
Molecular Regulation of Bud Regeneration from Callus of Hybrid Sweetgum (Liquidambar styraciflua × Liquidambar formosana)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Culture Conditions
2.2. RNA Extraction
2.3. mRNA Sequencing and Data Analysis
2.4. Differential Expression Analysis
2.5. GO and KEGG Enrichment Analysis
2.6. qRT-PCR Fluorescence Quantitative Analysis
3. Results
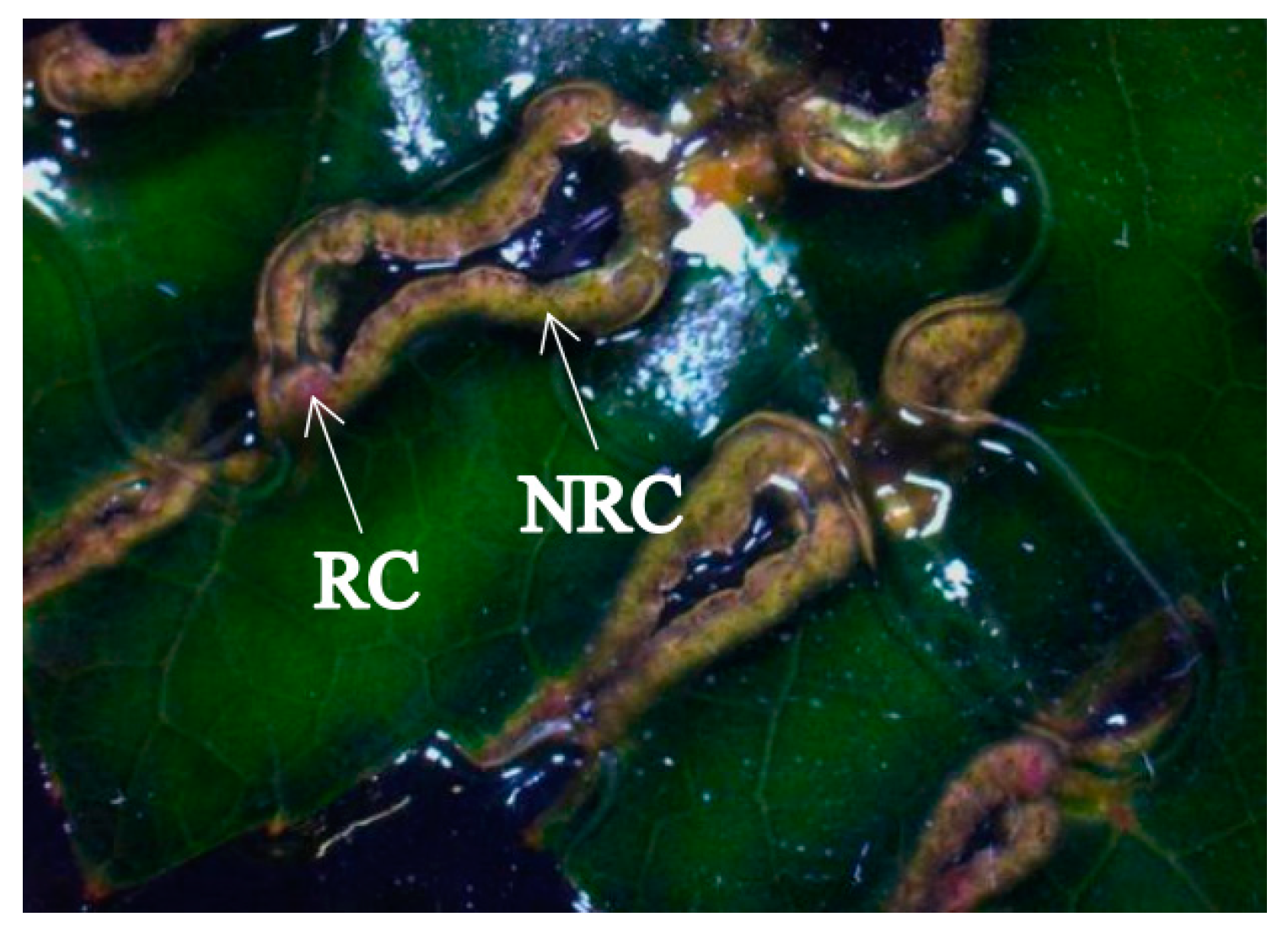
3.1. Phenotype Observation of Regenerable Calli (RC) and Non-Regenerable Calli (NRC)
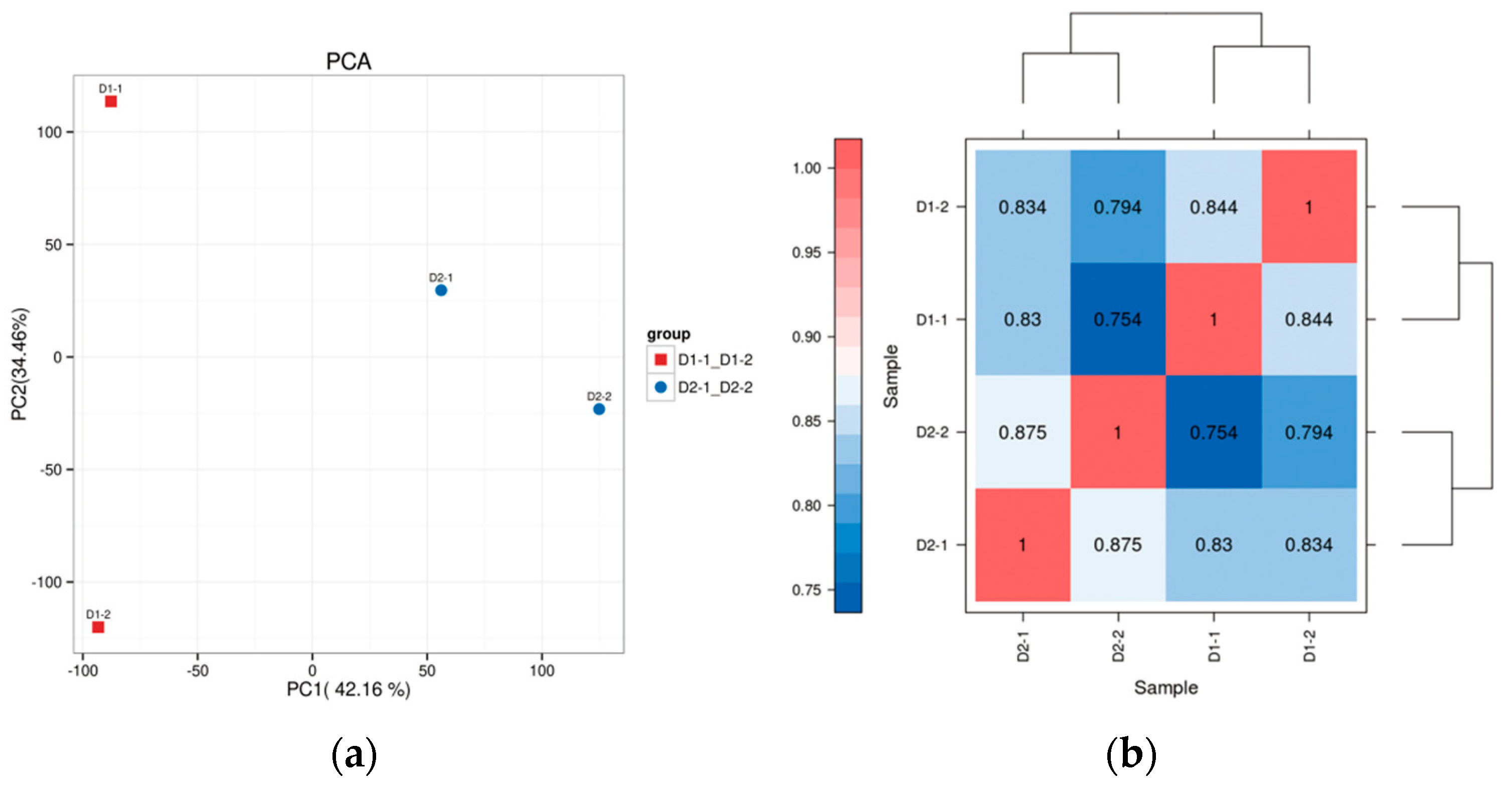
3.2. Transcriptome Sequence Alignment and Screening of Differentially Expressed Genes
3.3. GO Analysis on DEGs
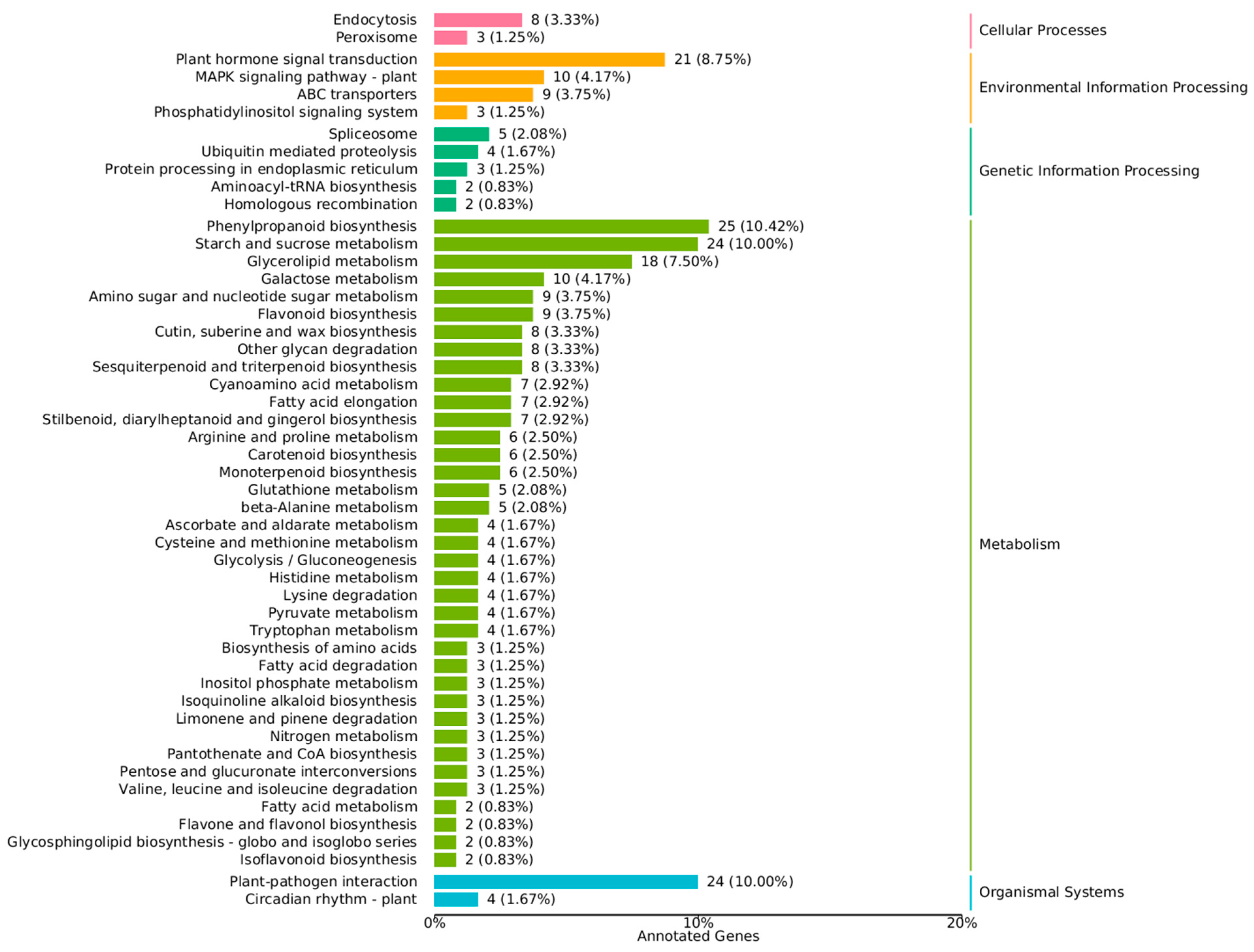
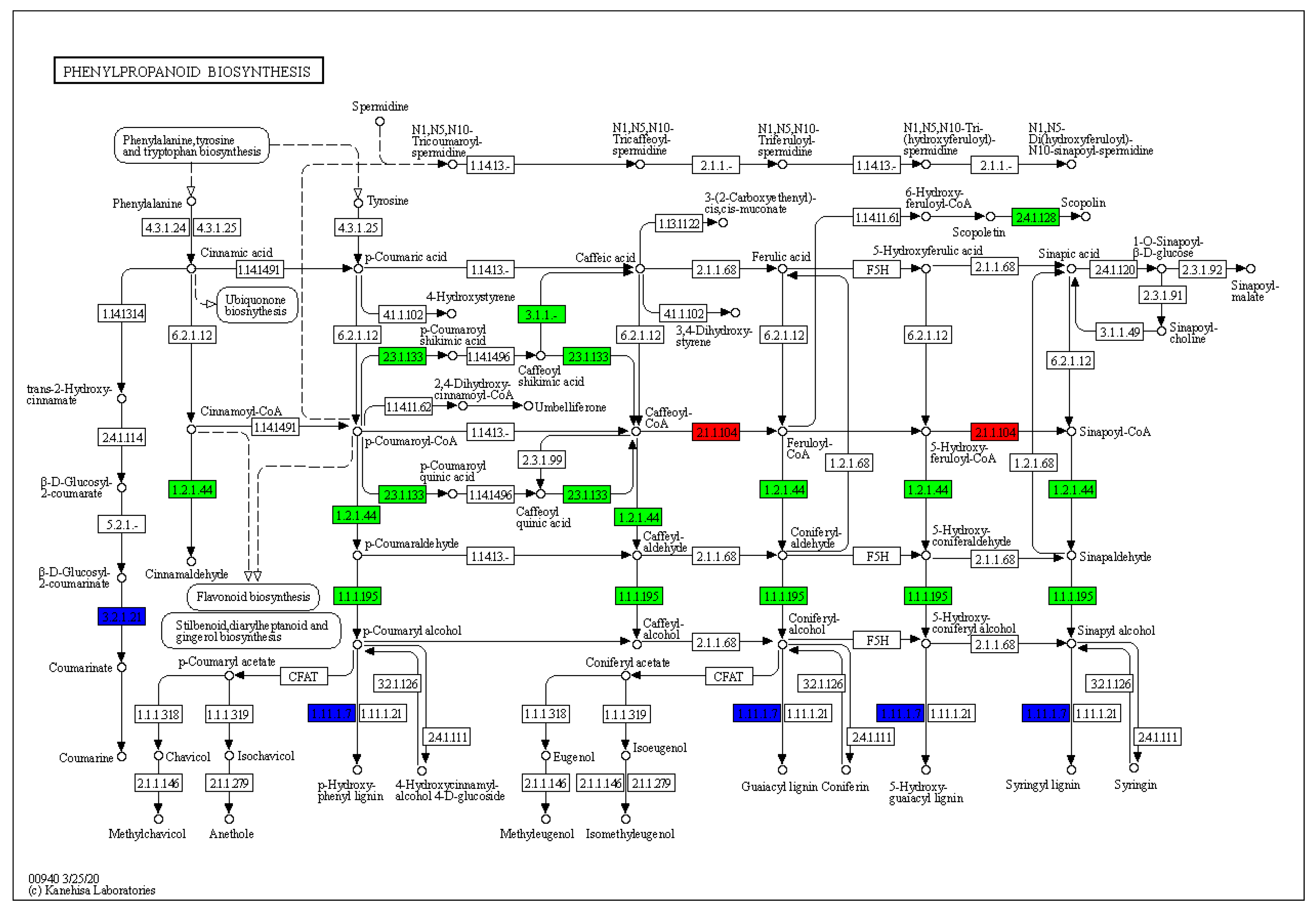
3.4. KEGG Analysis Revealed the DEG-Rich Metabolic Process
3.5. Plant-Hormone-Related Genes in NRC and RC
3.6. Differential Variable Shear Analysis
3.7. qRT-PCR Verification of Regenerative Gene in Hybrid Sweetgum Callus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gordon, S.P.; Heisler, M.G.; Reddy, G.V.; Ohno, C.; Das, P.; Meyerowitz, E.M. Pattern formation during de novo assembly of the Arabidopsis shoot meristem. Development 2007, 134, 3539–3548. [Google Scholar] [CrossRef]
- Su, Y.H.; Liu, Y.B.; Bai, B.; Zhang, X.S. Establishment of embryonic shoot-root axis is involved in auxin and cytokinin response during Arabidopsis somatic embryogenesis. Front. Plant Sci. 2015, 5, 792. [Google Scholar] [CrossRef]
- Wu, Y.L. Functional Studies on Structural Genes and Transcription Factors of Phenylpropane Pathway in Tea Plants; Anhui Agricultural University: Hefei, China, 2023. [Google Scholar]
- La, C.S.; Gouzerh, G.; Dhondt, S.; Hoffmann, L.; Fritig, B.; Legrand, M.; Heitz, T. Metabolic reprogramming in plant innate immunity: The contributions of phenylpropanoid and oxylipin pathways. Immunol. Rev. 2010, 198, 267–284. [Google Scholar]
- Huang, H.D. Molecular Mechanism Analysis of Arabidopsis KCS5 and KCS6/CER6 Synergistically Involved in Epidermal Wax Synthesis; Hubei University: Wuhan, China, 2022. [Google Scholar]
- Haslam, T.M.; Kunst, L. Extending the story of very-long-chain fatty acid elongation. Plant Sci. 2013, 210, 93–107. [Google Scholar] [CrossRef]
- Ketudat, C.; Mahong, B.; BAIYA, S. B-Glucosidases: Multitasking, moonlighting or simply misunderstood. Plant Sci. 2015, 241, 246–259. [Google Scholar] [CrossRef]
- Sampedro, J.; Valdivia, E.R.; Fraga, P.; Iglesias, N.; Revilla, G.; Zarra, I. Soluble and membrane-bound g-Glucosidases are involved in trimming the xylo-glucan backbone. Plant Physiol. 2017, 173, 1017–1030. [Google Scholar] [CrossRef]
- Yao, X.Z.; Chen, L.X.; Zhang, B.H.; Lv, L.T. The identification of tea tree BGLU gene families and expression pattern analysis. Seeds 2022, 9, 1–9. [Google Scholar]
- Roepke, J.; Gordon, H.; Neil, K.; Gidda, S.; Mullen, R.; Freixas, C.J.; Bray-Stone, D.; Bozzo, G.G. An Apoplastic β-Glucosidase is Essential for the Degradation of Flavonol 3-O-β-Glucoside- 7-O-α-Rhamnosides in Arabidopsis. Plant Cell Physiol. 2017, 58, 1030–1047. [Google Scholar] [CrossRef]
- Sun, H.H.; Xue, Y.M.; Lin, Y.F. Enhanced catalytic efficiency in quercetin-4'-glucoside hydrolysis of Thermotoga maritima β-glucosidase A by site-directed mutagenesis. J. Agric. Food Chem. 2014, 62, 6763–6770. [Google Scholar] [CrossRef]
- Kim, H.J.; Pesacreta, T.C.; Triplett, B.A. Cotton-fiber germin-like protein.II: Immunolocalization, purification and functional analysis. Planta 2004, 218, 525–535. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Lin, J.C.; Ren, Z.Z.; Chen, C.X.; Miki, D. Genome-wide distribution and functions of the AAE complex in epigenetic regulation in Arabidopsis. J. Integr. Plant Biol. 2021, 63, 707–722. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Zheng, L.; Ma, J.; Song, C.; An, N.; Zhang, D.; Zhao, C.; Qi, S.; Han, M. Genome-wide identification and expression profiling analysis of brassinolide signal transduction genes regulating apple tree architecture. Acta Physiol. Plant. 2017, 39, 1–19. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Corredoira, E.; Valladares, S.; Vieitez, A.M. Morphohistological analysis of the origin and development of somatic embryos from leaves of mature Quercus robur. Vitr. Cell. Dev. Biol.-Plant 2006, 42, 525–533. [Google Scholar] [CrossRef]
- Xu, Z.H.; Zhang, X.X.; Su, Y.H.; Hu, Y.X.; Xu, L.; Wang, J.W. Totipotency and regeneration of plant cells. Sci. China Life Sci. 2019, 49, 1282–1300. [Google Scholar]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, 593–601. [Google Scholar] [CrossRef]
- Zhou, Q.; Mao, P.; Luo, D.; Chai, X.T.; Deng, H.; Fang, Q.G.; Fang, L.F.; Nan, Z.B.; Wen, J.Q.; Liu, Z.P. Comparative transcriptome analyses reveal that the MsNST1 gene affects lignin synthesis in alfalfa (Medicago sativa L.). Crop J. 2022, 10, 1059–1072. [Google Scholar] [CrossRef]
- Young, H.K.; Gyung, H.H. Overexpression of cinnamyl alcohol dehydrogenase gene from sweetpotato enhances oxidative stress tolerance in transgenic Arabidopsis. Vitr. Cell. Dev. Biol.-Plant 2019, 55, 172–179. [Google Scholar]
- Zhao, D.Q.; Luan, Y.T.; Shi, W.B.; Zhang, X.Y.; Meng, J.S.; Tao, J. A Paeonia ostii caffeoyl-CoA O-methyltransferase confers drought stress tolerance by promoting lignin synthesis and ROS scavenging. Plant Sci. 2021, 303, 110765. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Ishida, A.; Suzuki, Y.; Aoki, Y.; Suzuki, S.; Enoki, S. Ethylene Induced by Sound Stimulation Enhances Anthocyanin Accumulation in Grape Berry Skin through Direct Upregulation of UDP-Glucose: Flavonoid 3-O-Glucosyltransferase. Cells 2021, 10, 2799. [Google Scholar] [CrossRef] [PubMed]
- Li, W.L.; Liu, T.T.; Chen, C.L.; Luo, X.H.; An, X.; Zhou, L.N.; Zhu, G.L.; Li, P.F. Progress in the mechanism of plant TCP protein. Mol. Plant Breed. 2023, 21, 4650–4658. [Google Scholar]
- Zhou, Y.; Xun, Q.Q.; Zhang, D.Z.; Lv, M.H.; Ou, Y.; Li, J. TCP transcription factors associate with PHYTOCHROME INTERACTING FACTOR 4 and CRYPTOCHROME 1 to regulate thermomorphogenesis in Arabidopsis thaliana. iScience 2019, 15, 600–610. [Google Scholar] [CrossRef]
- Han, X.; Yu, H.; Yuan, R.R.; Yang, Y.; An, F.J.; Qin, G.J. Arabidopsis transcription factor TCP5 controls plant thermomorphogenesis by positively regulating PIF4 activity. iScience 2019, 15, 611–622. [Google Scholar] [CrossRef]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, S.; Liu, Q.; Wu, K.; Zhang, J.; Wang, S.; Wang, Y.; Chen, X.; Zhang, Y.; Gao, C.; et al. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 2015, 47, 949–954. [Google Scholar] [CrossRef]
- Dai, X.H.; Wang, J.; Song, Y.G.; Liu, Z.H.; Xue, T.; Qiao, M.; Yu, Y.C.; Xin, W.; Xiang, F.N. Cytosine methylation of the FWA prooter promotes direct in vitro shoot regeneration in Arabidopsis thaliana. J. Integr. Plant Biol. 2021, 63, 1491–1504. [Google Scholar] [CrossRef]
- Miao, J.M. The Role of PtPLT1 and PtWOX5B in Poplar Bud Regeneration; Shandong Agricultural University: Tai’an, China, 2020. [Google Scholar]
- Chatfield, S.P.; Capron, R.; Severino, A.; Penttila, P.; Alfred, S.E.; Nahal, H.; Provart, N.J. Incipient stem cell niche conversion in tissue culture: Using a systems approach to probe early events in WUSCHEL-dependent conversion of lateral root primordia into shoot meristems. Plant J. Cell Mol. Biol. 2013, 73, 798–813. [Google Scholar] [CrossRef]
- Kaoru, S.; Jiao, Y.L.; Elliot, M.M. Arabidopsis regeneration from multiple tissues occurs via a root development pathway. Dev. Cell 2010, 18, 463–471. [Google Scholar]
- Shi, L. Study on WOX gene mining and its effect on genetic transformation efficiency and mechanism of action in wheat. Chin. Acad. Agric. Sci. 2022, 4, 157. [Google Scholar]
- Hao, Q.; Zhang, L.; Yang, Y.; Shan, Z.; Zhou, X.A. Genome-wide analysis of the WOX gene gamily and function exploration of GmWOX18 in soybean. Plants 2009, 8, 215. [Google Scholar] [CrossRef]
- Lowe, K.; La, R.M.; Hoerster, G.; Hastings, C.; Wang, N.; Chamberlin, M.; Wu, E.; Jones, T.; Gordon-Kamm, W. Rapid genotype “independent” Zea mays L. (maize) transformation via direct somatic embryogenesis. In vitro cellular & developmental biology. Plant J. Tissue Cult. Assoc. 2018, 54, 240–252. [Google Scholar]
- Mookkan, M.; Nelson-Vasilchik, K.; Hague, J.; Zhang, Z.Y.; Kausch, A.P. Selectable marker independent transformation of recalcitrant maize inbred B73 and sorghum P898012 mediated by morphogenic regulators BABY BOOM and WUSCHEL2. Plant Cell Rep. 2017, 36, 1477–1491. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D1-1 | D1-2 | D2-1 | D2-2 | |
|---|---|---|---|---|
| No. of total reads | 41,873,814 | 42,299,632 | 39,227,620 | 42,584,274 |
| No. of mapped reads | 32,813,900 (78.36%) | 32,438,906 (76.69%) | 30,517,548 (77.80%) | 33,318,882 (78.24%) |
| Unique mapped reads | 31,850,925 (76.06%) | 31,217,955 (73.80%) | 29,624,803 (75.52%) | 32,240,118 (75.71%) |
| Multiple map reads | 962,975 (2.30%) | 1,220,951 (2.89%) | 892,745 (2.28%) | 1,078,764 (2.53%) |
| % ≥ Q30 1 | 94.36 | 93.88 | 93.89 | 94.09 |
| Hormone | Total Gene | Number of Differentially Expressed Genes | Quantity Down Value | Quantity Up Value |
|---|---|---|---|---|
| Auxin | 292 | 15 | 14 | 1 |
| Abscisic acid | 197 | 13 | 13 | 0 |
| Brassinosteroid | 340 | 11 | 10 | 1 |
| Cytokinin | 108 | 1 | 1 | 0 |
| Ethylene | 184 | 10 | 8 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ai, Z.; Ma, H.; Zhang, T.; Chen, S.; Zhang, J. Molecular Regulation of Bud Regeneration from Callus of Hybrid Sweetgum (Liquidambar styraciflua × Liquidambar formosana). Forests 2023, 14, 1833. https://doi.org/10.3390/f14091833
Ai Z, Ma H, Zhang T, Chen S, Zhang J. Molecular Regulation of Bud Regeneration from Callus of Hybrid Sweetgum (Liquidambar styraciflua × Liquidambar formosana). Forests. 2023; 14(9):1833. https://doi.org/10.3390/f14091833
Chicago/Turabian StyleAi, Zhongyao, Haiyao Ma, Ting Zhang, Siyuan Chen, and Jinfeng Zhang. 2023. "Molecular Regulation of Bud Regeneration from Callus of Hybrid Sweetgum (Liquidambar styraciflua × Liquidambar formosana)" Forests 14, no. 9: 1833. https://doi.org/10.3390/f14091833
APA StyleAi, Z., Ma, H., Zhang, T., Chen, S., & Zhang, J. (2023). Molecular Regulation of Bud Regeneration from Callus of Hybrid Sweetgum (Liquidambar styraciflua × Liquidambar formosana). Forests, 14(9), 1833. https://doi.org/10.3390/f14091833