
Pan-Transcriptome Analysis of Willow Species from Diverse Geographic Distributions

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Transcriptome Sequencing
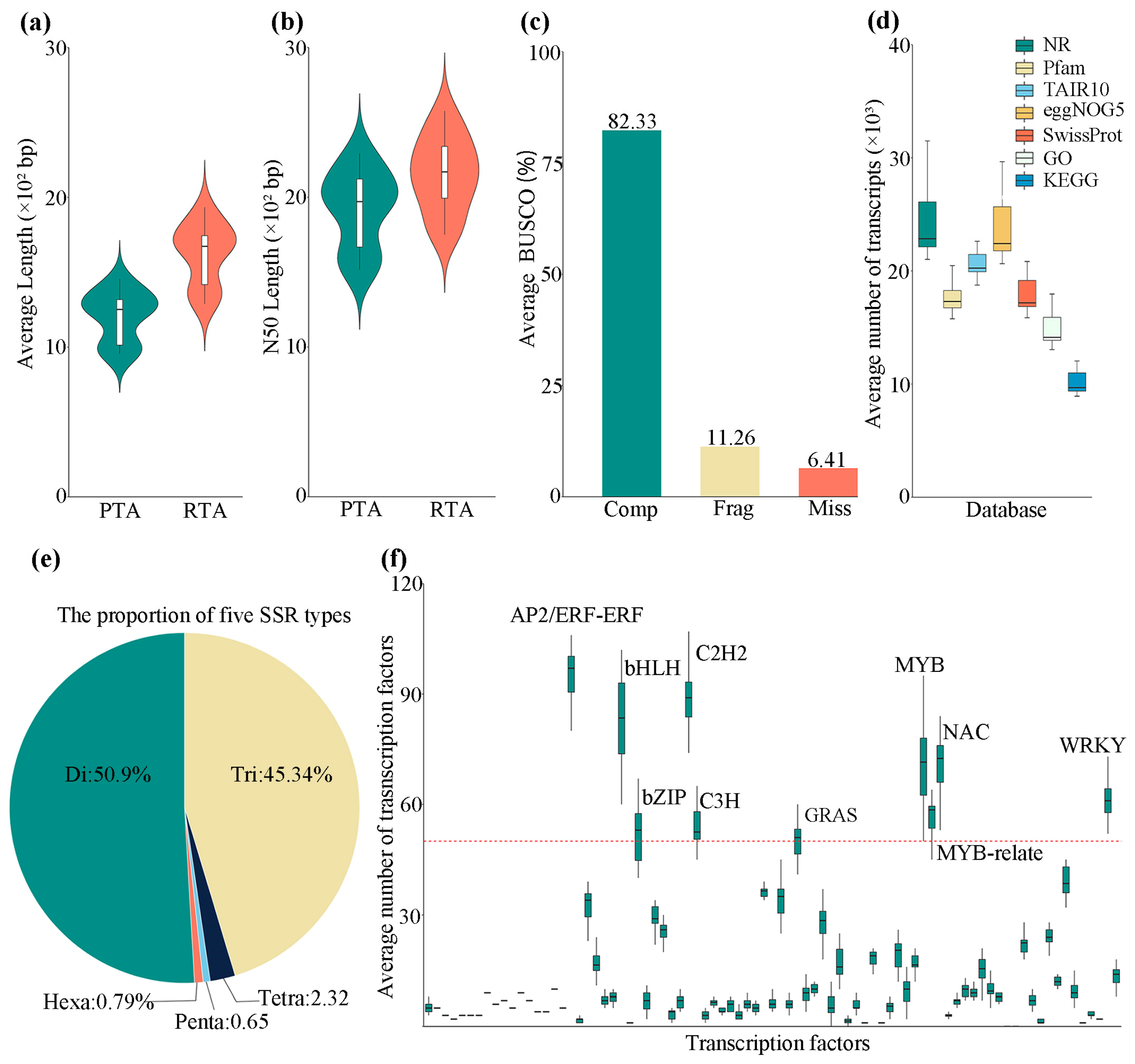
2.2. Transcriptome Assembly and Completeness Assessment
2.3. Functional Annotation of Transcripts
2.4. SSR and TF Identification
2.5. Gene Family Clustering and Evolutionary Analysis
2.6. Identification of One-to-One Orthologous Genes and Expression Analysis
3. Results
3.1. De Novo Assembly and Annotation of Willow Transcriptomes
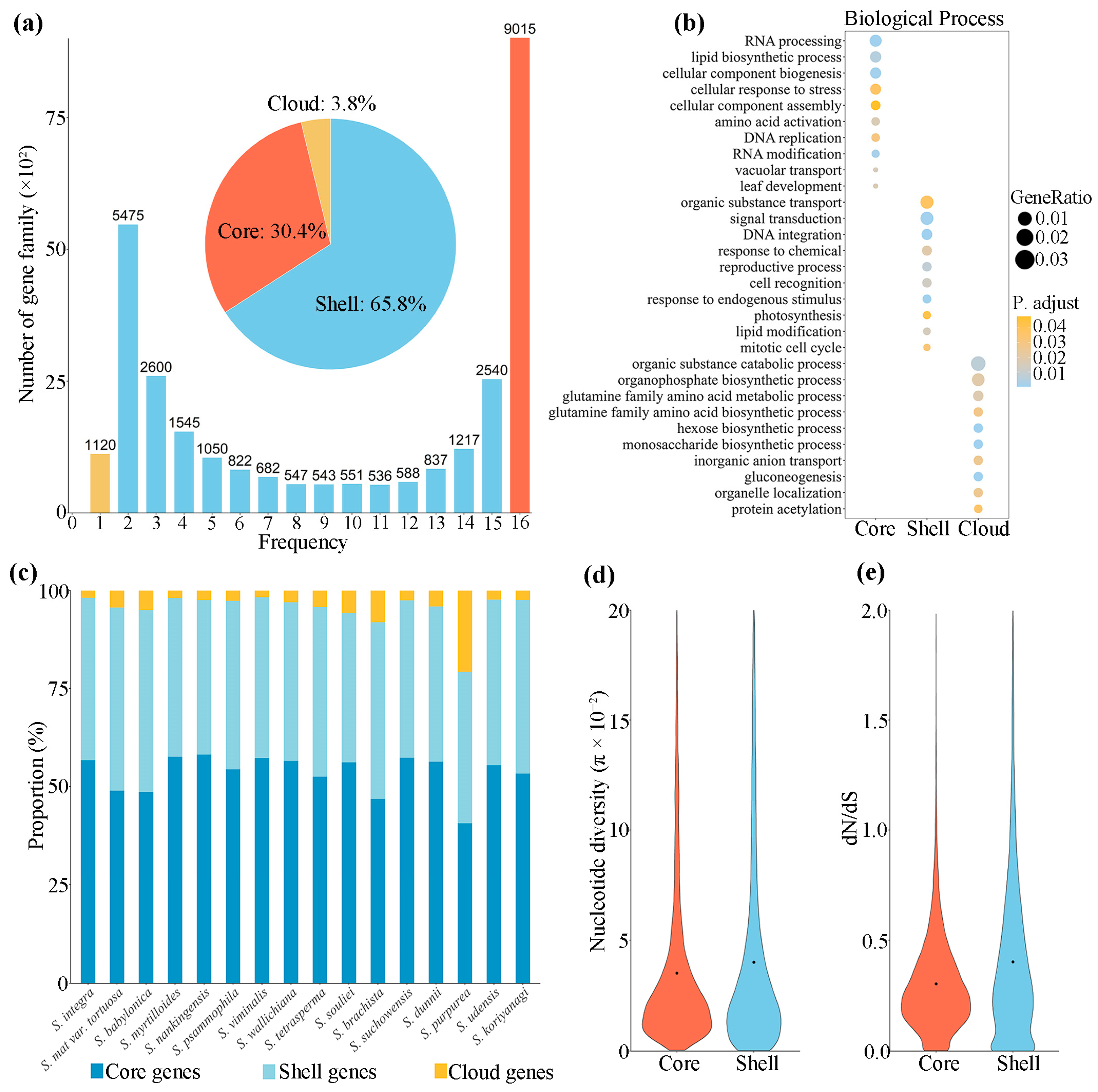
3.2. Characterization of Willow Pan-Transcriptome
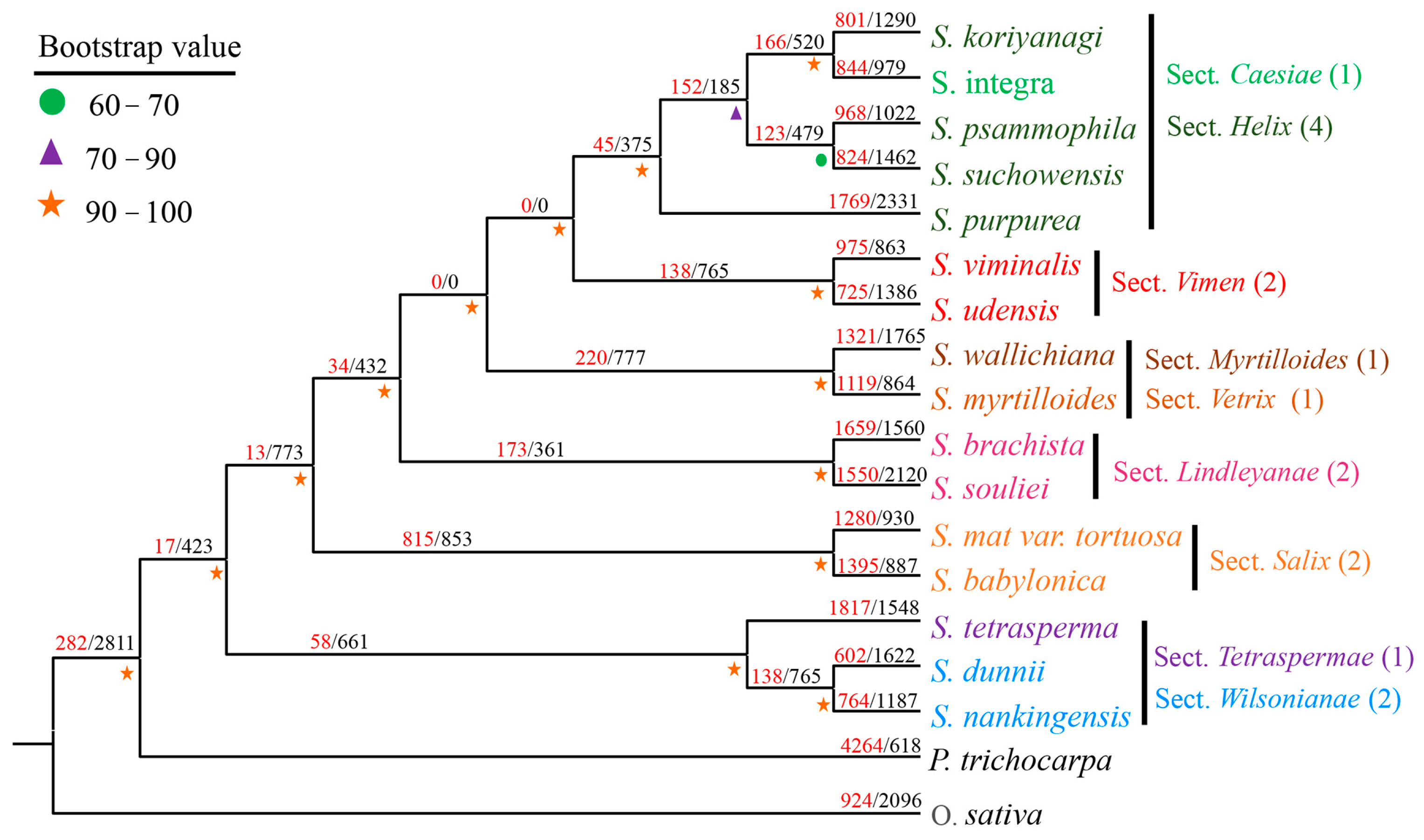
3.3. Phylogenetic Analysis of Willow Species
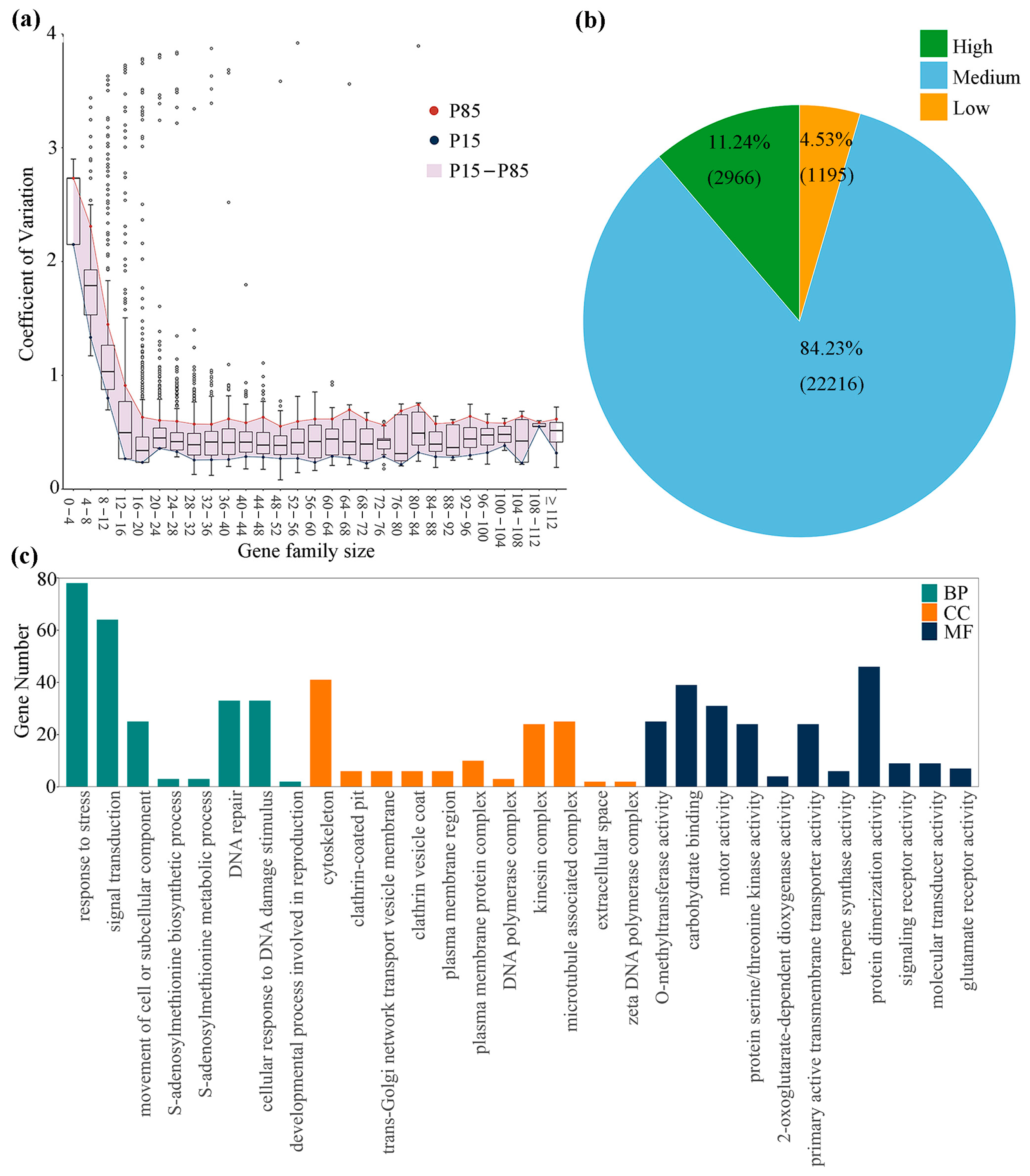
3.4. Variation in Gene Family Size among Willow Species
3.5. Clustering of Gene Families Based on Family Size
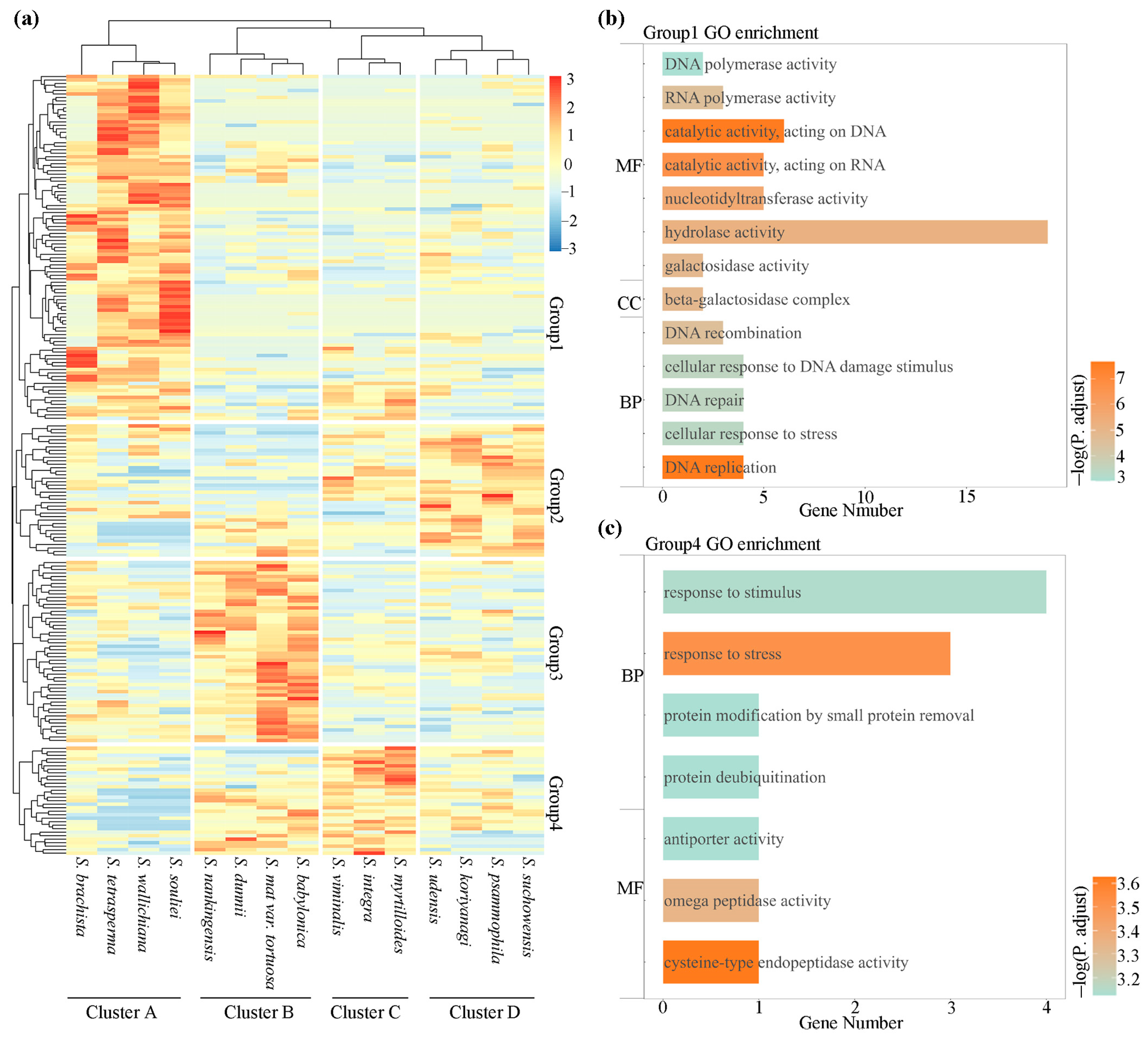
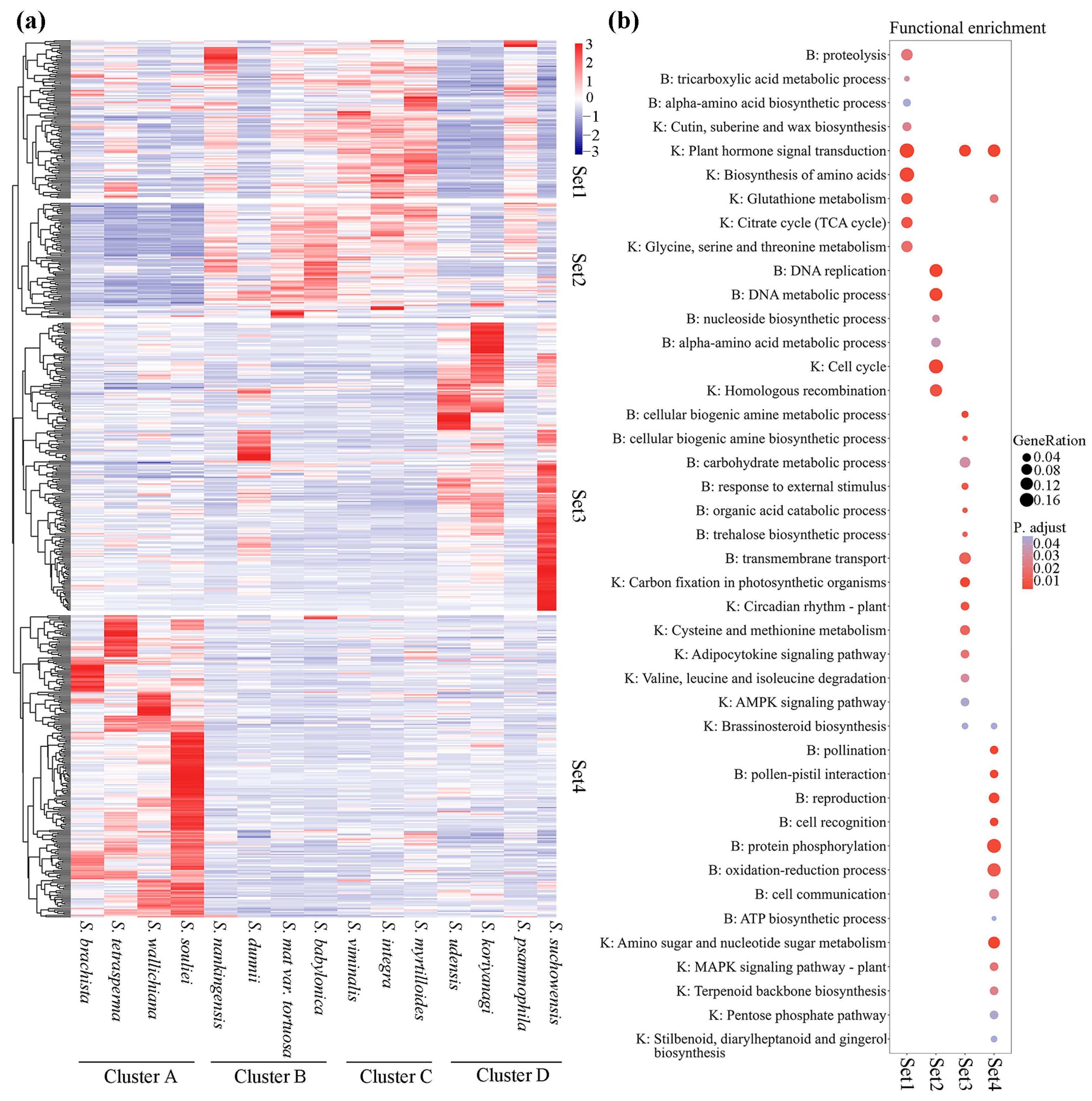
3.6. Expression Patterns of Orthologous Genes in Willow Species
4. Discussion
4.1. High-Quality Willow Pan-Transcriptome
4.2. Systematic and Taxonomic Significance of Salix
4.3. Potential Role of Gene Family Expansion in Environmental Adaptation of Willows
4.4. Relationship between Geographical Characteristics and Expression Patterns of Orthologs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Argus, G.W. The genus Salix (Salicaceae) in southeastern United States. Am. Soc. Plant Taxon. 1986, 9, 1–170. [Google Scholar] [CrossRef]
- Tawfeek, N.; Mahmoud, M.F.; Hamdan, D.I.; Sobeh, M.; Farrag, N.; Wink, M.; El-Shazly, A.M. Phytochemistry, pharmacology and medicinal uses of plants of the genus Salix: An updated review. Front. Pharmacol. 2021, 12, 593856. [Google Scholar] [CrossRef] [PubMed]
- Flora of North America Editorial Commite (Ed.) Flora of North America North of Mexico, Vol 7; Oxford University Press: New York, NY, USA, 2010. [Google Scholar]
- Argus, G.W. Infrageneric classification of Salix (Salicaceae) in the new world. Syst. Bot. Monogr. 1997, 52, 1–121. [Google Scholar] [CrossRef]
- Skvortsov, A.K. Willows of Russia and Adjacent Countries: Taxonomical and Geographical Revision; University of Joensuu: Joensuu, Finland, 1999. [Google Scholar]
- Warmiński, K.; Stolarski, M.J.; Gil, Ł.; Krzyżaniak, M. Willow bark and wood as a source of bioactive compounds and bioenergy feedstock. Ind. Crops Prod. 2021, 171, 113976. [Google Scholar] [CrossRef]
- Keoleian, G.A.; Volk, T.A. Renewable energy from willow biomass crops: Life cycle energy, environmental and economic performance. BPTS 2005, 24, 385–406. [Google Scholar] [CrossRef]
- Kuzovkina, Y.A.; Martin, W.; Marta, A.R.; John, C.; Sarah, H.; Ian, M.; Angelas, K.; Sviatlana, T.; Michel, L. Salix: Botany and Global Horticulture. Hortic. Rev. 2007, 34, 447–489. [Google Scholar] [CrossRef]
- Richardson, J.; Isebrands, J.G.; Ball, J.B. Ecology and physiology of poplars and willows. In Poplars and Willows: Trees for Society and the Environment; Richardson, J., Isebrands, J.G., Eds.; The Food and Agriculture Organization of United Nations and CABI: Rome, Italy, 2014; pp. 92–123. [Google Scholar] [CrossRef]
- Suda, Y.; Argus, G.W. Chromosome numbers of some north American Salix. Brittonia 1968, 20, 191–197. [Google Scholar] [CrossRef]
- Barcaccia, G.; Meneghetti, S.; Lucchin, M.; de-Jong, H. Genetic segregation and genomic hybridization patterns support an allotetraploid structure and disomic inheritance for Salix species. Diversity 2014, 6, 633–651. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Yang, Y.; Yin, T. The chromosome-scale assembly of the willow genome provides insight into Salicaceae genome evolution. Hortic. Res. 2020, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Almeida, P.; Proux-Wera, E.; Churcher, A.; Soler, L.; Dainat, J.; Pucholt, P.; Nordlund, J.; Martin, T.; Ronnberg-Wastljung, A.C.; Nystedt, B.; et al. Genome assembly of the basket willow, Salix viminalis, reveals earliest stages of sex chromosome expansion. BMC Biol. 2020, 18, 78. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, H.; Li, Y.; Chen, Y.; Liu, G.; Ye, M.; Yu, C.; Lian, B.; Zhong, F.; Jiang, Y.; et al. Genome sequencing and phylogenetic analysis of allotetraploid Salix matsudana Koidz. Hortic. Res. 2020, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Jia, K.H.; Zhang, R.G.; Wang, Y.; Shi, T.L.; Li, Z.C.; Zeng, S.W.; Cai, X.J.; Wagner, N.D.; Horandl, E.; et al. Chromosome-scale assembly of the genome of Salix dunnii reveals a male-heterogametic sex determination system on chromosome 7. Mol. Ecol. Resour. 2021, 21, 1966–1982. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Huang, Y.; Brachi, B.; Yun, Q.Z.; Zhang, W.; Lu, W.; Li, H.N.; Li, W.Q.; Sun, X.D.; Wang, G.Y.; et al. Genome-wide analysis of Cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nat. Commun. 2019, 10, 5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Marroni, F.; Pinosio, S.; Morgante, M. Structural variation and genome complexity: Is dispensable really dispensable? Curr. Opin. Plant Biol. 2014, 18, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Chai, S.; Yang, Z.; Zhang, Q.; Xin, H.; Xu, Y.; Lin, S.; Chen, X.; Yao, Z.; et al. Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 2022, 13, 682. [Google Scholar] [CrossRef]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Luo, H.; Xu, J.; Cruickshank, A.; Zhao, X.; Teng, F.; Hathorn, A.; Wu, X.; Liu, Y.; Shatte, T.; et al. Extensive variation within the pan-genome of cultivated and wild sorghum. Nat. Plants 2021, 7, 766–773. [Google Scholar] [CrossRef]
- Gonzalez, E.; Brereton, N.J.; Marleau, J.; Guidi Nissim, W.; Labrecque, M.; Pitre, F.E.; Joly, S. Meta-transcriptomics indicates biotic cross-tolerance in willow trees cultivated on petroleum hydrocarbon contaminated soil. BMC Plant Biol. 2015, 15, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, N.; Fan, L.; Cao, Y.; Ling, H.; Xu, G.; Zhou, J.; Chen, Q.; Tao, J. Comparison of two willow genotypes reveals potential roles of iron-regulated transporter 9 and heavy-metal ATPase 1 in cadmium accumulation and resistance in Salix suchowensis. Ecotoxicol. Environ. Saf. 2022, 244, 114065. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, D.G.; Taskiran, B.; Carlson, C.H.; Smart, L.B. Mapping the sex determination region in the Salix F1 hybrid common parent population confirms a ZW system in six diverse species. G3 2022, 12, jkac071. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazutaka, K.; Kazuharu, M.; Kei-ichi, K.; Takashi, M. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Michael, J.S. R8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinform. Appl. Note 2003, 19, 301–302. [Google Scholar]
- De-Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.P.; Contreras-Moreira, B.; Woods, D.P.; Des Marais, D.L.; Burgess, D.; Shu, S.Q.; Stritt, C.; Roulin, A.C.; Schackwitz, W.; Tyler, L.; et al. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat. Commun. 2017, 8, 2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcaccia, G.; Meneghetti, S.; Albertini, E.; Triest, L.; Lucchin, M. Linkage mapping in tetraploid willows: Segregation of molecular markers and estimation of linkage phases support an allotetraploid structure for Salix alba x Salix fragilis interspecific hybrids. Heredity 2003, 90, 169–180. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Liu, X.Y.; Guo, R.; Hu, K.R.; Cao, Y.; Dai, F. Comparative genomics and transcriptomics analysis reveals evolution patterns of selection in the Salix phylogeny. BMC Genom. 2019, 20, 253. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, A.; Shah, F.S.; Azhar, J.; Ahmad, S.; John, P. Pan-transcriptomics and its applications. In Pan-Genomics: Applications, Challenges, and Future Prospects; Soares, S.C., Sandip, T., Debmalya, B., Vasco, A., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 343–356. [Google Scholar] [CrossRef]
- Pucholt, P.; Sjodin, P.; Weih, M.; Ronnberg-Wastljung, A.C.; Berlin, S. Genome-wide transcriptional and physiological responses to drought stress in leaves and roots of two willow genotypes. BMC Plant Biol. 2015, 15, 244. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.M.; Yang, Q.; Xu, H.; Liu, Y.J.; Yang, H.L. Physiological and transcriptomic analysis provide novel insight into cobalt stress responses in willow. Sci. Rep. 2020, 10, 2308. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.P.; Li, J.Y.; Zhu, T.H.; Yang, H.J.; Zhuo, Z.H. Comparative transcriptome analysis of Salix cupularis under drought stress. Glob. Ecol. Conserv. 2021, 27, e01532. [Google Scholar] [CrossRef]
- Yanitch, A.; Brereton, N.J.B.; Gonzalez, E.; Labrecque, M.; Joly, S.; Pitre, F.E. Transcriptomic response of purple willow (Salix purpurea) to arsenic stress. Front. Plant Sci. 2017, 8, 1115. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Wang, F.; Wu, M.; Wang, S. ddRAD Sequencing-Based Scanning of Genetic Variants in Sargassum fusiforme. J. Mar. Sci. Eng. 2022, 10, 958. [Google Scholar] [CrossRef]
- Verma, P.; Tiwari, P.; Singh, R.; Raghubanshi, A.S. Effect of rainfall variability on tree phenology in moist tropical deciduous forests. Environ. Monit. Assess. 2022, 194, 537. [Google Scholar] [CrossRef]
- Barkalov, V.Y.; Kozyrenko, M.M. Phylogenetic relationships of Salix L. subg. Salix species (Salicaceae) according to sequencing data of intergenic spacers of the chloroplast genome and ITS rDNA. Russ. J. Genet. 2014, 50, 828–837. [Google Scholar] [CrossRef]
- Zhou, J.; Jiao, Z.; Guo, J.; Wang, B.S.; Zheng, J. Complete chloroplast genome sequencing of five Salix species and its application in the phylogeny and taxonomy of the genus. Mitochondrial DNA B Resour. 2021, 6, 2348–2352. [Google Scholar] [CrossRef] [PubMed]
- Hardig, T.M.; Anttila, C.K.; Brunsfeld, S.J. A phylogenetic analysis of Salix (Salicaceae) based on matk and ribosomal DNA sequence data. J. Bot. 2010, 2010, 197696. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Zhao, S.; Skvortsov, A.K. Salicaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press & Missouri Botanical Garden Press: Beijing, China, 1999. [Google Scholar]
- Li, M.M.; Wang, D.Y.; Zhang, L.; Kang, M.H.; Lu, Z.Q.; Zhu, R.B.; Mao, X.X.; Xi, Z.X.; Tao, M. Intergeneric relationships within the family Salicaceae s.l. based on plastid phylogenomics. Int. J. Mol. Sci. 2019, 20, 3788. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.L. Gene family evolution in green plants with emphasis on the origination and evolution of Arabidopsis thaliana genes. Plant J. 2013, 73, 941–951. [Google Scholar] [CrossRef]
- Zmienko, A.; Samelak, A.; Kozlowski, P.; Figlerowicz, M. Copy number polymorphism in plant genomes. Theor. Appl. Genet. 2014, 127, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Carretero-Paulet, L.; Librado, P.; Chang, T.H.; Ibarra-Laclette, E.; Herrera-Estrella, L.; Rozas, J.; Albert, V.A. High gene family turnover rates and gene space adaptation in the compact genome of the carnivorous plant Utricularia gibba. Mol. Biol. Evol. 2015, 32, 1284–1295. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Moore, B.M.; Panchy, N.L.; Meng, F.; Lehti-Shiu, M.D.; Shiu, S.H. Factors influencing gene family size variation among related species in a plant family, Solanaceae. Genome Biol. Evol. 2018, 10, 2596–2613. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Guan, Y.; Qiong, L.; Lu, S.; An, M.; Crabbe, M.J.C.; Qi, J.; Zhao, F.; Qiao, Q.; Zhang, T. Genomic analysis of field pennycress (Thlaspi arvense) provides insights into mechanisms of adaptation to high elevation. BMC Biol. 2021, 19, 143. [Google Scholar] [CrossRef]
- Xu, L.; Cao, M.; Wang, Q.; Xu, J.; Liu, C.; Ullah, N.; Li, J.; Hou, Z.; Liang, Z.; Zhou, W.; et al. Insights into the plateau adaptation of Salvia castanea by comparative genomic and WGCNA analyses. J. Adv. Res. 2022, 42, 221–235. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Landis, J.B.; Shen, J.; Zhang, H.; Kuang, T.; Sun, W.; Sun, J.; Tiamiyu, B.B.; Deng, T.; et al. Transcriptomes of Saussurea (Asteraceae) provide insights into high-altitude adaptation. Plants 2021, 10, 1715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Qiao, Q.; Novikova, P.Y.; Wang, Q.; Yue, J.; Guan, Y.; Ming, S.; Liu, T.; De, J.; Liu, Y.; et al. Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc. Natl. Acad. Sci. USA 2019, 116, 7137–7146. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.F.; Yu, Y.; Wen, J.; Huang, J.; Chen, J.P.; Li, J.; Zhou, S.D.; He, X.J. Phylogeny and highland adaptation of Chinese species in Allium section Daghestanica (Amaryllidaceae) revealed by transcriptome sequencing. Mol. Phylogenet. Evol. 2020, 146, 106737. [Google Scholar] [CrossRef]
- Lai, Z.F.; Yao, Y.F.; Lin, B.Z.; Lian, D.M.; Hong, J.J. Analysis of transcriptome response to low temperature stress in mesem-bryanthemum crystallinum Linn. Mol. Plant Breed. 2021, 19, 7348–7358. [Google Scholar] [CrossRef]
- Niu, Z.X.; Liu, L.J.; Pu, Y.Y.; Ma, L.; Wu, J.Y.; Hu, F.D.; Fang, Y.; Li, X.C.; Sun, W.C.; Wang, W.T.; et al. iTRAQ-based quantitative proteome analysis insights into cold stress of Winter Rapeseed (Brassica rapa L.) grown in the field. Sci. Rep. 2021, 11, 23434. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, Q.; Zhang, R.; Liu, M.; Wang, C.; Liu, Z.; Xiang, C.; Lu, X.; Zhang, X.; Li, X.; et al. Rootstock-scion exchanging mRNAs participate in the pathways of amino acids and fatty acid metabolism in cucumber under early chilling stress. Hortic. Res. 2022, 9, uhac031. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.X.; Yan, B.Y.; Hu, J.; Yang, C.P.; Li, Y.J.; Liu, J.P.; Liao, W.B. Transcriptome profiling of rubber tree (Hevea brasiliensis) discovers candidate regulators of the cold stress response. Genes Genom. 2018, 40, 1181–1197. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Section | Number of Reads | Assembled Transcript Base (bp) | Number of Transcripts | Average Length (bp) | N50 (bp) |
|---|---|---|---|---|---|---|
| S. integra a | Caesiae | 90,289,300 | 38,665,408 | 22,693 | 1704 | 2131 |
| S. matsudana a | Salix | 78,431,900 | 41,673,650 | 29,192 | 1428 | 2003 |
| S. babylonica a | Salix | 80,092,914 | 41,933,688 | 30,278 | 1385 | 1966 |
| S. myrtilloides a | Myrtilloides | 77,083,220 | 39,406,201 | 23,055 | 1709 | 2102 |
| S. nankingensis a | Wilsonianae | 76,977,414 | 40,332,554 | 21,839 | 1847 | 2333 |
| S. psammophila a | Helix | 79,908,594 | 40,737,203 | 24,351 | 1673 | 2208 |
| S. viminalis a | Vimen | 79,311,062 | 38,145,278 | 22,829 | 1671 | 2054 |
| S. wallichiana | Vetrix | 53,957,578 | 31,552,157 | 23,622 | 1336 | 1799 |
| S. tetrasperma | Tetraspermae | 53,257,842 | 34,366,064 | 26,654 | 1289 | 1751 |
| S. souliei | Lindleyanae | 52,893,684 | 31,923,835 | 23,807 | 1341 | 1806 |
| S. brachista | Lindleyanae | 122,921,910 | 48,523,659 | 29,540 | 1643 | 2421 |
| S. suchowensis | Helix | 96,017,502 | 40,259,406 | 22,317 | 1804 | 2357 |
| S. dunnii | Wilsonianae | 89,665,696 | 42,538,906 | 22,000 | 1934 | 2576 |
| S. purpurea | Helix | 157,916,440 | 60,455,260 | 33,295 | 1816 | 2499 |
| S. udensis | Vimen | 88,904,158 | 39,661,865 | 23,004 | 1724 | 2242 |
| S. koriyanagi | Helix | 88,098,190 | 39,836,066 | 23,808 | 1673 | 2223 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Z.; Chen, L.; Guo, Y.; Dai, X.; Yin, T.; Xue, L. Pan-Transcriptome Analysis of Willow Species from Diverse Geographic Distributions. Forests 2023, 14, 1182. https://doi.org/10.3390/f14061182
Yan Z, Chen L, Guo Y, Dai X, Yin T, Xue L. Pan-Transcriptome Analysis of Willow Species from Diverse Geographic Distributions. Forests. 2023; 14(6):1182. https://doi.org/10.3390/f14061182
Chicago/Turabian StyleYan, Zhenyu, Li Chen, Ying Guo, Xiaogang Dai, Tongming Yin, and Liangjiao Xue. 2023. "Pan-Transcriptome Analysis of Willow Species from Diverse Geographic Distributions" Forests 14, no. 6: 1182. https://doi.org/10.3390/f14061182
APA StyleYan, Z., Chen, L., Guo, Y., Dai, X., Yin, T., & Xue, L. (2023). Pan-Transcriptome Analysis of Willow Species from Diverse Geographic Distributions. Forests, 14(6), 1182. https://doi.org/10.3390/f14061182