; Dagher-Kharrat, M.B.; Pinosio, S.; Vendramin, G. Genomic exploration and molecular marker development in a large and complex conifer genome using RADseq and mRNAseq. Mol. Ecol. Resour. 2015, 15, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Clugston, J.A.; Kenicer, G.J.; Milne, R.; Overcast, I.; Wilson, T.C.; Nagalingum, N.S. RADseq as a valuable tool for plants with large genomes—A case study in cycads. Mol. Ecol. Resour. 2019, 19, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.; Zhao, W.; Wennström, U.; Gull, B.A.; Wang, X.-R. Parentage and relatedness reconstruction in Pinus sylvestris using genotyping-by-sequencing. Heredity 2020, 124, 633–646. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Li, X.; Ling, S.; Fu, Y.-X.; Hungate, E.; Shi, S.; Wu, C.-I. Estimating DNA polymorphism from next generation sequencing data with high error rate by dual sequencing applications. BMC Genom. 2013, 14, 1–9. [Google Scholar] [CrossRef]
- O'Leary, S.J.; Puritz, J.B.; Willis, S.C.; Hollenbeck, C.M.; Portnoy, D.S. These aren’t the loci you’e looking for: Principles of effective SNP filtering for molecular ecologists. Mol. Ecol. 2018, 27, 3193–3206. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2012, 13, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Pettersson, F.H.; Clarke, G.M.; Cardon, L.R.; Morris, A.P.; Zondervan, K.T. Data quality control in genetic case-control association studies. Nat. Protoc. 2010, 5, 1564–1573. [Google Scholar] [CrossRef]
- Pavan, S.; Delvento, C.; Ricciardi, L.; Lotti, C.; Ciani, E.; D’Agostino, N. Recommendations for choosing the genotyping method and best practices for quality control in crop genome-wide association studies. Front. Genet. 2020, 11, 447. [Google Scholar] [CrossRef]
- Mastretta-Yanes, A.; Arrigo, N.; Alvarez, N.; Jorgensen, T.H.; Piñero, D.; Emerson, B.C. Restriction site-associated DNA sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Mol. Ecol. Resour. 2015, 15, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-B.; Cober, E.R.; Morrison, M.J.; Marsolais, F.; Peterson, G.W.; Horbach, C. Patterns of genetic variation in a soybean germplasm collection as characterized with genotyping-by-sequencing. Plants 2021, 10, 1611. [Google Scholar] [CrossRef] [PubMed]
- Ulaszewski, B.; Meger, J.; Burczyk, J. Comparative analysis of SNP discovery and genotyping in Fagus sylvatica L. and Quercus robur L. using RADseq, GB.S.; and ddRAD methods. Forests 2021, 12, 222. [Google Scholar] [CrossRef]
- Yang, M.-H.; Li, Z.; Zhang, D.; Tang, X.; Zhang, B.; Zhang, D. DNA Isolation from Pinus massoniana Needles. J. Central South Univ. For. Tech. 2008, 28, 39–44. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Zimin, A.V.; Stevens, K.A.; Crepeau, M.; Puiu, D.; Wegrzyn, J.; Yorke, J.A.; Langley, C.H.; Neale, D.B.; Salzberg, S.L. An improved assembly of the loblolly pine mega-genome using long-read single-molecule sequencing. Gigascience 2017, 6, giw016. [Google Scholar]
- Ye, J.; McGinnis, S.; Madden, T.L. BLAST: Improvements for better sequence analysis. Nucleic. Acids. Res. 2006, 34, W6–W9. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 2014, 15, 1–13. [Google Scholar] [CrossRef]
- Peterson, G.W.; Dong, Y.; Horbach, C.; Fu, Y.-B. Genotyping-by-sequencing for plant genetic diversity analysis: A lab guide for SNP genotyping. Diversity 2014, 6, 665–680. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 27, 29–34. [Google Scholar] [CrossRef]
- Yang, M.-H.; Fu, Y.B. AveDissR: An R function for assessing genetic distinctness and genetic redundancy. Appl. Plant Sci. 2017, 5, 1700018. [Google Scholar] [CrossRef]
- Core, R.; Rdct, R.; Team, R.; Team, R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: http://www.R-project.org/ (accessed on 3 March 2022).
- Chen, C.; Mitchell, S.E.; Elshire, R.J.; Buckler, E.S.; El-Kassaby, Y.A. Mining conifers’ mega-genome using rapid and efficient multiplexed high-throughput genotyping-by-sequencing (GBS) SNP discovery platform. Tree Genet. Genomes. 2013, 9, 1537–1544. [Google Scholar] [CrossRef]
- Pan, J.; Wang, B.; Pei, Z.Y.; Zhao, W.; Gao, J.; Mao, J.F.; Wang, X.R. Optimization of the genotyping-by-sequencing strategy for population genomic analysis in conifers. Mol. Ecol. Resour. 2015, 15, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.; Christie, N.; Reynolds, S.M.; Marais, G.C.; Tii-Kuzu, Y.; Caballero, M.; Kampman, T.; Visser, E.A.; Naidoo, S.; Kain, D.; et al. A genome-wide SNP genotyping resource for tropical pine tree species. Mol. Ecol. Resour. 2022, 22, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.L.; Liechty, J.D.; Stevens, K.A.; Wu, L.S.; Loopstra, C.A.; Vasquez-Gross, H.A.; Dougherty, W.M.; Lin, B.Y.; Zieve, J.J.; Martínez-García, P.J.; et al. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics 2014, 196, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, O.; Burke, P.; Arkhipov, A.; Kuchma, N.; James, S.J.; Kovalchuk, I.; Pogribny, I. Genome hypermethylation in Pinus silvestris of Chernobyl: A mechanism for radiation adaptation? Mutat Res-Fund Mol. M. 2003, 529, 13–20. [Google Scholar] [CrossRef]
- Borthakur, D.; Busov, V.; Cao, X.H.; Du, Q.; Gailing, O.; Isik, F. Current status and trends in forest genomics. For. Res. 2022, 2, 11. [Google Scholar] [CrossRef]
- Bresadola, L.; Link, V.; Buerkle, C.A.; Lexer, C.; Wegmann, D. Estimating and accounting for genotyping errors in RAD-seq experiments. Mol. Ecol. Resour. 2020, 20, 856–870. [Google Scholar] [CrossRef]
- Pompanon, F.; Bonin, A.; Bellemain, E.; Taberlet, P. Genotyping errors: Causes, consequences and solutions. Nat. Rev. Genet. 2005, 6, 847–859. [Google Scholar] [CrossRef]
- Sork, V.L.; Fitz-Gibbon, S.T.; Puiu, D.; Crepeau, M.; Gugger, P.F.; Sherman, R.; Stevens, K.; Langley, C.H.; Pellegrini, M.; Salzberg, S.L. First draft assembly and annotation of the genome of a California endemic oak Quercus lobata Née (Fagaceae). G3 Genes Genomes Genet. 2016, 6, 3485–3495. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).