Comparative Genomics of Eight Complete Chloroplast Genomes of Phyllostachys Species


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Genome Sequencing
2.2. Genome Assembly and Annotation
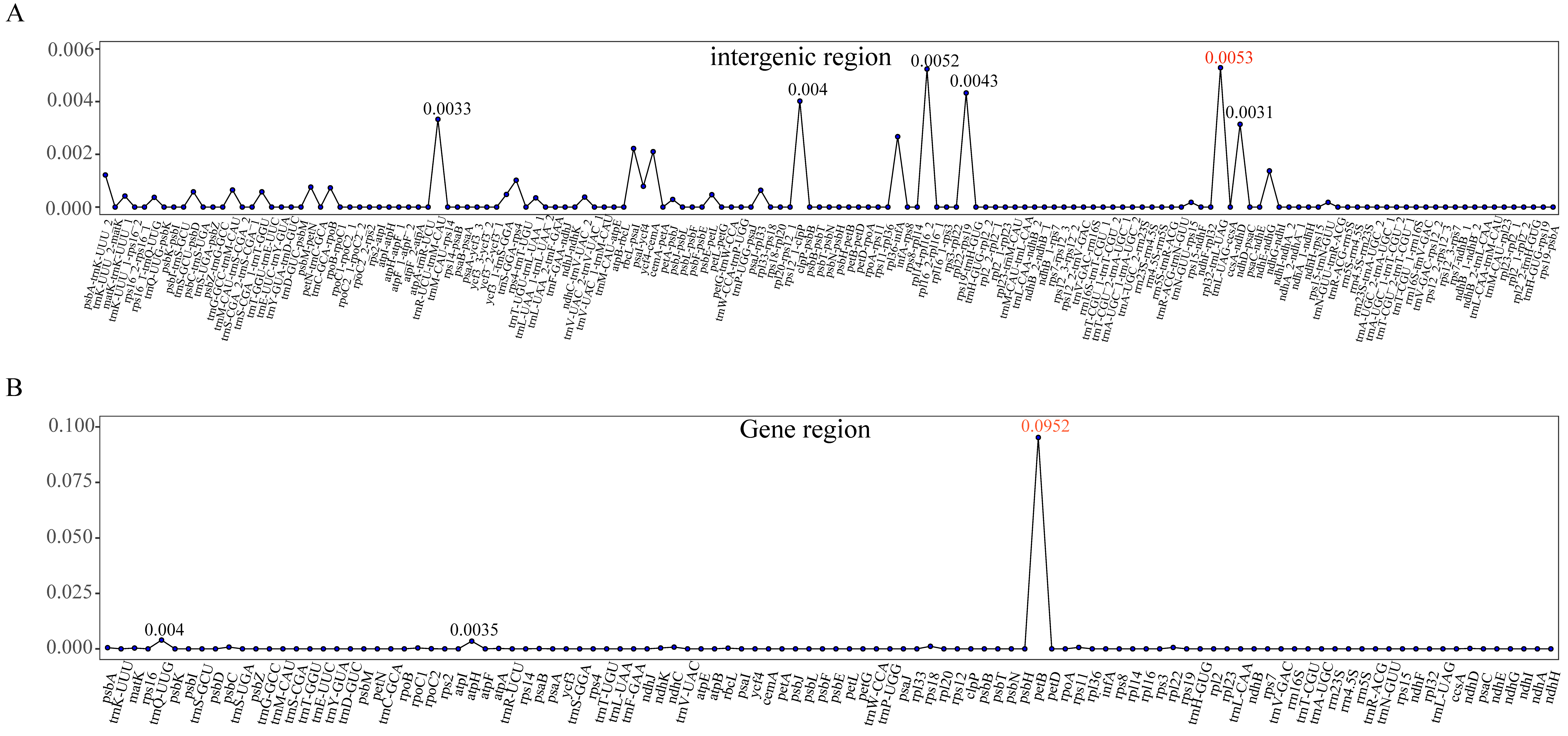
2.3. Comparative Genomic Analysis and Identification of Variation Hotspots
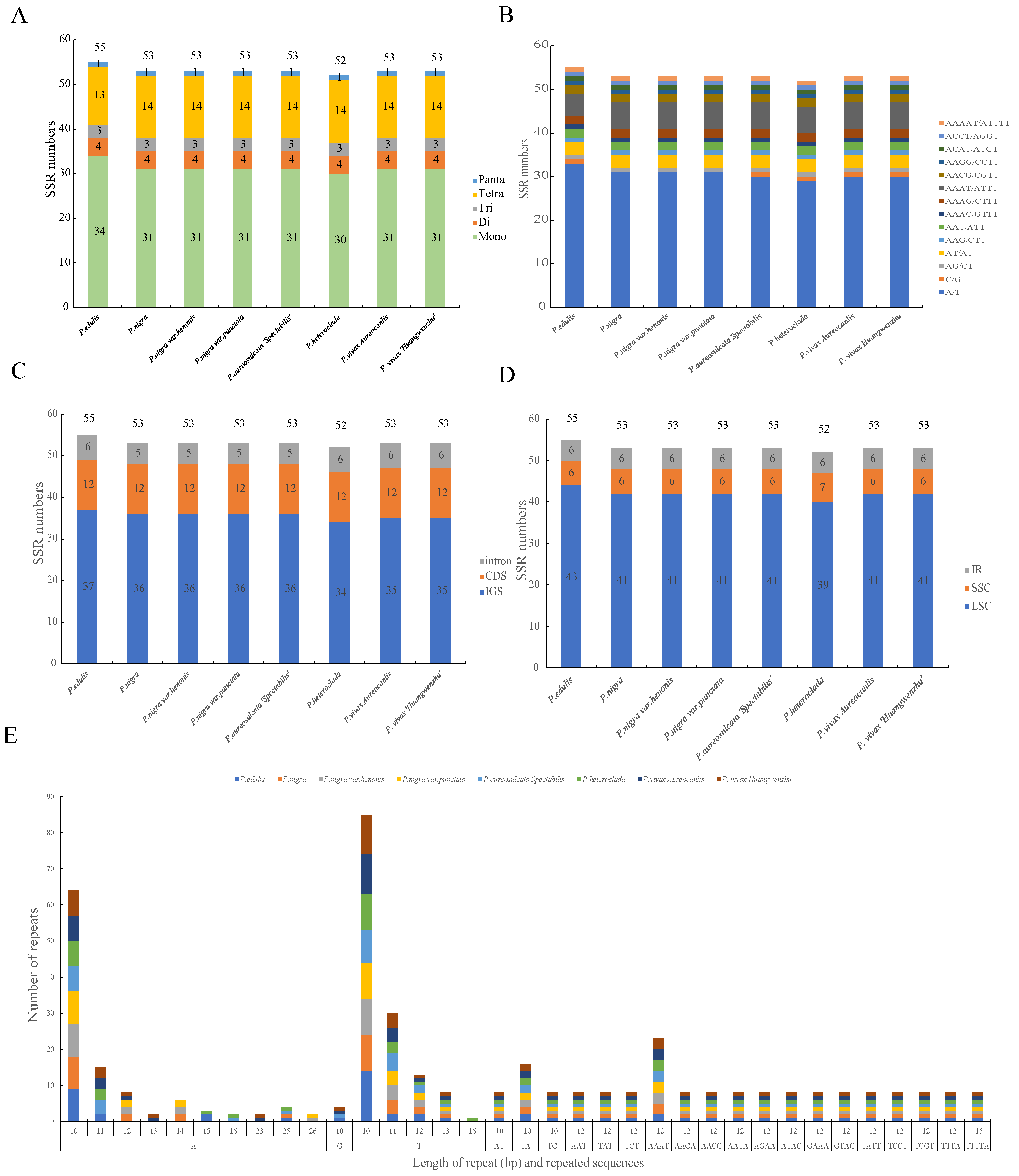
2.4. Characterization of Repeat Sequences and SSRs
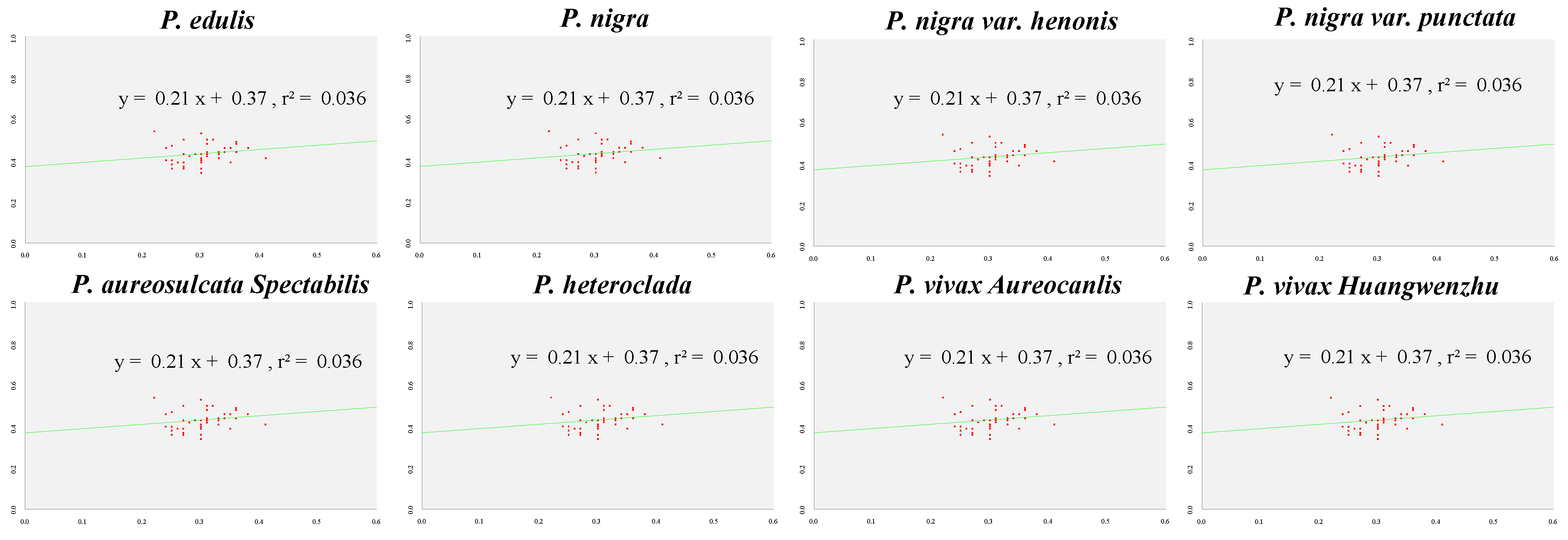
2.5. Codon Usage Analysis
3. Results
3.1. Chloroplast Genome Organization and Content
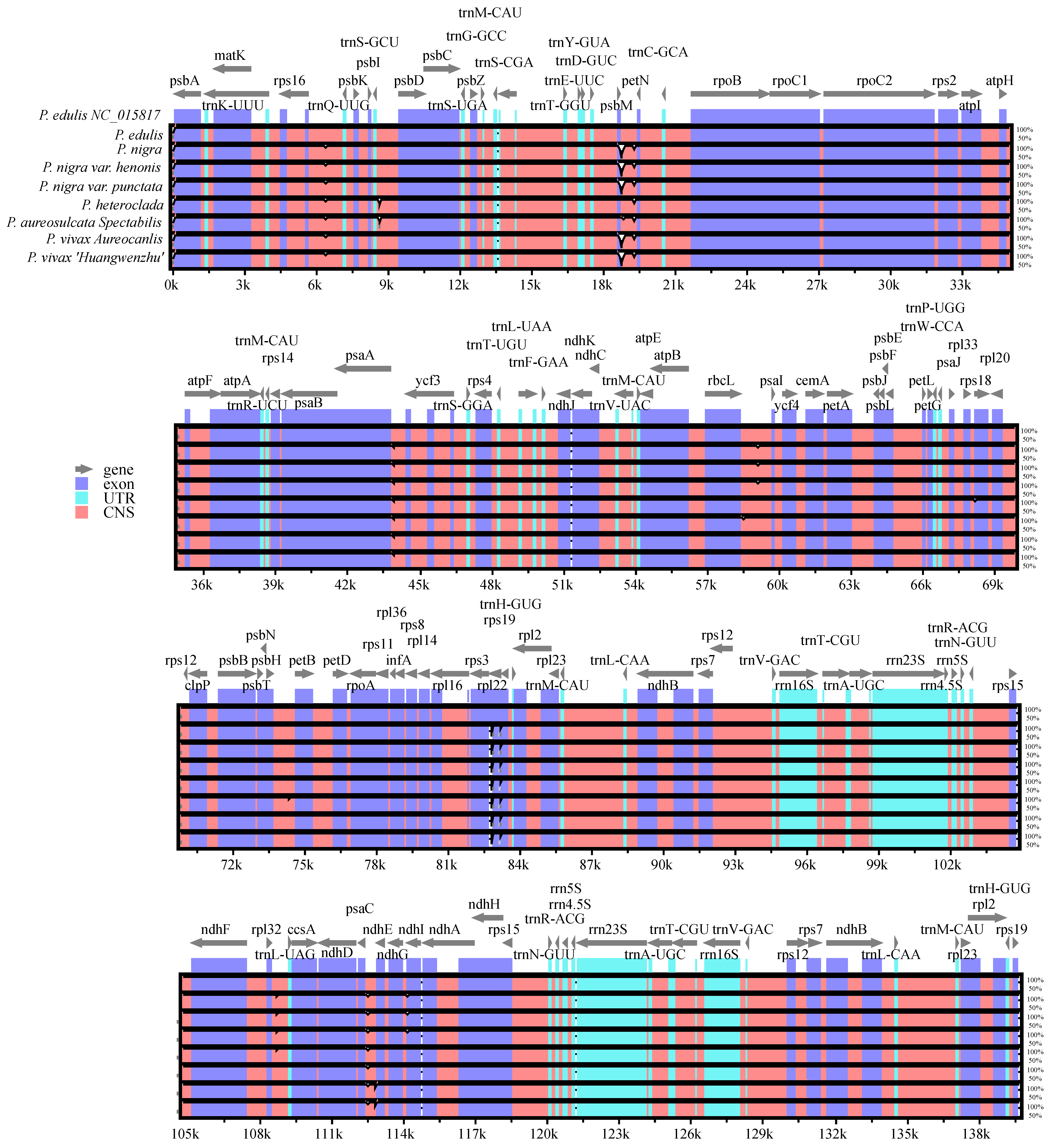
3.2. Genome Comparative Analyses
3.3. SSR and Repeat Sequences Analysis
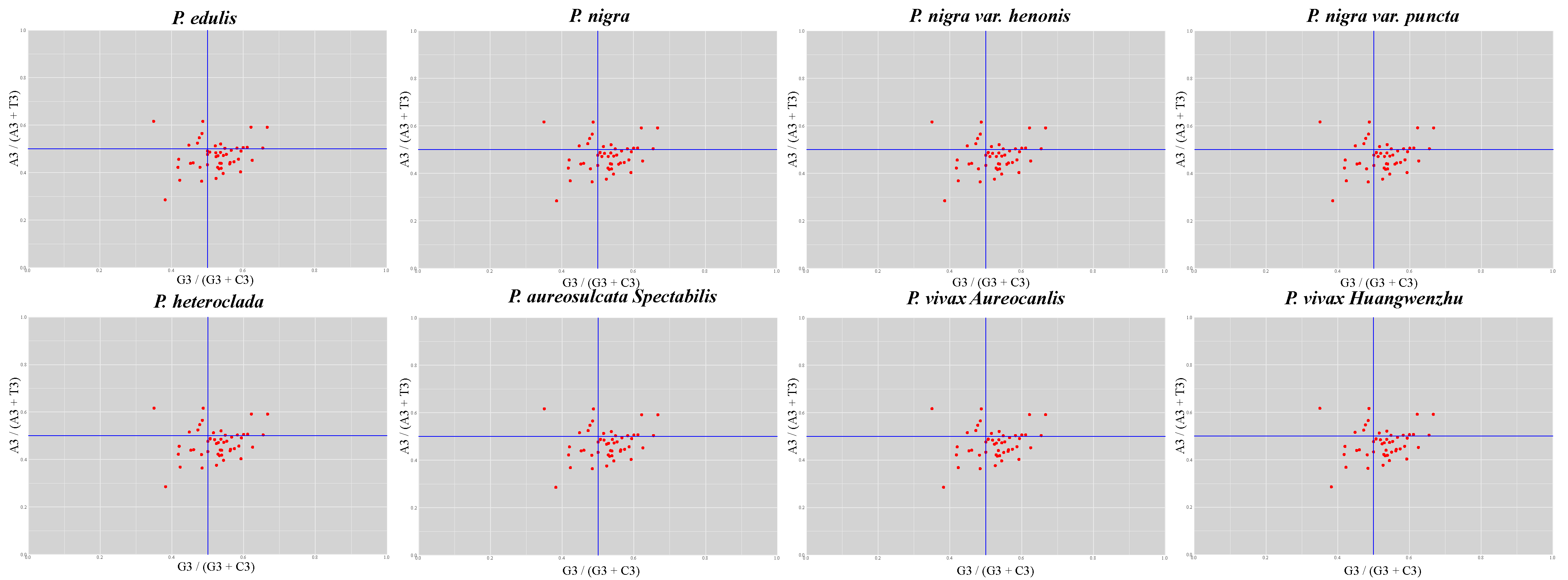
3.4. Codon Preference Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buchanan, B.B.; Gruissem, W.; Jones, R.L. Biochemistry and Molecular Biology of Plants, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, Y.; Gao, L. The complete chloroplast genome sequence of Phyllostachys heterocycla, a fast-growing non-timber bamboo (Poaceae: Bambusoideae). Conserv. Genet. Resour. 2017, 9, 217–219. [Google Scholar] [CrossRef]
- Pei, J.; Wang, Y.; Zhuo, J.; Gao, H.; Vasupalli, N.; Hou, D.; Lin, X. Complete chloroplast genome features of Dendrocalamus farinosus and its comparison and evolutionary analysis with other Bambusoideae species. Genes 2022, 13, 1519. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Yao, H.; Song, J.; Zhu, Y.; Liu, C.; Chen, S. Evaluating the feasibility of using candidate DNA barcodes in discriminating species of the large Asteraceae family. BMC Evol. Biol. 2010, 10, 324. [Google Scholar] [CrossRef]
- Wang, W.; Lanfear, R. Long-Reads Reveal That the Chloroplast Genome Exists in Two Distinct Versions in Most Plants. Genome Biol. Evol. 2019, 11, 3372–3381. [Google Scholar] [CrossRef]
- De Vries, J.; Archibald, J.M. Plastid genomes. Curr. Biol. 2018, 28, R336–R337. [Google Scholar] [CrossRef]
- Delannoy, E.; Fujii, S.; Colas des Francs-Small, C.; Brundrett, M.; Small, I. Rampant Gene Loss in the Underground Orchid Rhizanthella gardneri Highlights Evolutionary Constraints on Plastid Genomes. Mol. Biol. Evol. 2011, 28, 2077–2086. [Google Scholar] [CrossRef]
- Wu, C.; Chaw, S. Highly rearranged and size-variable chloroplast genomes in conifers II clade (cupressophytes): Evolution towards shorter intergenic spacers. Plant Biotechnol. J. 2014, 12, 344–353. [Google Scholar] [CrossRef]
- Wheeler, G.L.; Dorman, H.E.; Buchanan, A.; Challagundla, L.; Wallace, L.E. A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Appl. Plant Sci. 2014, 2, 1400059. [Google Scholar] [CrossRef]
- Wu, L.; Nie, L.; Wang, Q.; Xu, Z.; Wang, Y.; He, C.; Song, J.; Yao, H. Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae. Sci. Rep. 2021, 11, 14643. [Google Scholar] [CrossRef]
- Wang, Q.; Yue, J.; Yan, J. Research progress on maintaining chloroplast homeostasis under stress conditions: A review. Acta Biochim. Biophys. Sin. 2023, 55, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, L.; Lu, C. Chloroplast gene expression: Recent advances and perspectives. Plant Commun. 2023, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Dobrogojski, J.; Adamiec, M.; Lucinski, R. The chloroplast genome: A review. Acta Physiol. Plant. 2020, 42, 98. [Google Scholar] [CrossRef]
- Fages-Lartaud, M.; Hundvin, K.; Hohmann-Marriott, M.F. Mechanisms governing codon usage bias and the implications for protein expression in the chloroplast of Chlamydomonas reinhardtii. Plant J. 2022, 112, 919–945. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Shen, L.; Chen, S.; Qu, S.; Hou, N. Codon Usage Profiling of Chloroplast Genome in Juglandaceae. Forests 2023, 14, 378. [Google Scholar] [CrossRef]
- Clark, L.G.; Londono, X.; Ruiz-Sanchez, E. Bamboo taxonomy and habitat. In Bamboo: Tropical Forestry; Liese, W.K., Ed.; Springer: Cham, Switzerland, 2015; Volume 10, pp. 1–30. [Google Scholar]
- Ohrnberger, D. The Bamboos of the World: Annotated Nomenclature and Literature of the Species and the Higher and Lower Taxa; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Gielis, J.; Potters, G. An updated tribal and subtribal classification of the bamboos (Poaceae: Bambusoideae). In Proceedings of the IX World Bamboo Congress 2012, Antwerp, Belgium, 10–12 April 2012; pp. 3–27. [Google Scholar]
- Clark, L.G.; Oliveira, R.P. Diversity and Evolution of the New World Bamboos. In Proceedings of the World Bamboo Congress, Xalapa, Mexico, 14–18 August 2018. [Google Scholar]
- Zhou, G.; Meng, C.; Jiang, P.; Xu, Q. Review of carbon fixation in bamboo forests in China. Bot. Rev. 2011, 77, 262–270. [Google Scholar] [CrossRef]
- McCormac, D.J.; Litz, H.; Wang, J.X.; Gollnick, P.D.; Berry, J.O. Light-associated and processing-dependent protein binding to 5′ regions of rbcL mRNA in the chloroplasts of a C4 plant. J. Biol. Chem. 2001, 276, 3476–3483. [Google Scholar] [CrossRef]
- Mahapatra, K.; Mukherjee, A.; Suyal, S.; Dar, M.A.; Bhagavatula, L.; Datta, S. Regulation of chloroplast biogenesis, development, and signaling by endogenous and exogenous cues. Physiol. Mol. Biol. Plants 2024, 30, 167–183. [Google Scholar] [CrossRef]
- Wu, F.-H.; Kan, D.-P.; Lee, S.-B.; Daniell, H.; Lee, Y.-W.; Lin, N.-S.; Lin, C.-S. Complete nucleotide sequence of Dendrocalamus latiflorus and Bambusa oldhamii chloroplast genomes. Tree Physiol. 2009, 29, 847–856. [Google Scholar] [CrossRef]
- Ma, P.-F.; Zhang, Y.-X.; Guo, Z.-H.; Li, D.-Z. Evidence for horizontal transfer of mitochondrial DNA to the plastid genome in a bamboo genus. Sci. Rep. 2015, 5, 11608. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Ma, P.F.; Li, D.Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.V.; Grennan, C.P.; Duvall, M.R. Plastome sequences of two New World bamboos—Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)—Extend phylogenomic understanding of Bambusoideae. Am. J. Bot. 2012, 99, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Gao, L. The complete chloroplast genome sequence of Phyllostachys sulphurea (Poaceae: Bambusoideae). Mitochondrial DNA Part A 2016, 27, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, W.P.; Clark, L.G.; Attigala, L.; Ruiz-Sanchez, E.; Duvall, M.R. Evolution of the bamboos (Bambusoideae; Poaceae): A full plastome phylogenomic analysis. BMC Evol. Biol. 2015, 15, 50. [Google Scholar] [CrossRef]
- Ma, P.-F.; Zhang, Y.-X.; Zeng, C.-X.; Guo, Z.-H.; Li, D.-Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, J.; Yu, Z.-Y.; Li, J.-J.; Xu, M.-Y.; Guo, Q.-R. The complete chloroplast genome of Phyllostachys heteroclada f. solida (Poaceae). Mitochondrial DNA Part B 2021, 6, 566–567. [Google Scholar]
- Jie, Z.; Ya, H.; Zhaoyan, Y.; Zhou, J.; Hu, Y.; Yu, Y.; Li, J.; Xu, M.; Guo, Q. The complete chloroplast genome of a solid type of Phyllostachys nidularia (Bambusoideae: Poaceae), a species endemic to China. Mitochondrial DNA Part B 2021, 6, 978–979. [Google Scholar] [CrossRef]
- Liu, X.; Liu, L.; Li, L.; Yue, J. The complete chloroplast genome of Phyllostachys edulis f. tubiformis (Bambusoideae): A highly appreciated type of ornamental bamboo in China. Mitochondrial DNA Part B 2022, 7, 185–187. [Google Scholar] [CrossRef]
- Jj, D. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Depamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Li, H.; Guo, Q.; Xu, L.; Gao, H.; Liu, L.; Zhou, X. CPJSdraw: Analysis and visualization of junction sites of chloroplast genomes. PeerJ 2023, 11, e15326. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, L.; Wang, W.; Zhang, Z.; Du, H.; Qu, Z.; Li, X.-Q.; Xiang, H. Differences in codon usage bias between photosynthesis-related genes and genetic system-related genes of chloroplast genomes in cultivated and wild solanum species. Int. J. Mol. Sci. 2018, 19, 3142. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, B.; Li, B.; Zhou, Q.; Wang, G.; Jiang, X.; Wang, C.; Xu, Z. Comparative analysis of codon usage patterns in chloroplast genomes of six Euphorbiaceae species. PeerJ 2020, 8, e8251. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Yengkhom, S.; Uddin, A. Analysis of codon usage bias of chloroplast genes in Oryza species: Codon usage of chloroplast genes in Oryza species. Planta 2020, 252, 67. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yang, Q. An improved implementation of effective Number of Codons (N c). Mol. Biol. Evol. 2013, 30, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Huang, N.J.; Li, J.P.; Yang, G.Y.; Yu, F. Two plastomes of Phyllostachys and reconstruction of phylogenic relationship amongst selected Phyllostachys species using genome skimming. Mitochondrial DNA Part B 2020, 5, 69–70. [Google Scholar] [CrossRef]
- Gao, L.Q.; Li, Y.L.; Zhang, W.G.; Yang, G.Y. The complete chloroplast genome of Phyllostachys edulis f. curviculmis (Bambusoideae): A newly ornamental bamboo endemic to China. Mitochondrial DNA Part B 2021, 6, 941–942. [Google Scholar] [CrossRef]
- Hurt, E.; Günter, H. A cytochrome f/b6 complex of five polypeptides with plastoquinol-plastocyanin-oxidoreductase activity from spinach chloroplasts. Eur. J. Biochem. 1981, 3, 591–599. [Google Scholar] [CrossRef]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Depamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar]
- Kaila, T.; Chaduvla, P.K.; Saxena, S.; Bahadur, K.; Gahukar, S.J.; Chaudhury, A.; Sharma, T.R.; Singh, N.K.; Gaikwad, K. Chloroplast genome sequence of Pigeonpea (Cajanus cajan (L.) Millspaugh) and Cajanus scarabaeoides (L.) Thouars: Genome organization and comparison with other legumes. Front. Plant Sci. 2016, 7, 1847. [Google Scholar] [CrossRef]
- Choi, K.S.; Ogyeong, S.; Seonjoo, P. The chloroplast genome of Elaeagnus macrophylla and trnH duplication event in Elaeagnaceae. PLoS ONE 2015, 10, e0138727. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; dePamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Saski, C.; Lee, S.-B.; Hansen, A.K.; Daniell, H. Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): Evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 2011, 28, 835–847. [Google Scholar] [CrossRef]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T.; et al. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Qian, J.; Li, X.; Sun, Z.; Xu, X.; Chen, S. Complete chloroplast genome of medicinal plant Lonicera japonica: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 2017, 22, 249. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.-Y.; Wu, H.; Wang, Y.-L.; Gao, L.-M.; Zhang, S.-Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; A Rafalski, J. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef]
- Qin, Z.; Wang, Y.; Wang, Q.; Li, A.; Hou, F.; Zhang, L. Evolution analysis of simple sequence repeats in plant genome. PLoS ONE 2015, 10, e0144108. [Google Scholar] [CrossRef]
- Tautz, D.; Renz, M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res. 1984, 12, 4127–4138. [Google Scholar] [CrossRef]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Iram, S.; Hayat, M.Q.; Tahir, M.; Gul, A.; Abdullah; Ahmed, I. Chloroplast genome sequence of Artemisia scoparia: Comparative analyses and screening of mutational hotspots. Plants 2019, 8, 476. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yao, W.; Guo, C.; Bian, L.; Ding, Y.; Lin, S. Chloroplast Genome Variation and Phylogenetic Analyses of Seven Dwarf Ornamental Bamboo Species. Forests 2022, 13, 1671. [Google Scholar] [CrossRef]
- Gao, L.; Yi, X.; Yang, Y.-X.; Su, Y.-J.; Wang, T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: Insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 2009, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Cavalier-Smith, T. Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 2002, 12, R62–R64. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2017, 7, 2054. [Google Scholar] [CrossRef]
- Wei, X.; Li, X.; Chen, T.; Chen, Z.; Jin, Y.; Malik, K.; Li, C. Complete chloroplast genomes of Achnatherum inebrians and comparative analyses with related species from Poaceae. FEBS Open Bio 2021, 11, 1704–1718. [Google Scholar] [CrossRef]
- Vieira, L.N.; Faoro, H.; Rogalski, M.; de Freitas Fraga, H.P.; Cardoso, R.L.A.; de Souza, E.M.; de Oliveira Pedrosa, F.; Nodari, R.O.; Guerra, M.P. The complete chloroplast genome sequence of Podocarpus lambertii: Genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 2014, 9, e90618. [Google Scholar] [CrossRef]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef]
- Quax, T.E.; Claassens, N.J.; Söll, D.; van der Oost, J. Codon bias as a means to fine-tune gene expression. Mol. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef]
- Tuller, T.; Waldman, Y.Y.; Kupiec, M.; Ruppin, E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. USA 2010, 107, 3645–3650. [Google Scholar] [CrossRef]
- Uddin, A. Indices of codon usage bias. J. Proteomics Bioinform. 2017, 10, 1000e34. [Google Scholar] [CrossRef]
- Tang, D.; Wei, F.; Cai, Z.; Wei, Y.; Khan, A.; Miao, J.; Wei, K. Analysis of codon usage bias and evolution in the chloroplast genome of Mesona chinensis Benth. Dev. Genes Evol. 2021, 231, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Deng, P.; Feng, K.; Liu, P.; Du, X.; You, F.M.; Weining, S. Comparative analysis of codon usage patterns in chloroplast genomes of the Asteraceae family. Plant Mol. Biol. Rep. 2014, 32, 828–840. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | P. edulis | P. nigra | P. nigra var. punctata | P. aureosulcata spectabilis | P. heteroclada | P. vivax aureocanlis |
|---|---|---|---|---|---|---|
| Total size (bp) | 139,679 | 139,708 | 139,709 | 139,701 | 139,669 | 139,715 |
| LSC length (bp) | 83,213 | 83,233 | 83,234 | 83,223 | 83,199 | 83,220 |
| IR length (bp) | 21,798 | 21,798 | 21,798 | 21,798 | 21,798 | 21,798 |
| SSC length (bp) | 12,870 | 12,879 | 12,879 | 12,882 | 12,874 | 12,899 |
| Total genes | 108 | 108 | 108 | 108 | 108 | 107 |
| Protein-coding genes | 76 | 76 | 76 | 76 | 76 | 75 |
| tRNA genes | 28 | 28 | 28 | 28 | 28 | 28 |
| rRNA genes | 4 | 4 | 4 | 4 | 4 | 4 |
| Total GC (%) | 38.9 | 38.9 | 38.9 | 38.9 | 38.9 | 38.9 |
| LSC of GC (%) | 36.97 | 36.98 | 36.98 | 36.97 | 36.98 | 36.97 |
| IR of GC (%) | 44.22 | 44.22 | 44.22 | 44.22 | 44.22 | 44.22 |
| SSC of GC (%) | 33.17 | 33.15 | 33.15 | 33.14 | 33.15 | 33.14 |
| Accession number | PQ325516 | PQ325517 | PQ325523 | PQ325519 | PQ325520 | PQ325521 |
| Gene Group | Gene Name |
|---|---|
| Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| NADPH dehydrogenase | ndhA a, ndhB ac, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Cytochrome b/f complex | petA, petB, petD, petG, petL, petN |
| ATP synthase | atpA, atpB, atpE, atpF a, atpH, atpI |
| Rubisco | rbcL |
| Large ribosomal units | rpl14, rpl16 a, rpl2 ac, rpl20, rpl22, rpl23 c, rpl32, rpl33, rpl36 |
| Small ribosomal units | rps11, rps12 bc, rps14, rps15 c, rps16 a, rps18, rps19 c, rps2, rps3, rps4, rps7 c, rps8 |
| RNA polymerase sub-units | rpoA, rpoB, rpoC1, rpoC2 a |
| Ribosomal RNA | rrn16S c, rrn23S c, rrn4.5S c, rrn5S c |
| Transfer RNA | trnA-UGC ac, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnH-GUG c, trnK-UUU a, trnL-CAA c, trnL-UAA a, trnL-UAG, trnM-CAU c, trnN-GUUc, trnP-UGG, trnQ-UUG, trnR-ACG c, trnR-UCU, trnS-CGA a, trnS-GCU, trnS-GGA, trnS-UGA, trnT-CGU ac, trnT-GGU, trnT-UGU, trnV-GAC c, trnV-UAC a, trnW-CCA, trnY-GUA |
| Maturase | matK |
| Protease | clpP |
| Envelope membrane protein | cemA |
| c-Type cytochrome synthesis | ccsA |
| Translation initiation factor | infA |
| Hypothetical genes reading frames | ycf3 b, ycf4 |
| Species | Codon No. | GC1 | GC2 | GC3 | GC | CAI | ENC |
|---|---|---|---|---|---|---|---|
| P. edulis | 16,433 | 0.4778 | 0.3934 | 0.3089 | 0.3934 | 0.167 | 50.40 |
| P. aureosulcata spectabilis | 16,433 | 0.4777 | 0.3933 | 0.309 | 0.3933 | 0.167 | 50.42 |
| P. heteroclada | 16,433 | 0.4777 | 0.3934 | 0.309 | 0.3934 | 0.167 | 50.42 |
| P. nigra var. henonis | 16,433 | 0.4777 | 0.3933 | 0.3091 | 0.3934 | 0.167 | 50.42 |
| P. nigra var. punctata | 16,433 | 0.4777 | 0.3933 | 0.3091 | 0.3934 | 0.167 | 50.42 |
| P. nigra | 16,433 | 0.4777 | 0.3933 | 0.3091 | 0.3934 | 0.167 | 50.42 |
| P. vivax aureocanlis | 16,433 | 0.4777 | 0.3932 | 0.309 | 0.3933 | 0.167 | 50.42 |
| P. vivax ‘Huangwenzhu’ | 16,433 | 0.4777 | 0.3932 | 0.309 | 0.3933 | 0.167 | 50.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Liu, G.; Liu, C. Comparative Genomics of Eight Complete Chloroplast Genomes of Phyllostachys Species. Forests 2024, 15, 1785. https://doi.org/10.3390/f15101785
Li G, Liu G, Liu C. Comparative Genomics of Eight Complete Chloroplast Genomes of Phyllostachys Species. Forests. 2024; 15(10):1785. https://doi.org/10.3390/f15101785
Chicago/Turabian StyleLi, Guolei, Guohua Liu, and Changlai Liu. 2024. "Comparative Genomics of Eight Complete Chloroplast Genomes of Phyllostachys Species" Forests 15, no. 10: 1785. https://doi.org/10.3390/f15101785
APA StyleLi, G., Liu, G., & Liu, C. (2024). Comparative Genomics of Eight Complete Chloroplast Genomes of Phyllostachys Species. Forests, 15(10), 1785. https://doi.org/10.3390/f15101785