Abstract
Adventitious bud regeneration is an effective means of rapid propagation of plants, which can be used in the study of plant development and genetic transformation. It can be divided into direct and indirect adventitious bud regeneration. Of the two kinds of adventitious bud regeneration mentioned, indirect adventitious bud regeneration includes callus formation in vitro and organ regeneration. In the process of callus formation, some cells acquire the pluripotency of tissue regeneration, which is the key to regeneration of adventient buds. It is not clear which molecular processes and genetic factors are involved in establishing cellular pluripotency. The object of the study is hybrid sweetgum (Liquidambar styraciflflua × L. formosana). At present, the reproductive efficiency of hybrid sweetgum is low and the reproductive cycle is long. Improving its reproductive efficiency by improving the differentiation speed of callus may be a decent approach. In order to explore the mechanism of pluripotency acquisition during forming hybrid sweetgum callus, we used RNA-seq to perform transcriptomic analysis of the regenerable calli (RC) and non-regenerable (NRC) calli of hybrid sweetgum. A dataset of differentially expressed genes (DEG) was obtained and several genes probably involved in bud formation were analyzed to explain the molecular processes of acquiring and maintaining pluripotency. In this study, a total of 665 significantly expressed DEGs were identified in the RC and NRC of hybrid sweetgum, among which, 585 differentially expressed genes were up-regulated and 80 differentially expressed genes were down-regulated. GO, KEGG analysis and qRT-PCR results showed phenylpropanoid is a key factor regulating the bud regeneration of hybrid sweetgum; WOX1, WOX11, BGLU12 and BGLU13 were also important regulatory factors. These results provide a pivotal reference point for future sweetgum propagation research.
1. Introduction
Plant regeneration refers to the process wherein damaged cells, tissues or organs respond by self-repair or structural replacement. The basis of regeneration is the pluripotency of plant cells. Pluripotency refers to the ability of cells or tissues, that already have a certain form and function, to exhibit a variety of other forms and functions due to changes in internal or external conditions. Adventitious bud regeneration is not only an effective means of rapid propagation of plants, but can can be used in the study of plant development and genetic transformation. The study of adventitious bud regeneration may be beneficial in cultivating rare species. Adventitious bud regeneration usually goes through four stages: the acquisition of cell pluripotency, the formation of a primary meristem of a bud, the establishment of a restrictive bud primordium, the germination and elongation of a bud. Many plant explants cultured in vitro have the ability to regenerate buds, roots, etc., and the related genetic regulation mechanism also attracts our attention.
The key event in shoot regeneration is the formation of meristems [1,2] and its transcription and hormones that regulate the formation of meristem. Genes involved in meristem formation may be partly regulated by epigenetic mechanisms. Phenylpropanoid biosynthesis is a key pathway of the transition from primary metabolism to secondary metabolism. It can regulate the synthesis of catechins, flavonoids, anthocyanins, lignin, lignans and other compounds. These compounds play an important role in plant growth and development and in response to biotic and abiotic stresses [3,4]. β-Keto-acyl-CoA synthase (KCS) members are responsible for catalyzing different elongation reactions and these members have substrate recognition specificity and organ tissue expression specificity [5,6]. In many plants, the β-glucosidase (BGLU) gene is present in chromosomes in the form of gene family and is involved in physiological processes such as cell wall lignylation, regulation of enzyme activity, signal transduction and hormone activation [7,8,9]. BGLU12 controls flavonoid utilization [10]. The BGLU family widely participates in many important physiological processes such as plant hormone signal activation and secondary metabolism [8,11]. Studies have shown that meristem formation regulatory genes, such as WOX5, WOX7, WOX11 and WOX14 of the WUSCHEL-related Homeobox (WOX) family, are also related to acquiring the ability of bud regeneration [12].
In addition, some studies have found that genic heterochromatin is required for the proper expression of the relevant genes. Disrupting either element of the AAE complex-genic heterochromatin module leads to RNA processing abnormalities and inhibits gene expression [13].
In this study, the isolated leaves of hybrid sweetgum were used. The leaves were cultured on a specific differentiation medium and callus was formed. At the same time, auxin (2,4-dichlorophenoxyacetic acid [2,4-D]) in the culture medium induced bud regeneration. However, the molecular regulatory mechanism of bud regeneration in hybrid sweetgum is unclear and has not been reported. In this study, transcriptome analysis was used to explore the molecular processes and key regulatory factors involved in the acquisition of somatic cells pluripotency, so as to further provide the reproductive efficiency of hybrid sweetgum.
2. Materials and Methods
2.1. Plant Materials and Culture Conditions
The material of this study was the cross of North American sweetgum as mother and Chinese sweetgum as father, and the first generation of hybrid sweetgum was obtained. The experiment was carried out by selecting the hybrid type, “HT-8”, which had good character and strong regeneration ability. Take the second and third functional top leaves of the sterile seedling as explants material (different strains of the homologous plants). In this study, about 240 slices were collected and half of them were used for sampling. After mechanical damage, the leaves were placed on the regeneration medium with the back side up for induction culture. In addition to organic salts, inorganic salts, water, sucrose and plant gels necessary for material growth, the differentiation medium also contains three hormones, the types and concentrations of which are TDZ (0.1 g/mL), 6-BA (0.8 g/mL) and NAA (0.1 g/mL). The temperature of the tissue culture room was controlled at 25 ± 2 °C, the illumination intensity of the fluorescent lamp was about 1200 lx, and the illumination time was 16 h/d. After 20 days of inoculation, the regenerative callus that produced indefinite buds and the non-regenerative callus that did not produce indefinite buds were, respectively, peeled off under a type microscope, and the next experiment was carried out.
2.2. RNA Extraction
The RC and NRC of hybrid sweetgum were selected for transcriptome sequencing. Two biological replicates were set for each developmental stage of sampling (just budding and without budding after a 30-day wait), with a total of 4 samples. Each sample was quickly frozen in liquid nitrogen after 0.2 g was taken and stored at −80 °C until RNA was extracted. RNA prep Pure Plant Kit (Tiangen, Beijing, China) was used for RNA extraction.
2.3. mRNA Sequencing and Data Analysis
Each sample used 1 µg of RNA as the input material for sample preparation. Illumina’s NEBNext Ultra RNA Library preparation Kit was used to generate RNA-SEQ libraries as recommended by the manufacturer. After sequencing, raw data were filtered to remove adapter contamination and low-quality readings by (1) removing reads containing connectors; (2) low-quality reads (including those with N ratio greater than 10%) were removed; the number of bases with mass value Q ≤ 10 accounted for more than 50% of the reads of the whole read). The high-quality clean data obtained after series of quality control above were provided in FASTQ format. All clean readings were then compared to the hybrid Amblygum reference genome using HISAT2 2.2.1 [14] tool software.
2.4. Differential Expression Analysis
DESeq2 [15] was used for differential expression analysis. The corrected p-values were obtained using Benjamini and Hochberg’s method for controlling the error detection rate. Corrected p-Values analyzed via DESeq2 Genes with 0.01 and multiple changes of ≥2 were designated as differentially expressed. Significantly differentially expressed genes (DEGs) were screened in the comparison, and gene expression levels were standardized using the [fragment per kilobase transcript per million fragment mapping (FPKM)] method [16].
2.5. GO and KEGG Enrichment Analysis
Gene ontology (GO) enrichment analysis of differentially expressed genes (DEG) was applied by using clusterProfiler R software 4.0 package. Enrichment analysis used hypergeometric tests to look for GO entries that were significantly enriched compared to the whole genomic background. The enrichment p-value for each GO term was calculated and corrected. We believe that the GO terms of P value (corrected for error discovery rate) ≤0.05 were significantly enriched. In addition, we used the KOBAS [17] database and the clusterProfiler software 4.0 to analyze the enrichment of differentially expressed genes in the KEGG pathways.
2.6. qRT-PCR Fluorescence Quantitative Analysis
Real-time fluorescent quantitative polymerase chain reaction (qRT-PCR) was performed on RNA samples used for sequencing library preparation using the CFX Connect® real-time PCR assay system (CFX Connect, Bio-Rad, Munich, Germany) to verify RNA-SEQ results. RNA was extracted using EasyPure Plant RNA Kit, and then 1 µg total RNA was reverse-transcribed into cDNA. Each PCR reaction mixture contained 10 µL Universal Blue qPCR SYBR Green Master Mix (Shanghai Yesen Biotechnology Co., Ltd., Shanghai, China), 1 µL template cDNA, 0.4 µL forward primer, 0.4 µL reverse primer and 8.2 µL dd H2O to a final volume of 20 µL was repeated three times per gene. The amplification conditions of qRT-PCR were as follows: 95 °C for 30 s, 1 cycle; 95 °C 5 s + 60 °C 20 s, 40 cycles; melting 65–95 °C. The internal reference gene is the EF1-r gene of apple [18]. Relative gene expression was calculated using the 2−ΔΔCt method [19].
3. Results
3.1. Phenotype Observation of Regenerable Calli (RC) and Non-Regenerable Calli (NRC)
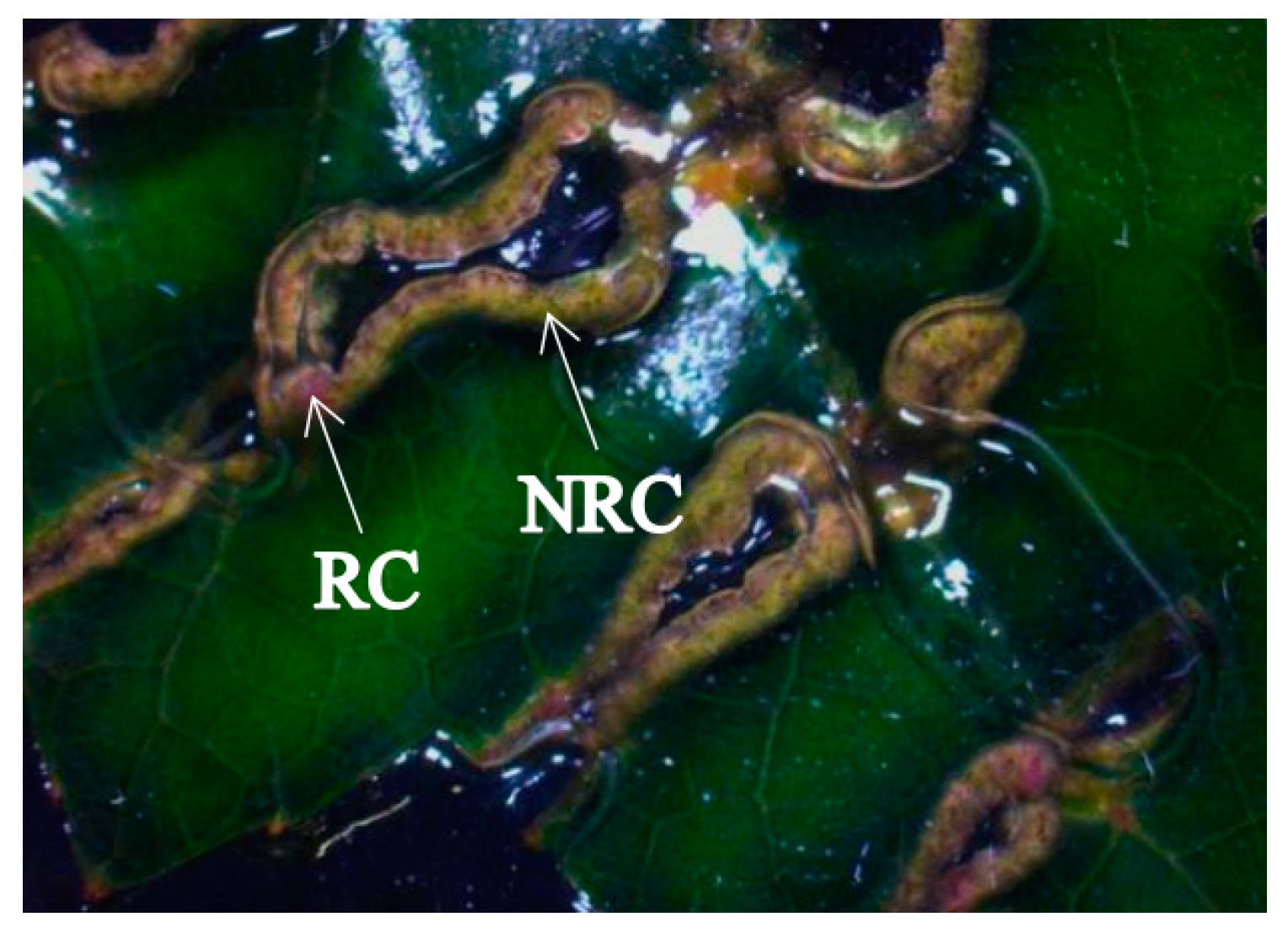
After 20 days of induction, some of the callus showed a pink surface with a convex surface, which gradually changed into indefinite buds and then regenerated into plants. We called this callus regenerable calli. Part of the callus was always green and there was no change or bulge until 30 days later, and it gradually browned and could not be regenerated into new plants. This kind of callus was called non-regenerable calli (Figure 1).
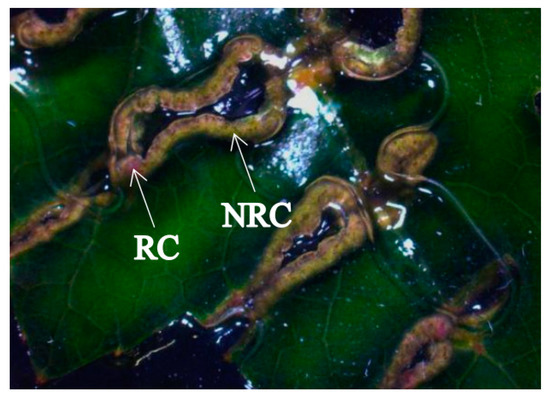
Figure 1.
Regenerable calli (RC) and non-regenerable calli (NRC) of hybrid sweetgum. The macroscopic appearance of RC and NRC.
3.2. Transcriptome Sequence Alignment and Screening of Differentially Expressed Genes
In order to explore the mechanism of pluripotency acquisition during callus formation in hybrid sweetgum, we performed transcriptomic sequencing on 4 cDNA libraries constructed from regenerable pluripotency callus and non-regenerable non-pluripotency callus. In the following Table 1, we use D1-1, D1-2, D2-1, D2-2 to represent the four samples of the study, D1-1, D1-2 as renewable callus, D2-1, D2-2 as non-renewable callus. A total of 24.81 Gb reads were obtained from the transcriptome sequencing results after filtering and comparison, and the clean data of each sample was 5.86 Gb.

Table 1.
Statistical table of sequence alignment results of sample sequencing data with the selected reference genome.
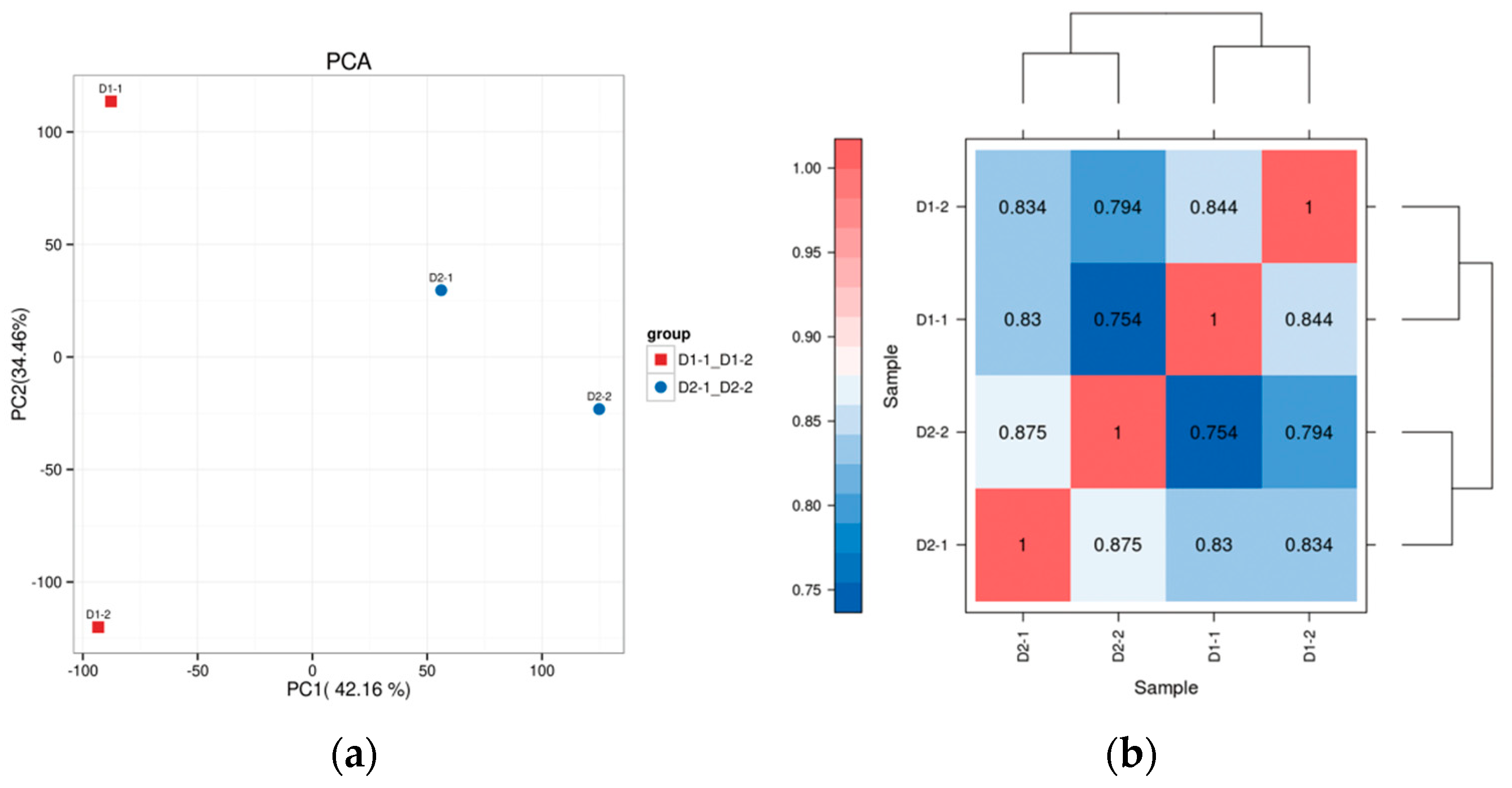
Principal component analysis (PCA) results showed that the two replicates of regenerable calli and non-regenerable calli could be clearly distinguished, as shown in Figure 2a. In the transcriptome comparison of regenerable calli and non-regenerable calli in hybrid sweetgum, a total of 665 significantly expressed DEGs were identified, of which 585 differentially expressed genes were up-regulated and 80 differentially expressed genes were down-regulated. Genes with similar expression patterns may have the same function. In order to visually display the expression differences of genes in different groups and to explore new functional genes, hierarchical clustering analysis was carried out on all the differentially expressed genes selected. Genes with the same or similar expression patterns in different samples were clustered and displayed through heat maps. Thus, the potential functions of genes with similar expression patterns could be found according to genes with known functions. The heat map of gene clustering expression in this project is presented in Figure 2b.
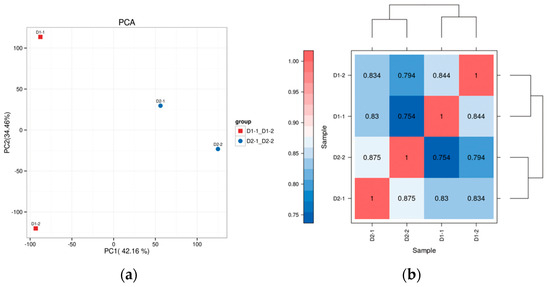
Figure 2.
(a) PCA plot of transcript abundance of two different types of callus. (b) Hierarchical clustering of differentially expressed genes. Differential genes were divided into two clusters by cluster analysis and showed opposite expression patterns in pluripotent and non-pluripotent calli.
The analysis of differentially expressed genes showed that there were significant differences in the transcriptomes of regenerable calli and non-regenerable calli. In the follow-up statistics, it was found that among the top 15 genes of regenerable calli with the most significant up-regulation changes, 9 genes were related to multiple metabolic pathways and 5 genes were related to enzyme activity and metabolism compared with non-regenerable calli. Among the 15 genes with the most significant down-regulation changes, 5 genes were related to the mechanism of transcriptional regulation and there was one transcription factor, bHLH.
In addition, we identified significant differences in the expression of eight genes of the WOX family, six genes were up-regulated and two genes were down-regulated in regenerable calli compared with non-regenerable calli. There were significant differences in the expression of three genes in the BHLH family. Compared with NRC, the expression of two genes was up-regulated and one gene was down-regulated.
3.3. GO Analysis on DEGs
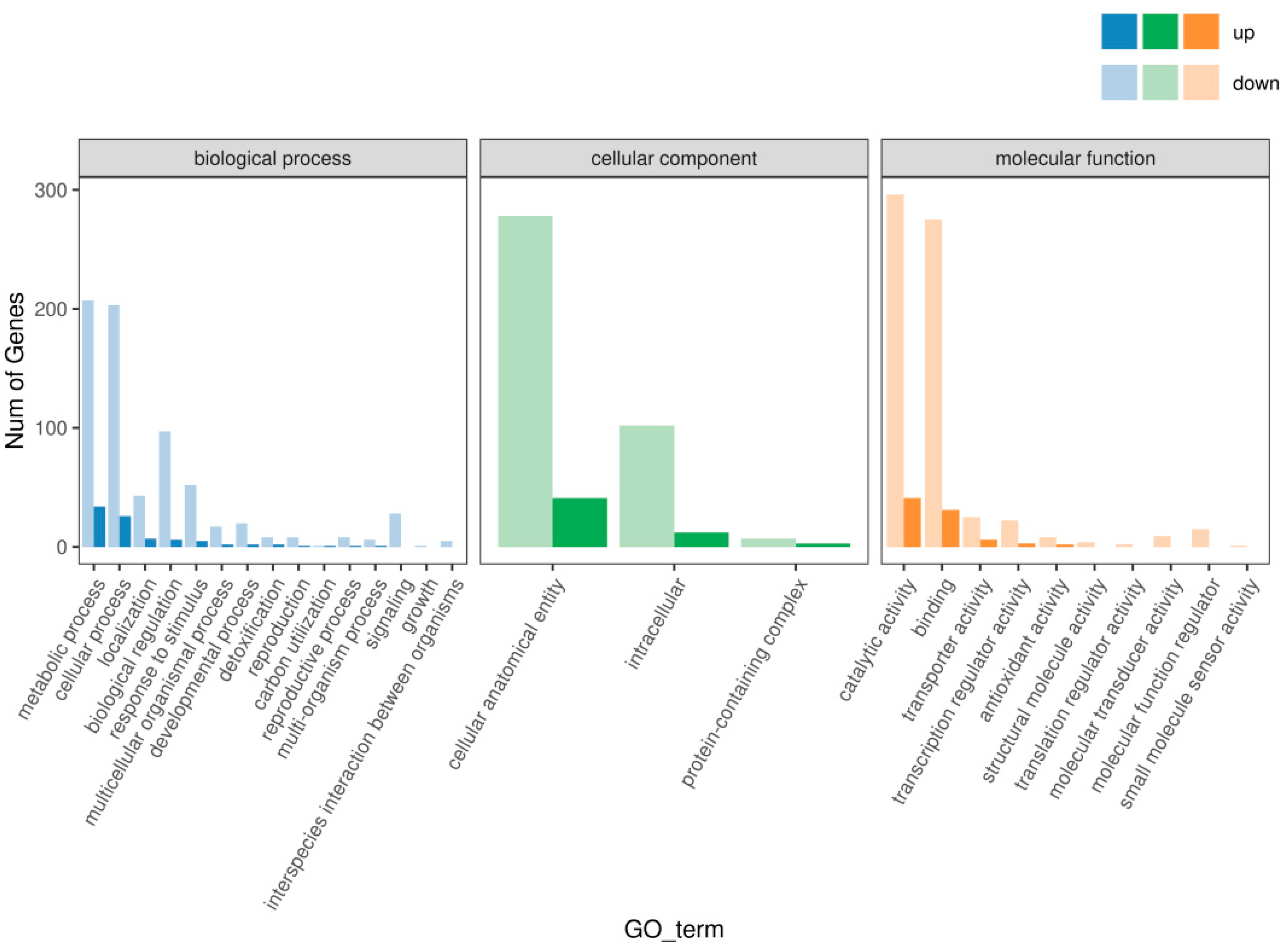
After GO enrichment analysis, 792, 443 and 740 differential genes were enriched into three categories: biological process (BP), cell component (CC) and molecular function (MF), respectively. According to the GO functional enrichment results, 22, 3 and 16 GO terms were significantly enriched into biological processes, cell components and molecular functions, respectively, as shown in Figure 3. A large number of genes were downregulated in metabolic process and cellular process, suggesting that the RC of hybrid sweetgum had a more vigorous metabolism and growth process.
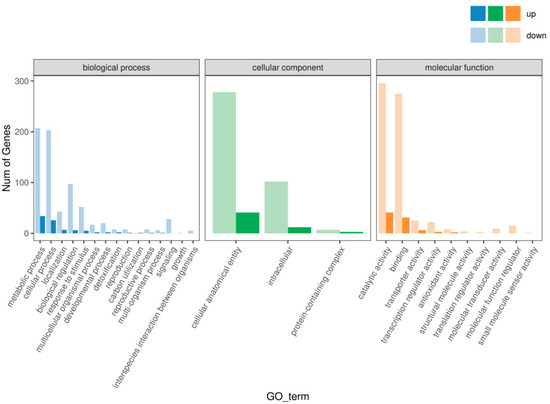
Figure 3.
Statistical diagram of GO enrichment classification of differential genes.
In addition, in terms of molecular function, a large number of up-regulated genes were enriched in the transcriptional regulation process of the hybrid sweetgum regenerable calli, indicating that the hybrid sweetgum regenerable calli may have more complex transcriptional factor regulation and expression than the hybrid sweetgum non-regenerable calli. On the level of biological processes, the top three noted differential genes were 241 metabolic processes (GO:0008152), with 34 genes up-regulated and 207 genes down-regulated. There were 229 cellular processes (GO:0009987), with 26 up-regulated genes and 203 down-regulated genes. Biological regulation had (GO:0065007) 103, with 6 up-regulated genes and 97 down-regulated genes. At the cell component level, the top three annotated differential genes were cellular anatomical entities (GO:0071944), which had 319, with 41 genes up-regulated and 278 genes down-regulated. Intracellular processes had (GO:0005622) 114 genes, with 12 up-regulated and 102 down-regulated. Protein-containing complexes (GO:0043234) had 10, with three genes up-regulated and seven genes down-regulated. On the molecular functional level, the top two most prominent annotated differential genes were related to catalytic activity with 337 (GO:0003824), 41 genes up-regulated and 296 genes down-regulated. Binding (GO:0005488) had 306, with 31 genes up-regulated and 275 genes down-regulated.
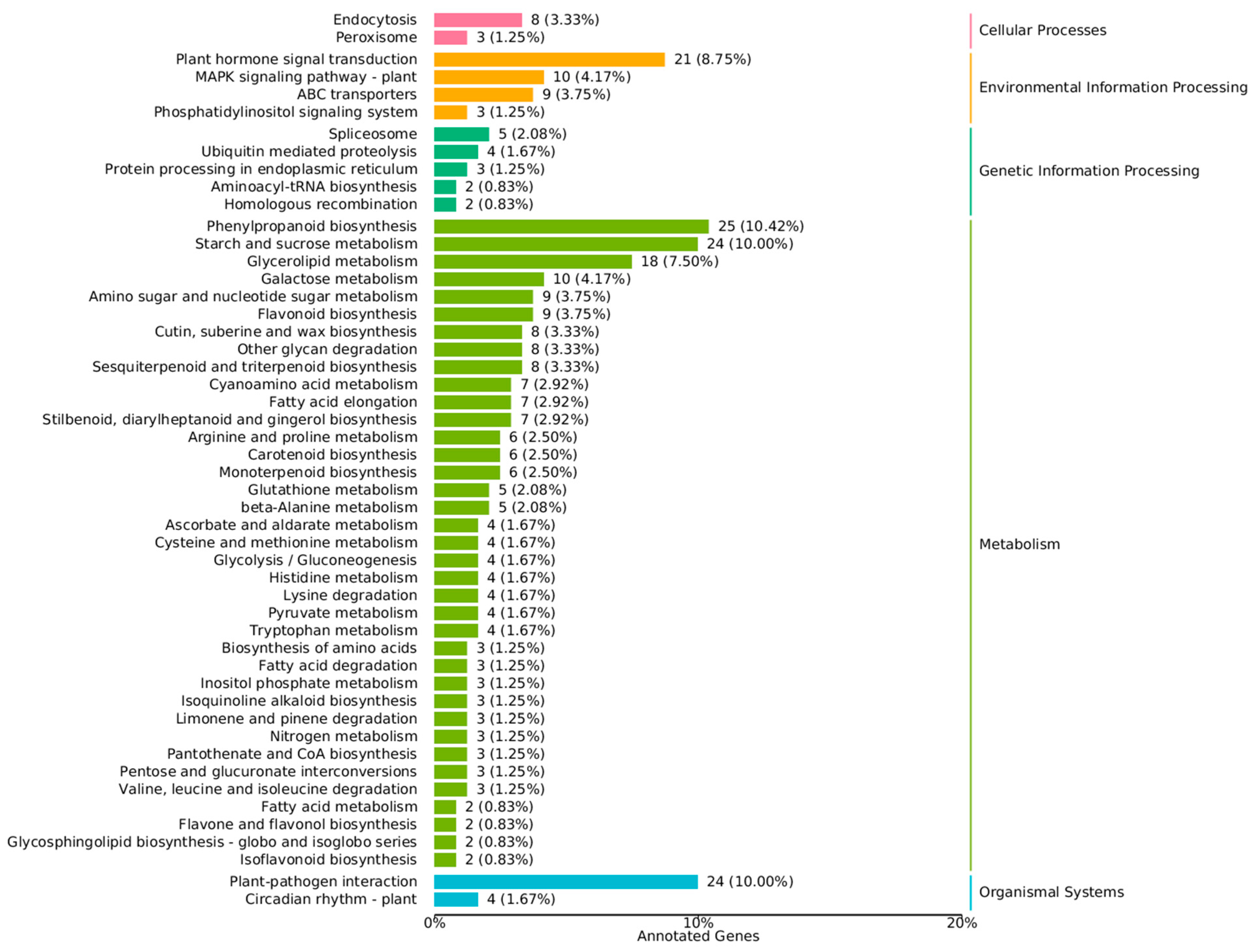
3.4. KEGG Analysis Revealed the DEG-Rich Metabolic Process
Changes in gene expression underlie the differences in biological and metabolic functions between regenerable calli and non-regenerable calli [20,21]. To investigate the biological pathways of differential genes, we used the KEGG database to classify the functions of differential genes, with a focus on biological pathways. A total of 665 genes were used to annotate 50 KEGG pathways, of which 197 differentially expressed genes were enriched on 20 KEGG pathways. Acorrding to Figure 4, in the up-regulated genome, there was little difference in the enrichment status of each pathway. In down-regulated genomes, differential genes were mainly enriched in metabolism-related pathways, which mainly include phenylpropanoid biosynthesis, starch and sucrose metabolism, glycerolipid metabolism, plant hormone signal transduction, flavonoid biosynthesis and so on. This was similar to the result of the GO annotation. The results showed that phenylpropanoid biosynthesis (10.42%), starch and sucrose metabolism (10.00%), glycerolipid metabolism (7.50%). The synthesis of some secondary metabolites may play an important role in the reproducibility of the callus. In addition, a large number of differentially expressed down-regulated genes were enriched in plant hormone signal transduction. This suggests that plant hormones may play an important role in maintaining callus reproducibility.
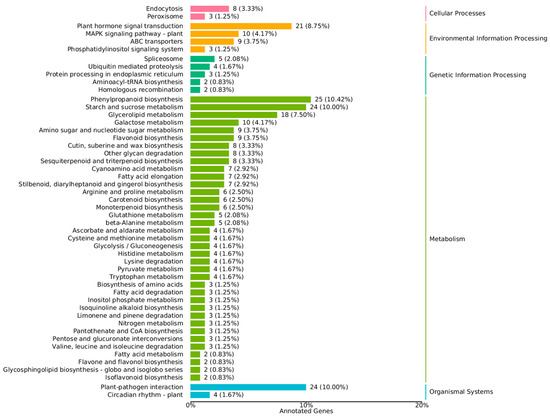
Figure 4.
KEGG classification map of differentially expressed genes.
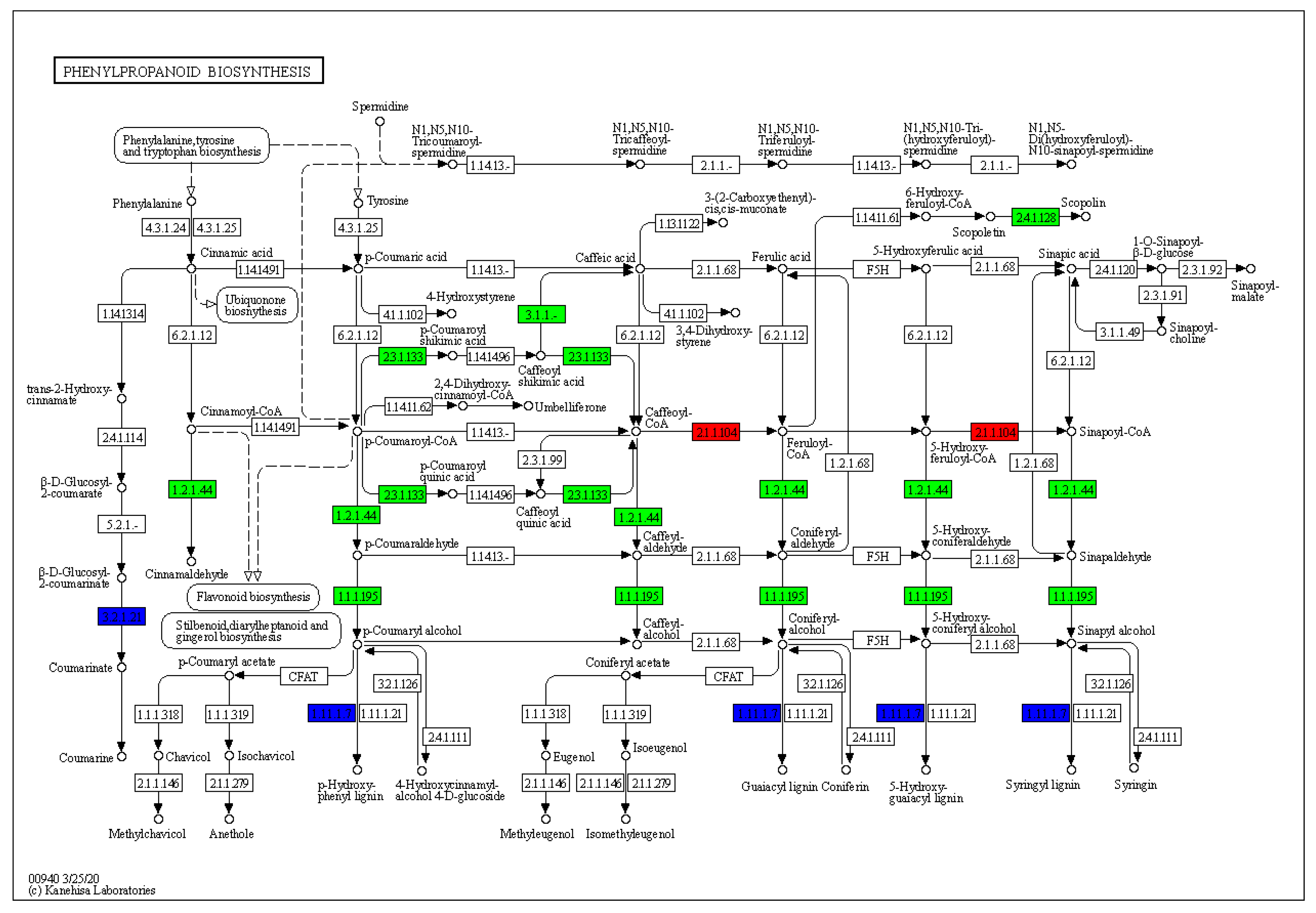
Both up-regulated and down-regulated differential genes were highly enriched in phenylpropanoid biosynthesis and differentially expressed genes were most significantly enriched in this pathway. We annotated the phenylpropanoid biosynthesis pathway (Figure 5). It was found that a large number of differentially expressed genes were involved in the biosynthesis of phenylpropanoid. The next step was to investigate the genes involved in this pathway and examine the functions of these genes to see if they were involved in bud regeneration. The differentially expressed genes were cinnamyl alcohol dehydrogenase 6 (EVM0021779), cinnamoyl-Coa reductase 1 (EVM0001636), peroxidase (such as peroxidase 3 (EVM0000076)), caffeoyl-CoA O-methyltransferase (EVM0002205), shikimate O-hydroxycinnamoyltransferase (such as shikimate O-hydroxycinnamoyltransferase-like (EVM0001797)), beta-glucosidase (such as beta-glucosidase 24-like (EVM0005769)), caffeoylshikimate esterase (EVM0024020) and UDP-glucose flavonoid 3-O-glucosyltransferase (such as UDP-glucose flavonoid 3-O-glucosyltransferase 7-like (NewGene_309)) and so on.
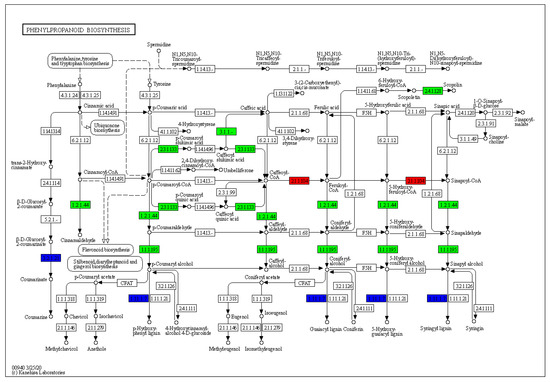
Figure 5.
Expression of phenylpropanoid biosynthesis pathway gene in the callus of hybrid sweetgum.
Compared to the control group, the enzymes labeled in the red box were associated with up-regulated genes, and the enzymes labeled in the green box were associated with down-regulated genes. The enzymes labeled in blue are associated with both up-regulated and down-regulated genes. The mapped genes include cinnamyl alcohol dehydrogenase 6 [1.1.1.195], cinnamoyl-CoA reductase 1 [1.2.1.44], peroxidase (such as peroxidase 3) [1.11.1.7], caffeoyl-CoA O-methyltransferase [2.1.1.104], shikimate O-hydroxycinnamoyltransferase (such as shikimate O-hydroxycinnamoyltransferase-like) [2.3.1.133], beta-glucosidase (such as beta-glucosidase 24-like) [3.2.1.21], caffeoylshikimate esterase [3.1.1.-] and UDP-glucose flavonoid 3-O-glucosyltransferase (such as UDP-glucose flavonoid 3-O-glucosyltransferase 7-like) [2.4.1.128] and so on.
3.5. Plant-Hormone-Related Genes in NRC and RC
Genes involved in plant hormone signaling and biosynthesis are critical for the transition from NRC to RC. In total, we annotated 941 genes associated with plant hormones in the transcriptome data. Among the down-regulated genes, 21 (2.23%) were enriched in plant hormone signal transduction. Furthermore, we identified genes associated with plant hormones among the differentially expressed genes. The statistics found that auxin was the most correlated with 15, of which, 14 were significantly down-regulated (Table 2). These results indicate that auxin played an important role in bud regeneration, in addition to abscisic acid, brassinosteroid, cytokinin and ethylene, all of which had higher expression levels in the RC.
Table 2.
Statistical table of the number of differential genes related to hormones.
3.6. Differential Variable Shear Analysis
Variable splicing is closely related to the formation of important traits or diseases. The study of variable splicing with significant differences in expression under different conditions is helpful in accurately finding the transcripts related to traits and further exploring the changes in variable splicing patterns under different conditions and the mechanism of action in important traits or biological functions. rMATS software 4.1.2 [22] was used to perform differential variable shear analysis on different samples (within different differential groups) and the transcripts with differential variable shear between samples included two types: the exon inclusion isoform and the exon skipping isoform, the rMATS statistical model defined the number of reads that were uniquely matched to the two types of transcripts as the inclusion level of the cut site. Then, p-value was calculated with a likelihood ratio test to represent the difference in IncLevel (inclusion level) between the two groups of samples, and the FDR value was obtained by correcting p-value with the Benjamini Hochberg algorithm. To visualize each differential variable splicing, you can use the rmats2sashimiplot software4.1.2 to visualize the differential variable splicing results. The results show that the number of differential genes involved in exon jumping events was large on chromosomes 13, 14 and 15, while the number of differential genes involved in exon selective jumping events was the largest on chromosomes 5 and 14. However, the influencing mechanism of differentially variable splicing events on the gene expression regulation and cellular processes related to bud regeneration remains to be explored.
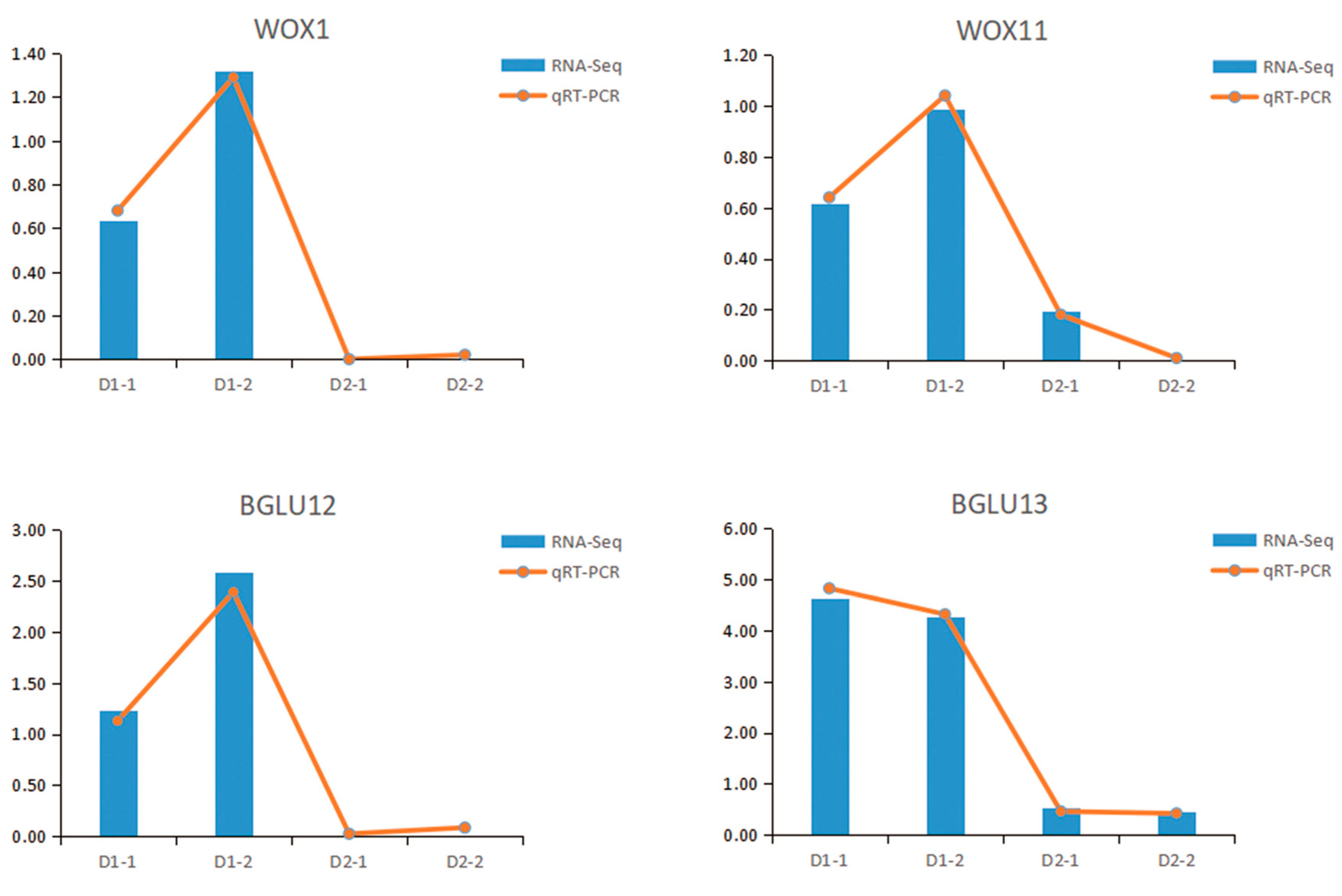
3.7. qRT-PCR Verification of Regenerative Gene in Hybrid Sweetgum Callus
In order to test the reliability of transcriptome data and key genes, some genes, including WOX1 (EVM0007971) and BGLU13 (EVM0005769), were selected for qRT-PCR gene expression verification. The trend of qRT-PCR was consistent with the transcriptome data, which also demonstrated a high correlation between RNA-seq and qRT-PCR data (Figure 6).
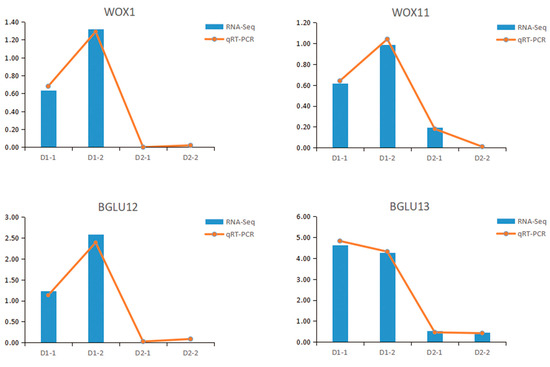
Figure 6.
Transcriptome expression (FPKM value) and qRT-PCR value of four genes in hybrid sweetgum. Relative expression values were calculated using the 2−ΔΔCᴛ method with apple EF1-α as a housekeeping gene. D1-1, D1-2 are renewable callus; D2-1, D2-2 are non-renewable callus.
4. Discussion
It is worth mentioning that several genes in the phenylpropanoid biosynthesis pathway were noticed when analysing KEGG classification map phase [23]. As the second-most-abundant natural polymer, lignin plays an essential role in plant development. Cinnamyl alcohol dehydrogenase (CAD) is the enzyme in the last step of lignin biosynthetic pathway and is involved in the generation of lignin monomers [24]. In a study of Paeonia ostii, caffeoyl-CoA O -methyltransferasePoCCoAOMT was found to be a vital gene in promoting lignin synthesis [25]. The upregulation of UDP-glucose flavonoid 3-O-glucosyltransferase can promote anthocyanin accumulation [26].
In the study on the mechanism of the Arabidopsis TCP protein [27], it was found that the TCP transcription factor had functional redundancy. For example, high temperature promoted the release of AtTCP17 from the CRY1-TCP17 complex, which then bound to the PIF4 gene promoter region to activate transcription and expression, and could also interact with the PIF4 protein. Enhanced transcriptional activation of downstream target genes, YUC8 and IAAI19 [28]; AtTCP5 can also bind and transcriptionally activate the expression of PIF4 and interact with PIF4 protein to increase its transcriptional activity on downstream genes [29]. Different expressions of BGLU gene in tea plants were found in PEG-induced drought stress, salt stress treatment/methyl jasmonate treatment and cold treatment, indicating that the CsBGLU gene plays an important role in growth and development of tea plants and in the response to abiotic stress [9]. GW8 (GRAIN WIDTH8)/OsSPL16 positively regulates rice cell proliferation. High expression of GW8 (grain width8)/OSSPL16 can promote cell proliferation, increase grain width and grain filling and improve rice yield. However, when the function is lost, the slender grain type is formed and the appearance quality of rice is improved [30]. Subsequently, it was found that GW8/OsSPL16, as a transcription factor, could directly bind to the GW7 promoter of the granulotype gene and inhibit its expression [31]. In this study, transcriptome and differentially variable cut analysis were used to find that phenylpropanoid was the key factor regulating the bud regeneration of hybrid sweetgum, which is of great significance in improving the reproductive efficiency of hybrid sweetgum.
Gene families and their functions are evolutionarily conserved. AtWOX9 was found to be an important regulatory factor in the bud regeneration of Arabidopsis thaliana [32]. In the study of poplar bud regeneration, PtWOX5B was found to be an important regulatory factor [33,34,35]. TaWOX10 was found to be an important regulatory factor in the callus differentiation of wheat [36,37,38,39]. In this study, we found that WOX1 and WOX11 were also important regulatory factors in the bud regeneration of hybrid sweetgum. Similarly, BGLU12, BGLU13 were differentially expressed in the RC and NRC; the BGLU gene family also plays an important role in the regulation of bud regeneration in many plants, and we speculated that the gene family and its function are conservative, which indicates that the study may have guiding significance for improving the reproductive efficiency of other plants.
5. Conclusions
In this study, the genome of hybrid sweetgum has revealed the presence of many differentially expressed genes. Phenylpropanoid is a key factor regulating the bud regeneration of hybrid sweetgum. We performed KEGG analysis on differentially expressed genes and focused on the pathway genes that might be involved in bud regeneration; we selected several genes in the phenylpropanoid biosynthesis pathway that might be associated with bud regeneration. Then, based on the published role of their gene families, the mechanism of their influence was speculated. The transcriptome and qRT-PCR results show that the expression of these genes had a certain specificity, which preliminarily revealed the function of these selected genes in somatic embryogenesis. These results provide a pivotal reference point for future bioinformatic analyses and in-depth investigations into the profound and intricate function of WOX and BGLU genes in bud regeneration.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/f14091833/s1.
Author Contributions
Investigation, H.M. and T.Z.; Data curation, H.M.; Writing—review & editing, Z.A. and J.Z.; Supervision, J.Z.; Project administration, S.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 32271836), the National Forestry and Grassland Administration Promotion Project of China (2020133102) and the Fundamental Research Funds for the Central Universities (2019ZY39).
Data Availability Statement
Publicly available datasets were analyzed in this study. These data can be found here: Transcriptome data.xls (Supplementary Materials).
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Gordon, S.P.; Heisler, M.G.; Reddy, G.V.; Ohno, C.; Das, P.; Meyerowitz, E.M. Pattern formation during de novo assembly of the Arabidopsis shoot meristem. Development 2007, 134, 3539–3548. [Google Scholar] [CrossRef]
- Su, Y.H.; Liu, Y.B.; Bai, B.; Zhang, X.S. Establishment of embryonic shoot-root axis is involved in auxin and cytokinin response during Arabidopsis somatic embryogenesis. Front. Plant Sci. 2015, 5, 792. [Google Scholar] [CrossRef]
- Wu, Y.L. Functional Studies on Structural Genes and Transcription Factors of Phenylpropane Pathway in Tea Plants; Anhui Agricultural University: Hefei, China, 2023. [Google Scholar]
- La, C.S.; Gouzerh, G.; Dhondt, S.; Hoffmann, L.; Fritig, B.; Legrand, M.; Heitz, T. Metabolic reprogramming in plant innate immunity: The contributions of phenylpropanoid and oxylipin pathways. Immunol. Rev. 2010, 198, 267–284. [Google Scholar]
- Huang, H.D. Molecular Mechanism Analysis of Arabidopsis KCS5 and KCS6/CER6 Synergistically Involved in Epidermal Wax Synthesis; Hubei University: Wuhan, China, 2022. [Google Scholar]
- Haslam, T.M.; Kunst, L. Extending the story of very-long-chain fatty acid elongation. Plant Sci. 2013, 210, 93–107. [Google Scholar] [CrossRef]
- Ketudat, C.; Mahong, B.; BAIYA, S. B-Glucosidases: Multitasking, moonlighting or simply misunderstood. Plant Sci. 2015, 241, 246–259. [Google Scholar] [CrossRef]
- Sampedro, J.; Valdivia, E.R.; Fraga, P.; Iglesias, N.; Revilla, G.; Zarra, I. Soluble and membrane-bound g-Glucosidases are involved in trimming the xylo-glucan backbone. Plant Physiol. 2017, 173, 1017–1030. [Google Scholar] [CrossRef]
- Yao, X.Z.; Chen, L.X.; Zhang, B.H.; Lv, L.T. The identification of tea tree BGLU gene families and expression pattern analysis. Seeds 2022, 9, 1–9. [Google Scholar]
- Roepke, J.; Gordon, H.; Neil, K.; Gidda, S.; Mullen, R.; Freixas, C.J.; Bray-Stone, D.; Bozzo, G.G. An Apoplastic β-Glucosidase is Essential for the Degradation of Flavonol 3-O-β-Glucoside- 7-O-α-Rhamnosides in Arabidopsis. Plant Cell Physiol. 2017, 58, 1030–1047. [Google Scholar] [CrossRef]
- Sun, H.H.; Xue, Y.M.; Lin, Y.F. Enhanced catalytic efficiency in quercetin-4'-glucoside hydrolysis of Thermotoga maritima β-glucosidase A by site-directed mutagenesis. J. Agric. Food Chem. 2014, 62, 6763–6770. [Google Scholar] [CrossRef]
- Kim, H.J.; Pesacreta, T.C.; Triplett, B.A. Cotton-fiber germin-like protein.II: Immunolocalization, purification and functional analysis. Planta 2004, 218, 525–535. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Lin, J.C.; Ren, Z.Z.; Chen, C.X.; Miki, D. Genome-wide distribution and functions of the AAE complex in epigenetic regulation in Arabidopsis. J. Integr. Plant Biol. 2021, 63, 707–722. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Zheng, L.; Ma, J.; Song, C.; An, N.; Zhang, D.; Zhao, C.; Qi, S.; Han, M. Genome-wide identification and expression profiling analysis of brassinolide signal transduction genes regulating apple tree architecture. Acta Physiol. Plant. 2017, 39, 1–19. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Corredoira, E.; Valladares, S.; Vieitez, A.M. Morphohistological analysis of the origin and development of somatic embryos from leaves of mature Quercus robur. Vitr. Cell. Dev. Biol.-Plant 2006, 42, 525–533. [Google Scholar] [CrossRef]
- Xu, Z.H.; Zhang, X.X.; Su, Y.H.; Hu, Y.X.; Xu, L.; Wang, J.W. Totipotency and regeneration of plant cells. Sci. China Life Sci. 2019, 49, 1282–1300. [Google Scholar]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, 593–601. [Google Scholar] [CrossRef]
- Zhou, Q.; Mao, P.; Luo, D.; Chai, X.T.; Deng, H.; Fang, Q.G.; Fang, L.F.; Nan, Z.B.; Wen, J.Q.; Liu, Z.P. Comparative transcriptome analyses reveal that the MsNST1 gene affects lignin synthesis in alfalfa (Medicago sativa L.). Crop J. 2022, 10, 1059–1072. [Google Scholar] [CrossRef]
- Young, H.K.; Gyung, H.H. Overexpression of cinnamyl alcohol dehydrogenase gene from sweetpotato enhances oxidative stress tolerance in transgenic Arabidopsis. Vitr. Cell. Dev. Biol.-Plant 2019, 55, 172–179. [Google Scholar]
- Zhao, D.Q.; Luan, Y.T.; Shi, W.B.; Zhang, X.Y.; Meng, J.S.; Tao, J. A Paeonia ostii caffeoyl-CoA O-methyltransferase confers drought stress tolerance by promoting lignin synthesis and ROS scavenging. Plant Sci. 2021, 303, 110765. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Ishida, A.; Suzuki, Y.; Aoki, Y.; Suzuki, S.; Enoki, S. Ethylene Induced by Sound Stimulation Enhances Anthocyanin Accumulation in Grape Berry Skin through Direct Upregulation of UDP-Glucose: Flavonoid 3-O-Glucosyltransferase. Cells 2021, 10, 2799. [Google Scholar] [CrossRef] [PubMed]
- Li, W.L.; Liu, T.T.; Chen, C.L.; Luo, X.H.; An, X.; Zhou, L.N.; Zhu, G.L.; Li, P.F. Progress in the mechanism of plant TCP protein. Mol. Plant Breed. 2023, 21, 4650–4658. [Google Scholar]
- Zhou, Y.; Xun, Q.Q.; Zhang, D.Z.; Lv, M.H.; Ou, Y.; Li, J. TCP transcription factors associate with PHYTOCHROME INTERACTING FACTOR 4 and CRYPTOCHROME 1 to regulate thermomorphogenesis in Arabidopsis thaliana. iScience 2019, 15, 600–610. [Google Scholar] [CrossRef]
- Han, X.; Yu, H.; Yuan, R.R.; Yang, Y.; An, F.J.; Qin, G.J. Arabidopsis transcription factor TCP5 controls plant thermomorphogenesis by positively regulating PIF4 activity. iScience 2019, 15, 611–622. [Google Scholar] [CrossRef]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, S.; Liu, Q.; Wu, K.; Zhang, J.; Wang, S.; Wang, Y.; Chen, X.; Zhang, Y.; Gao, C.; et al. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 2015, 47, 949–954. [Google Scholar] [CrossRef]
- Dai, X.H.; Wang, J.; Song, Y.G.; Liu, Z.H.; Xue, T.; Qiao, M.; Yu, Y.C.; Xin, W.; Xiang, F.N. Cytosine methylation of the FWA prooter promotes direct in vitro shoot regeneration in Arabidopsis thaliana. J. Integr. Plant Biol. 2021, 63, 1491–1504. [Google Scholar] [CrossRef]
- Miao, J.M. The Role of PtPLT1 and PtWOX5B in Poplar Bud Regeneration; Shandong Agricultural University: Tai’an, China, 2020. [Google Scholar]
- Chatfield, S.P.; Capron, R.; Severino, A.; Penttila, P.; Alfred, S.E.; Nahal, H.; Provart, N.J. Incipient stem cell niche conversion in tissue culture: Using a systems approach to probe early events in WUSCHEL-dependent conversion of lateral root primordia into shoot meristems. Plant J. Cell Mol. Biol. 2013, 73, 798–813. [Google Scholar] [CrossRef]
- Kaoru, S.; Jiao, Y.L.; Elliot, M.M. Arabidopsis regeneration from multiple tissues occurs via a root development pathway. Dev. Cell 2010, 18, 463–471. [Google Scholar]
- Shi, L. Study on WOX gene mining and its effect on genetic transformation efficiency and mechanism of action in wheat. Chin. Acad. Agric. Sci. 2022, 4, 157. [Google Scholar]
- Hao, Q.; Zhang, L.; Yang, Y.; Shan, Z.; Zhou, X.A. Genome-wide analysis of the WOX gene gamily and function exploration of GmWOX18 in soybean. Plants 2009, 8, 215. [Google Scholar] [CrossRef]
- Lowe, K.; La, R.M.; Hoerster, G.; Hastings, C.; Wang, N.; Chamberlin, M.; Wu, E.; Jones, T.; Gordon-Kamm, W. Rapid genotype “independent” Zea mays L. (maize) transformation via direct somatic embryogenesis. In vitro cellular & developmental biology. Plant J. Tissue Cult. Assoc. 2018, 54, 240–252. [Google Scholar]
- Mookkan, M.; Nelson-Vasilchik, K.; Hague, J.; Zhang, Z.Y.; Kausch, A.P. Selectable marker independent transformation of recalcitrant maize inbred B73 and sorghum P898012 mediated by morphogenic regulators BABY BOOM and WUSCHEL2. Plant Cell Rep. 2017, 36, 1477–1491. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).