Abstract
Jujube (Ziziphus jujuba Mill.) stands as a pivotal fruit tree with significant economic, ecological, and social value. Recent years have witnessed remarkable strides in multi-omics-based biological research on jujube. This review began by summarizing advancements in jujube genomics. Subsequently, we provided a comprehensive overview of the integrated application of genomics, transcriptomics, and metabolomics to explore pivotal genes governing jujube domestication traits, quality attributes (including sugar synthesis, terpenoids, and flavonoids), and responses to abiotic stress and discussed the transcriptional regulatory mechanisms underlying these traits. Furthermore, challenges in multi-omics research on jujube biological traits were outlined, and we proposed the integration of resources such as pan-genomics and sRNAome to unearth key molecules and regulatory networks influencing diverse biological traits. Incorporating these molecules into practical breeding strategies, including gene editing, transgenic approaches, and progressive breeding, holds the potential for achieving molecular-design breeding and efficient genetic enhancement of jujube.
1. Introduction
Jujube (Ziziphus jujuba Mill.), commonly known as Chinese date, is a member of the Rhamnaceae family, comprising 170 species and 12 varieties within the Ziziphus genus globally [1]. Originating from wild jujube in the middle and lower reaches of the Yellow River in China [2], cultivated jujube has been a staple for over 4000 years [3]. Presently, jujube trees are cultivated in 47 countries, spanning tropical and subtropical regions in Europe, Asia, along the Silk Road (India, Iran, Russia, and the Middle East), and even the United States [4]. This fruit tree holds immense economic, ecological, and social significance. In China alone, the cultivation area for jujube exceeds three million hectares, yielding over seven million tons [5]. This has translated into an annual output value of approximately USD 6 to 10 billion, based on a field-picking price ranging from USD 0.8 to 1.3 per kilogram over the past five years.
Cultivated jujube trees, derived from the wild Ziziphus jujuba var. spinosa (2n = 2x = 24) through prolonged selection [6,7], exhibit distinctive characteristics. These plants reach heights of 3 to 10 m, featuring elliptical or ovate leaves (2 to 4 cm wide and 2.5 to 5.5 cm long) and yellow-green flowers with five petals, sepals, and anthers, respectively [8]. Jujube demonstrates robust adaptability to various soil environments, particularly thriving under drought and salt-alkali stress [9]. Rich in nutrients such as polysaccharides, amino acids, ascorbic acid, triterpene acids, flavonoids, phenolic acids, and minerals across its roots, stems, leaves, flowers, and fruits [10], this plant’s active molecules hold therapeutic potential for diverse ailments. For instance, triterpene acids exhibit antioxidant, anti-inflammatory, antibacterial, and anti-cancer properties [11,12]. The appealing crispness, delightful flavor, and high nutritional value of jujube fruits have captured the interest of both consumers and breeders [13]; ongoing research delves into understanding the formation of key traits and the metabolic synthesis of active molecules.
The intricate process involved in shaping jujube biological traits and accumulating active molecules encompasses numerous complexities. Flower bud differentiation, for instance, relies on a dynamic balance of environmental factors, internal nutrients, and various endogenous hormones, resulting in swift and multi-state differentiation [14,15]. Jujube fruit growth and development is divided into four stages, young fruit, expansion, hard core, and maturity, influencing shape, size, peel color, and intrinsic qualities (nutrients, flavor, and soluble solids) [16]. The formation of intrinsic quality correlates with the synthesis of numerous primary and secondary metabolites. In jujube genetic transformation development, modified engineering breeding of jujube will be an important direction. Thus, studying the mechanisms governing the molecular and chemical aspects of these key traits presents an opportunity for innovative and advanced methodologies.
In recent years, the advent of next-generation sequencing has revolutionized plant biology research, offering a cost-effective solution, even in non-model systems [17]. Second-generation sequencing data correlation analysis, particularly, has successfully unraveled molecular mechanisms governing the formation of multiple crop traits and the synthesis of biological components [18,19,20]. These methodologies have found widespread application in jujube studies. The analysis of key genes associated with biological traits and their regulatory networks lays a foundational groundwork for a comprehensive understanding of the molecular mechanisms driving jujube’s biological traits and the pathways of active substance synthesis. This research also contributes novel biomarkers for breeding initiatives.
To the best of our knowledge, a systematic and comprehensive review focusing on the transcriptional regulatory mechanisms of jujube biological traits based on multi-omics remains limited. Our objective is to provide an overview of recent advancements in understanding the key genes that regulate jujube domestication traits, quality traits (such as sugar, terpenoid, and flavonoid synthesis), and responses to abiotic stress. Leveraging data from the genome, transcriptome, and metabolome, we explore the molecular regulatory mechanisms governing these traits. Furthermore, we discuss the challenges associated with multi-omics research in deciphering the formation of biological traits in jujube and highlight the potential applications of identified key genes in breeding programming.
2. Progress in Genome Sequencing and Assembly of Jujube Genomes
Recent years have witnessed intensive research on jujube plant genomics, driven by the advancement of second-generation sequencing technology and plant genomics. In 2014, Liu et al. [21] achieved a milestone by completing the whole genome map of the fresh food cultivar “Dongzao”. Utilizing whole-genome shotgun and BAC sequencing methods along with a de novo genome assembly strategy, they unveiled a genome of 437.65 Mb, hosting 32,808 annotated coding genes, with 23,996 genes distributed across 12 chromosomes. In addition, 410 rRNA, 1209 tRNA, 286 snRNA, and 272 miRNAs were annotated, as well as 204.91 Mb of repeated sequences, accounting for 46.82% of the “Dongzao” genome, among which Transposon (Tn) sequences account for the vast majority of these repeats. Transcriptome analysis of 15 jujube tissues elucidated specific properties and molecular mechanisms, providing valuable genetic information for molecular breeding of jujube and genetic improvement in Rhamnaceae fruit trees (Table 1). Similarly, in 2016, Huang et al. [22] employed whole-genome shotgun sequencing and a de novo genome assembly strategy to analyze the genome (351.1 Mb) of the highly heterozygous dry jujube cultivar “Junzao” (voucher number: NWA-FU-Junzao001). They annotated 27,443 coding genes, with 91.2% distributed across 12 chromosomes. Repeated sequences (136.2 Mb) accounted for 38.8% of the genome size, predominantly represented by Gypsy and Copia retrotransposons (Table 1). Further analysis, combined with population structure assessment, revealed a complex genetic structure due to extensive hybridization between jujube and its wild species. The study also identified key genes subject to human domestication selection, regulating organic acid and sugar content in jujube fruits, offering insights into jujube genome evolution, population structure, and domestication. In 2021, Shen et al. [23,24] analyzed the genome of sour jujube as wild ancestor species of Chinese jujube (Z. jujuba Mill. var. spinosa), revealing a genome of 406 Mb containing 25,089 predicted genes, 150 rRNA, 1181 tRNA, and 420 non-coding RNAs (ncRNAs). Repeated sequences totaled 204.91 Mb (Table 1), providing a valuable gene pool for jujube stress response and fruit flavor improvement. In 2023, Yang et al. utilized PacBio HiFi, ONT ultra-long, and Hi-C sequencing technologies to re-upgrade the previously published “Dongzao” genome [25]. This upgrade achieved a telomere-to-telomere gapless assembly of 12 chromosomes, with an assembled genome size of 393 Mb. All 12 chromosomes contained complete telomeric sequences, and centromeres are jointly identified via long tandem repeats (more than 100 bp) and Hi-C interactions. The improved genome assembly enhances our understanding of jujube genomes, particularly in the context of jujube evolution. These comprehensive jujube plant genome maps not only contribute to fundamental research in jujube breeding and improvement but also expand the scope and depth of molecular breeding investigations.

Table 1.
Summary of the genome sequences of Chinese jujube cultivars and sour jujube.
3. Genomics Improves the Construction of High-Density Genetic Linkage Maps of Jujube
With the unveiling of the jujube genome and advancements in sequencing technology, high-coverage genetic maps for jujube have been progressively constructed. For instance, Zhao et al. [26] utilized restriction-site associated DNA RAD Tag sequencing technology to establish an inter-specific F1 population genetic map for “JMS2” × “Xing 16”. This map incorporates 2748 RAD markers spanning 913.87 cM, with an average distance between markers of 0.34 cM. Employing genotyping by sequencing (GBS) technology, Zhang et al. [27] crafted a “Dongzao” × “Zhongningyuanzao” genetic linkage map, consisting of 2540 SNP marker sites, totaling 1456.73 cM, and displaying an average genetic distance of 0.88 cM. Zhang et al. [28] employed RAD-seq technology on 99 F1 generations of “Dongzao” × “Yingshanhong” hybrids, constructing a genetic map encompassing 4669 markers, with a total length of 2643.79 cM and an average genetic distance of 0.57 cM. The map revealed 117 QTLs controlling growth-related traits, including plant height, ground diameter, and leaf area. Wang et al. also adopted GBS simplified genome sequencing for the F1 generation of 103 strains of “Dongzao” × “Jinsi4”, resulting in the creation of a high-density genetic linkage map named “D-map” [29]. This map encompassed 3792 markers, spanning 2167.5 cM, with an average genetic distance of 0.358 cM, and identified 27 QTLs for leaf traits and three QTLs for needle length. As resequencing technology progressed, high-density marker genetic linkage maps became more prevalent. For example, Yan et al. [20] utilized 140 F1 strains of “JMS2” × “Jiaocheng” as a mapping population, constructing a genetic map with 8.684 markers using whole-genome re-sequencing (WGRS) technology. The total length of the map is 1713.22 cM with an average marker interval of 0.2 cM. This map revealed 31 QTLs related to leaf traits, shedding light on 4, 8, 14, and 5 QTLs contributing to leaf length, leaf width, leaf shape index, and leaf area, respectively. Further investigation of genes in the QTL region identified that four genes (LOC107418196, LOC107418241, LOC107417968, and LOC112492570) play roles in cell division and cell wall integrity. Guo et al. [30] employed 165 F1 strains of “JMS2” × “Xing16”, utilizing WGRS technology to create a genetic bin map containing 116,312 markers. The total length of this map is 1074.33 cM with an average genetic distance of 0.45 cM. The map identified 15 QTLs related to fruit size, unveiling 113 candidate genes linked to fruit size, encompassing roles in cell division, cell wall metabolism, phytohormone synthesis (ABA, IAA, and auxin), and the encoding of enzymes and transcription factors. These high-density genetic maps serve as invaluable tools for future QTL analysis, candidate gene identification, map-based gene cloning, comparative mapping, and marker-assisted selection (MAS) in jujube.
4. Domestication-Related Genes Identified Based on Jujube Genomics
Based on the analysis of jujube genomics, it has been elucidated that the domestication of jujube involved a progression from sour jujube, transitioning from wild to semi-wild, and ultimately to cultivated forms. The origin center of this domestication is Xi’an, China, spreading from the west to the southeast [30]. Throughout this evolutionary process, various traits of jujube underwent selective domestication, encompassing aspects such as environmental adaptation [23], fruit shape [23], kernel shape [31], bearing-shoot length [31], sweetness/sourness of fruits [22,30], number of leaves per bearing shoot [31], presence of prickles on bearing shoots [31], seed-setting rate [31], fruit weight, and postharvest shelf life of fleshy fruits [23]. Some functional genes associated with these domestication traits have been identified. For instance, Huang et al. [22] conducted a population genome analysis, revealing 1372 genes within the selective sweep regions of the jujube genome. Among these, four genes played pivotal roles in organic acid metabolism, NADP-dependent malic enzyme (NADP-ME, Zj.jz006119090), pyruvate kinase (PK, Zj.jz006429010), isocitrate dehydrogenase (IDH, Zj.jz003285012), and aconitate hydratase (ACO, Zj.jz013123003). Sixteen genes related to sugar metabolism were also uncovered, including sucrose synthase (SUSY, Zj.jz031941019)), phosphoglucomutase (Zj.jz021807003), 6-phosphofructokinase (Zj.jz010621015), and thirteen sugar transporters (Zj.jz034227050, Zj.jz042571026, Zj.jz036789032 et al.). Expression profile analysis indicated that the encoding genes NADP-ME and PK exhibited higher expression levels in the wild compared to cultivated jujube fruits, while a sugar transporter (SUT, Zj.jz042571026) showed lowered expression in the wild compared to cultivated jujubes, suggesting their potential roles in regulating sweetness/sourness during jujube domestication (Table 2).
Table 2.
Summary of candidate genes for domesticated jujube traits.
Shen et al. [23] conducted a comparative analysis of the jujube genome, revealing multiple selection signals, including 421 genes in the wild group and 415 in the semi-wild group, many of which are implicated in environmental adaptation. Notably, histidine kinase 4 (Zijuj10G0113500) exhibited selection signals associated with responses to salt stress, water deprivation, cold, abscisic acid, sucrose stimulus, phosphate starvation, and toxic substances, with a distinct peak on chromosome 10 (Table 2). Guo et al. (2020) [31] performed a genome-wide association study (GWAS) and selective sweep analysis on 350 materials, identifying candidate genes potentially regulating various domestication traits in jujube. Examples include fruit shape- and kernel shape-related genes (ZjFS3, Zj.jz044531027), genes governing the number of leaves per bearing shoot (NLBS, Zj.jz003639032), genes related to prickles on bearing shoots (ZjHDG2, Zj.jz044447010; ZjOVA4, Zj.jz006119092; and ZjRAD51D, Zj.jz001293012), and genes associated with the postharvest shelf life of fleshy fruits (ZjPG, Zj.jz044553003). Guo et al. (2021) [30] employed genomic analysis of different jujube wild and cultivated germplasms to identify the ZjPOD1 (Zj.jz015743041) gene related to the seed-setting rate, the “Early flowering 3” (Zj.jz044531027) gene related to flowering time, and pinpointed a fruit weight gene ZjDA3 (Zj.jz038707057) and a fruit size gene FW2.2/CNR1 (Zj.jz029849045) (Table 2). Notably, ZjDA3 exhibited a negative correlation with fruit weight through expression analysis and transgene verification in tomatoes. These identifications of genes related to jujube domestication traits contribute rich genomic resources not only for jujube but also for other horticultural plants.
5. Molecular Regulation Identification of Jujube Biology Traits Based on Multi-Omics
In recent years, the swift advancement of omics technology has emerged as a potent tool for the precise genetic breeding of fruit trees [32,33]. The genomics of jujube has established a robust foundation for comprehending the intricate biological traits of this fruit. The integration of multi-omics technology has further yielded valuable insights into various aspects of jujube, including fruit quality-related traits such as sugar/acid accumulation and metabolism, the synthesis of functional components in jujube fruits (triterpenoids and flavonoids), and abiotic stress responses.
5.1. Sugar and Organic Acid Accumulation and Metabolism
The sugar content in jujube fruits serves as a pivotal indicator influencing fruit quality, taste, and market value. Extensive studies have revealed that sucrose, glucose, and fructose constitute the primary sugars in jujube fruits [34,35], with their accumulation intricately linked to metabolic and transport processes. Conventionally, sucrose is initially transported from the sieve element-companion cell complex (SE-CC) to pulp cells through plasmodesmata (PD) or into the intercellular space via transporters (SUT and SWEET). Subsequently, a portion of sucrose is transported into the pulp cell cytoplasm through specific transporters (SUT and SWEET), while the remaining sucrose undergoes enzymatic breakdown into fructose and glucose facilitated by extracellular cell wall invertase (cwINV). These monosaccharides are then further transported into the cytoplasm through membrane monosaccharide transporters (MSTs). Within the cytoplasm, sucrose can undergo reversible conversion into fructose and uridine diphosphate glucose (UDPG) via sucrose synthase (SUSY) or be converted into fructose and glucose via neutral/alkaline invertase (nINV). Further phosphorylation by fructokinase (FK) and hexokinase (HK) results in the formation of fructose-6-phosphate (F6P) and glucose-6-phosphate (G6P). F6P and UDPG can subsequently resynthesize sucrose through the actions of sucrose-phosphate synthase (SPS) and sucrose–phosphate phosphatase (SPP). Sucrose and monosaccharides synthesized in the cytoplasm find their way into the vacuole through specific transporters or SWEET proteins on the tonoplast, where they are stored in the form of fructans [36,37,38,39] (Figure 1).
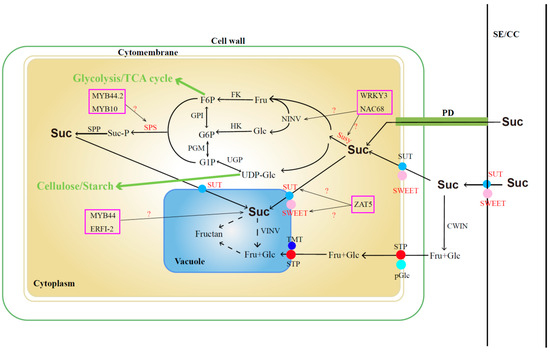
Figure 1.
Sucrose metabolic pathway. The marked color enzymes mean that some relative genes have been identified; TF in the color frame means the candidate transcript factors are involved in sucrose synthesis. Suc: sucrose; Glu: glucose; Fru: fructose; SE/CC: sieve element–companion cell complex; PD: plasmodesmata; SUT: sucrose transporter; SWEET: sugars will eventually be exported transporter; cwINV: cell wall invertase; STP: sugar transport proteins; pGlc: plasma membrane glucose transporters; TMT: tonoplast monosaccharide transporters; vINV: vacuolar invertase; MSTs: membrane monosaccharide transporter; UDPG: uridine diphosphate glucose; SUSY: sucrose synthase; nINV: neutral/alkaline invertase; FK: fructokinase; HK: hexokinase; F6P: fructose-6-phosphate; G6P: glucose-6-phosphate; SPS: sucrose–phosphate synthase; SPP: sucrose–phosphate phosphatase.
In recent years, some key genes that are involved in sugar accumulation and metabolism pathways in jujube were identified based on multi-omics. Zhang et al. (2016) [34] identified 83 sugar transporter genes through whole-genome analysis and demonstrated that ZjSUC2 (scaffold32501.138) and ZjSWEET2 (scaffold15029.70) play roles in promoting sucrose accumulation in fruits. Additionally, monosaccharide transporter genes such as ZjSTP12 (scaffold37921.140), ZjSTP16 (scaffold44531.58), ZjpGlc3 (scaffold4979.175), SWEET15 (scaffold40945.111), ZjSWEET20 (scaffold44739.8), and ZjTMT2 (scaffold39911.7) contribute significantly to sugar accumulation in fruits. In another study by Zhang et al. (2018) [40], the presence of numerous plasmodesmata between the SE/CC complex and surrounding phloem parenchyma cells during the white-ripening stage of cultivated jujube suggested that “the homogeneous unloading pathway has greater transport capacity than the exoplast pathway” [41,42]. This implies that the homogenous unloading pathway through plasmodesmata is more conducive to sugar accumulation. Key genes, including ZjSUT2 (scaffold32501.138) and ZjSWEET11 (scaffold15029.70), were also identified as crucial in the sucrose-accumulation process. The expression levels of three sucrose phosphate synthase genes (ZjSPS1, scaffold1731.36; ZjSPS2, scaffold19851.46; and ZjSS2, scaffold41823.29) were found to be positively correlated with sucrose content in fruits [43]. Yang et al. (2023) [44] identified 19 ZjSWEET genes based on gene family analysis and observed that the expression levels of 2 genes (ZjSWEET11, LOC107404505 and ZjSWEET18, LOC107417626) progressively increased during fruit development, peaking at the full red fruit stage. Several other ZjSWEET genes were found to play vital roles in abiotic stress conditions. While the identification of these genes provides a foundation for enhancing jujube quality, further exploration of their transcriptional regulatory mechanisms is warranted.
Various transcription factors (TFs) play pivotal roles in regulating sugar accumulation and fruit ripening by modulating the expression of genes associated with sugar metabolism and transport. For instance, FaMYB44.2 exerts a negative regulatory effect on soluble sugar content in strawberries by inhibiting the expression of FaSPS3, a critical gene for sucrose accumulation. Additionally, the interaction between FaMYB10 and FaMYB44.2 contributes to sucrose accumulation in mature strawberry fruits [45]. In dragon fruit, HpWRKY3 participates in soluble sugar accumulation by transcriptionally activating the sugar metabolism genes HpINV2 and HpSuSy1 [46]. Meanwhile, in watermelon, ClNAC68 positively mediates sugar accumulation by inhibiting ClINV and ClGH3.6 [47]. In citrus, the transcription factor CitZAT5 (ZINC FINGER OF ARABIDOPSIS THALIANA) regulates CitSUS5 and CitSWEET6, influencing sucrose metabolism and fructose transportation [48]. Furthermore, the oriental melon fruit transcription factor CmERFI-2 represses CmMYB44 expression, leading to increased sucrose levels [49]. The functional characterization of these transcription factors provides a solid foundation for investigating the molecular regulation of sugar accumulation and metabolism in jujube fruit.
Organic acids, important flavor nutrients in jujube fruits, also play a crucial role in adjusting the jujube’s taste [50]. Zhao et al. [34,35] identified 13 organic acid components, including malic acid, citric acid, quinic acid, and tartaric acid, in wild jujube, Chinese jujube, and other types of jujube fruits. Among them, Ziziphus jujuba var. spinosa is characterized by malic acid and citric acid as primary components, while jujube fruit is dominated by malic acid and quinic acid. Key enzymes involved in organic acid metabolism include citrate synthase (CS), aconitase (Aco), NAD+-malate dehydrogenase (NAD+-MDH), NAD+-isocitrate dehydrogenase (NAD+-IDH), NADP+-isocitrate dehydrogenase (NADP+-IDH), NADP+-malic enzyme (NADP+-ME), malate synthase (MS), fumarase (FUM), phosphoenolpyruvate carboxylase (PEPC), and phosphoenolpyruvate carboxy kinase (PEPCK), among others [51]. Transcriptome analysis of jujube fruits from the low-acid and low-sugar variety “Jing 39” and the high-acid and low-sugar variety “Heigeda” unveiled potential key genes involved in malic acid and citric acid metabolism, including ZjPK (LOC107421991), ZjPDC1 (LOC107411322), ZjPDC2 (LOC107430892), ZjCS (LOC107416375), ZjACO (LOC107424464), ZjMDH (LOC107422883), ZjNAD-IDH (LOC107424540), ZjNADP-ME (LOC107432838), ZjNAD-MDH (LOC107413281), ZjGS1 (LOC107423244), and ZjGS2 (LOC107435118) [52]. The aluminum-activated malate transferase (ALMT) ZjALMT4 (Zj.jz000565069) was implicated in malate accumulation, with ZjWRKY7 (Zj.jz007819051) binding to the ZjALMT4 promoter to activate its transcriptional expression and promote malate accumulation [53]. However, compared with sweet jujube research, lots of work is still needed to explore the molecular regulation mechanism of acid accumulation or metabolism in jujube fruits.
5.2. Terpenoid Biosynthesis
Jujube fruits, leaves, roots, and seeds boast a rich content of tetracyclic and pentacyclic triterpenes, including ocyanic acid, floric acid, betulinic acid, oleanolic acid, ursolic acid, oleanolic acid, and 3-ketoacid [54,55]. Many of these compounds exhibit sedative and anticancer effects [56]. Terpenoids in plants are synthesized through two main pathways: the mevalonate pathway (MVA pathway) and the 2-methylerythritol 4-phosphate pathway (MEP pathway), but jujube primarily utilizes the MVA pathway for terpenoid synthesis. In the MVA pathway, two acetyl-CoA molecules serve as starting substrates, forming acetoacetyl-CoA with the aid of acetoacetyl-CoA thiolase (AACT). Subsequently, hydroxy methyl glutaryl-CoA synthase (HMGS) transforms acetoacetyl-CoA into 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA). Hydroxy methyl glutaryl-CoA reductase (HMGR) and NADPH then catalyze the conversion of HMG-CoA into mevalonic acid (MVA). This MVA undergoes a series of enzymatic reactions, including the action of mevalonate kinase (MK), forming mevalonate-5-phosphate (MVAP). Then, phosphomevalonate kinase (PMK) forms mevalonate-5-diphosphate (MVAPP), and finally, mevalonate pyrophosphate decarboxylase (MPD) generates isopentenyl pyrophosphate (IPP), which is generated under the influence of farnesyl pyrophosphate synthase (FPS). IPP, along with dimethylallyl pyrophosphate (DMAPP), is converted into farnesyl pyrophosphate through the catalysis of FPS. Squalene synthase (SQS) then transforms FPP into squalene (SQ), and squalene epoxygenase (SQE) catalyzes the synthesis of 2,3-oxysqualene [57,58,59,60]. Cyclization, hydroxylation, and glycosylation of 2,3-oxysqualene lead to the formation of various terpenoids, mediated by enzymes such as oxysqualene cyclase (OSC), cytochrome P450 (CYP450), and glycosyltransferase (UGT) [61]. Essential enzymes for triterpene biosynthesis include HMGR, FPS, SQS, SQE, and OSCs, in which SQS and SQE play a crucial role in synthesizing squalene and 2,3-oxysqualene, key intermediates for various triterpenes. Furthermore, oxidized squalene cyclases (OSCs) and several cytochrome P450s catalyze the conversion of 2,3-oxosqualene precursors into diverse triterpenes. In jujube, Wen et al. (2022) [62] conducted a comprehensive study using metabolite profiling and transcriptomic data analysis on cultivated jujube, which revealed that terpenoids were mainly concentrated in ZjSQS1 (evm.model.Contig42.302), ZjP450/1 (evm.model.Contig66.109), ZjP450/3 (evm.model.Contig5.527), ZjOSC1 (evm.model.Contig63.27), ZjFPS (evm.model.Contig34.195), and ZjAACT2 (evm.model.Contig37.1.115), representing key candidate genes for terpene synthesis in various jujube tissues. Specifically, ZjFPS, ZjSQS2 (evm.model.Contig75.307), and ZjP450/3 emerged as crucial candidate genes for triterpene synthesis. Wen (2023) [63] further explored the role of ZjWRKY18 (evm.model.Contig24.0.57) in activating ZjHMGR (evm.model.Contig21.0.64 and evm.model.Contig112.45) and ZjFPS, promoting triterpene accumulation. Wen et al. (2023) [64] analyzed enzymes related to terpenoid synthesis through transcriptome analysis and identified ZjFPS, ZjSQS, and the corresponding transcription factors, ZjMYB39 (evm.model.Contig108.375) and ZjMYB4 (evm.model.Contig23.4.315), as key genes for the biosynthesis of jujube triterpenoids. However, the corresponding transcription factor of ZjSQE is still unclear (Figure 2).
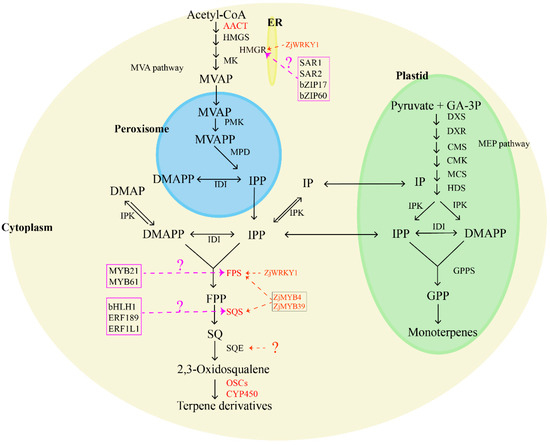
Figure 2.
Biosynthesis pathway of terpenoids in jujube. The marked color enzymes mean that some relative genes have been identified; TF in the color frame means the candidate transcript factors are involved in terpenoid synthesis. AACT: acetoacetyl-CoA thiolase; HMGS: hydroxy methyl glutaryl-CoA synthase; HMGR: hydroxy methyl glutaryl-CoA reductase; MK: mevalonate kinase; MVAP: mevalonate-5-phosphate; PMK: phosphomevalonate kinase; MVAPP: mevalonate-5-diphosphate; MPD: mevalonate pyrophosphate decarboxylase; IPP: isopentenyl pyrophosphate; DMAPP: dimethylallyl pyrophosphate; IDI: isopentane diphosphate isomerase; FPS: farnesyl pyrophosphate synthase; FPP: farnesyl pyrophosphate; SQS: squalene synthase; SQ: squalene; SQE: squalene epoxygenase; OSCs: oxysqualene cyclases; CYP450: cytochrome P450.
Many transcription factors (TFs) have been confirmed to have roles in regulating terpene accumulation by affecting the expression of genes related to terpene synthesis in other plants, such as WRKY [65,66], bHLH [67,68,69], AP2/ERF [70,71,72], MYB [73,74,75,76,77], and bZIP [78,79,80]. Notable examples of these regulators include PnbHLH1, which controls the biosynthesis of triterpene saponins in Panax notoginseng [67,81], ERF189, an ethylene response factor implicated in the biosynthesis of steroidal glycoalkaloids in potatoes and tomatoes [81,82], and SmERF1L1, which regulates the biosynthesis of tanshinones in Salvia miltiorrhiza Bunge [83]. In Medicago truncatula, the bHLH transcription factors TSAR1 and TSAR2 have been identified as key players in the regulation of non-hemolytic and hemolytic triterpene saponins, respectively [84]. Birch triterpene synthesis is influenced by BpMYB21 and BpMYB61, acting as essential regulatory factors [85], while bZIP17 and bZIP60 play regulatory roles in triterpene acid synthesis in alfalfa [78]. The functional characterization of these transcription factors provides a clue for exploring the molecular regulation of terpene synthesis in jujube fruit.
5.3. Flavonoid Biosynthesis
Jujube is renowned for its richness in flavonoids [86], which play pivotal roles in plant growth, development, and stress resistance. The flavonoid content in jujube fruit is intricately linked to the tissue development stage of the fruit. Notably, the total flavonoid content exhibits a continuous decline as the jujube fruit develops and matures, with a rapid decline as it approaches maturity [87]. Anthocyanins as crucial flavonoid compounds in jujube fruits undergo biosynthesis primarily through the phenylalanine metabolic pathway, in which various enzymes play important roles, such as chalcone synthase (CHS), chalcone isomerase (CHI), flavanone-3-hydroxylase (F3H), flavonoid-3’-hydroxylase (F3’H), flavonoid- 3’.5’-hydroxylase, dihydroflavonol-4-reductase (DFR), anthocyanidin synthase (ANS), and uridine diphosphate glucose-flavonoid glucosyl transferase (UFGT) [57]. To explore the molecular mechanisms of jujube flavonoids synthesis, a comparative analysis of total flavonoids and total flavanol contents in the peel and pulp of “Junzao” and “Jishanbanzao” cvs. was conducted, along with identification of two potential key structural genes, F3H (Zj.jz028857033) and F3’H (Zj.jz032893025), suggesting their crucial roles in flavonoid synthesis and accumulation. The gene LAR was also implicated as a potential key regulatory gene in the flavanol pathway [88]. Metabolomic and transcriptomic analyses of jujube peel at different developmental stages unveiled 158 flavonoids, with cyanidin-3-O-rutinoside and peonidin-3,5-O-diglucoside as primary anthocyanins; meanwhile, ZjANS (Zj.jz022481171) and ZjUGT79B1 (Zj.jz027401002) were identified as key genes in the anthocyanin process, and three transcription factors, ZjMYB5 (Zj.jz005919060), ZjTT8 (Zj.jz001627021), and ZjWDR3 (Zj.jz043343182), were found to play pivotal roles in the anthocyanin synthesis in jujube [89]. The varieties “Fengmiguan” (FMG) and “Tailihong” (TLH), known for containing three and seven anthocyanins, respectively, exhibit a regulatory role in fruit coloration. Additionally, the interaction between ZjFAS2 and ZjSHV3 was found to regulate the color of red jujube fruits [90]. Moreover, a transcriptome analysis of Ziziphus mauritiana Lam. and Z. jujuba Mill identified four MYB genes (ZjMYB13, ZjMYB44, ZjMYB50, and ZjMYB56) as candidate key genes in the regulation of flavonoid biosynthesis. Notably, MYB44 was implicated in the biosynthetic pathways of flavonoids during fruit coloration in Ziziphus jujuba Mill. [91]. Wang et al. (2023) [92] constructed a network model comprising 1620 genes highly correlated with proanthocyanidin and catechin content. They identified 16 hub genes encoding 9 transcription factor families (MYB, bHLH, ERF, BZIP, NAC, SBP, MIKC, HB, WRKY), providing a foundation for further research on the molecular regulation of jujube flavonoid synthesis.
5.4. Abiotic Stress Response
Throughout the growth process of jujube trees, they frequently encounter abiotic stresses such as temperature fluctuations, light variations, salinity, and soil conditions, which can lead to a decline in both yield and quality of jujubes. Particularly in northern regions, where winter temperatures often drop significantly, jujube trees are susceptible to various factors, with severe low temperatures capable of causing the entire plant to perish. Soil salinization further poses a significant constraint on jujube production in certain areas, particularly in Xinjiang, where excessive fertilization has led to secondary salinization. To find the genes responding to abiotic stress in jujube, several genes responding to abiotic stress in jujube have been identified based on genome-wide analysis. For example, ZjCNGC2 (LOC107423657) exhibited significant downregulation, while it was highly induced under cold, salt, and alkali stress. It was found that the regulation of ZjCNGC2 by cAMP treatment and microtubule changes, along with its interaction with ZjMAPKK4, plays a role in the response to cold stress [93]. Four ZjBAM genes, ZjBAM1 (Zj.jz015515046), ZjBAM2 (Zj.jz044849113), ZjBAM5 (Zj.jz040945107), and ZjBAM6 (Zj.jz220022001), were significantly upregulated under severe drought conditions, as determined by transcriptome and expression pattern analysis [94]. ZjCML13 (LOC107424831) played a crucial role in regulating the difference in cold resistance between “Dongzao” and its autotetraploid [95]. Additionally, ZjWRKY family members, ZjAP2/ERF family members, ZjNAC family members, and ZjbZIP family members in jujube were also identified and analyzed to determine their function. Among these transcript factors, ZjDREB1 (LOC107404402) could enhance the freezing resistance of jujube via a transgenic experiment verification [96]. ZjWRKY27 (LOC107426111) responded strongly to drought and salt stress, presenting itself as a candidate target gene for salt tolerance breeding [97], and ZjWRKY6 (LOC107426870) and ZjWRKY65 (LOC107435508) in sour jujube were found to enhance plant salt tolerance by increasing the antioxidant enzyme activity, reducing malondialdehyde accumulation, and maintaining K+/Na+ homeostasis [98]. Additionally, ZjNAC6 (LOC107409987), ZjNAC35 (LOC107423780), ZjNAC47 (LOC107428022), and ZjNAC55 (LOC107433097) were found to be involved in responding to ABA, cold, drought, and salt stress. Finally, ZjNAC3 (LOC107416163), ZjNAC32 (LOC107421097), and ZjNAC35 (LOC107423780) were associated with responses to tissue aging [99]. However, the transcription factors corresponding to the identified genes that respond to abiotic stress remain unclear. Additionally, the targets of the identified transcription factors involved in abiotic stress are limited. Resolving these issues will enhance the exploration of the molecular mechanisms governing the jujube’s response to abiotic stress.
6. Conclusions and Perspectives
The intricate genetic background and expansive cultivation regions of jujube pose significant challenges to the analysis of its genetic mechanisms governing various biological traits. Advancements in jujube genome research have catalyzed the emergence of functional genomics, enabling the realization of GWAS and the creation of high-density genetic maps. Concurrently, the application of multi-omics research methodologies, such as transcriptomics, proteomics, and metabolomics, facilitates the exploration of potential target genes and metabolic processes associated with quality traits, including nutrient accumulation in jujube. This, in turn, provides valuable insights for enhancing the quality of jujube. To further investigate linked markers or candidate genes linked to complex agronomic traits, such as the accumulation of vital active ingredients and stress resistance, there is an anticipation that this research will advance genetic improvements in jujube (Figure 3). Nevertheless, in comparison to multi-omics studies on other crops, there is a notable scarcity of such research on jujube and a deficiency in exploring critical agronomic traits of jujube using pan-genomics, epigenomics, single-cell resequencing, and other omics approaches.
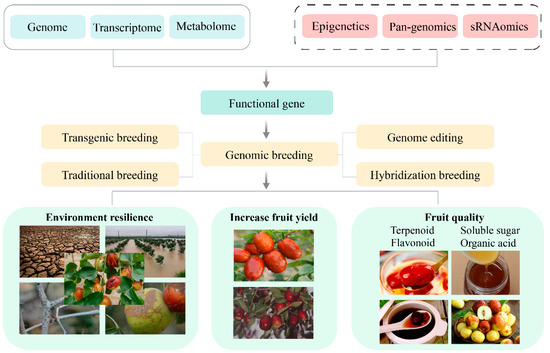
Figure 3.
Integrated approach to jujube genomic breeding strategies.
At present, the pan-genome method stands out as a revolutionary approach in functional genomics research, offering a more comprehensive alternative to single reference genomes. This innovation transcends the limitations of relying on a single reference genome by capturing all genes or genome sequences within a population [100]. The pan-genome has become a forefront and focal point in plant genomics research, facilitating a deeper understanding of the diversity and variability inherent in plant genomes. This, in turn, furnishes functional genomics research with a wealth of genetic information, surpassing the capabilities of traditional single reference genomes.
While pan-genomes have been established for more than 16 plants, including rice, corn, and cotton [101,102,103], the pan-genome of jujube remains unreported. In the future, the application of pan-genomics to jujube can provide a more accurate depiction and analysis of its genomic characteristics. By comparing and analyzing genome sequences and variations across different jujube varieties, germplasm resources, or populations, researchers can delve into the genes and regulatory elements linked to crucial jujube traits. This comprehensive exploration establishes a holistic molecular regulatory network, fostering the analysis of biological traits and contributing to the genetic enhancement of jujube.
As the study of jujube genomics progresses, an increasing number of jujube cultivar genomes will be sequenced, enabling a more precise comprehension of the genetic traits of diverse jujube varieties. This knowledge can be strategically applied to jujube breeding through selective breeding, presenting innovative methods and ideas for enhancing both yield and quality. However, realizing these advancements necessitates further enhancements in the sequenced genome information of jujube varieties, the establishment of a more accurate functional genomics research platform, and the exploration of interaction mechanisms between genes of different varieties and environmental factors.
Numerous studies have underscored the pivotal role of sRNAomics in various plant biological processes, encompassing developmental regulation, disease resistance, and environmental adaptation [104,105,106]. Investigating sRNAs in the context of jujube holds substantial promise for gaining profound insights into the molecular mechanisms underpinning jujube development, regulatory pathways, chromatin modifications, and stress responses. This approach bears potential value and application prospects in comprehending the biological traits of jujube. Firstly, scrutinizing sRNAs in the jujube genome facilitates a deep understanding of its development processes and regulatory mechanisms. By employing sRNA sequencing and analysis, highly expressed sRNAs and their target genes at different developmental stages of jujube can be identified and studied, unraveling the functional roles of sRNA in the developmental regulation of jujube. Secondly, the defensive role of sRNA is of paramount importance to jujube’s disease resistance. sRNA is instrumental in plant defense responses against viruses and transposons. Analyzing sRNAs associated with viral infection in jujube plants allows for the identification of the response mechanisms against viral infections, offering insights that can aid in developing disease-resistant varieties or enhancing the disease resistance of jujube. Furthermore, sRNA participates in the regulation of chromatin modifications and gene expression. Studying sRNAs linked to chromatin modifications enhances our understanding of the regulatory mechanisms governing chromosome structure and gene expression in jujube plants, shedding light on the role of sRNAs in genome stability and genetic expression. Lastly, sRNA emerges as a crucial player in the responses of jujube to both biotic and abiotic stresses. Analysis of sRNAs related to stress responses unveils the regulatory mechanisms employed by jujube plants against environmental stress. This knowledge provides theoretical support for research on jujube stress resistance and genetic improvement.
Gene-editing technology represents a significant advancement in the biological domain, allowing for precise modifications in plants. Among these technologies, CRISPR/Cas9 stands out as a prominent gene-editing tool [107]. In contrast to traditional transgenic methods, CRISPR/Cas9 not only enables the knockout of a single gene, leading to functional loss, but also facilitates the knock-in and modification of a single gene at the epigenetic and transcriptional levels. The application of CRISPR/Cas9 technology in editing the jujube genome holds great potential for reshaping various jujube traits. In the future, utilizing CRISPR/Cas9 technology for jujube genome editing could bring about transformations in traits such as enhancing yield, improving quality, fortifying disease resistance, increasing stress tolerance, and boosting the fruiting rate of modern cultivated jujube trees. Targeted editing of specific genes identified in previous studies offers avenues for regulating and optimizing the growth and development of jujube plants, thereby enhancing key traits like drought resistance and salt tolerance. For instance, knocking out genes such as the pollen-directed defective gene (POD1) [30] and two homologous genes associated with early flowering 3 (EF3), speculated to regulate the flowering time of jujube trees, significantly improves the fruit setting rate, thereby promoting increased fruit yield. Furthermore, precise editing of functional modules within key genes allows for the optimization of the nutritional composition of jujube. When coupled with traditional breeding methods, this approach maximizes advantages, ensuring safety, efficiency, and sustainability in breeding practices, aligning them more closely with market demands.
In summary, the future of jujube research holds great promise as multi-omics resources, encompassing transcriptomics, metabolomics, pan-genomics, and epigenomics, and can be harnessed and integrated. This comprehensive approach allows for a deeper understanding of the biological traits of jujube, facilitating the exploration of key molecules and regulatory networks that govern each trait. Leveraging genome-editing methods, transgenics, and progressive breeding, researchers can propel the breeding process toward the development of new jujube varieties characterized by high yield, diverse fruit flavors and quality, and robust environmental adaptability (Figure 3).
Author Contributions
Conceptualization, S.Z. and G.-P.Z.; writing preparation, L.F., Z.Z., Y.L. and H.Y.; writing—original draft preparation, S.Z., G.-P.Z. and Z.C.; writing—review and editing, S.Z., M.Z., G.-P.Z. and F.G.; supervision, F.G. and G.-P.Z.; funding acquisition and structure design and manuscript modification, S.Z. and Z.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the doctoral research program project of the Henan Institute of Science and Technology and Science and Technology Research Projects of Anyan (2022C01GX022) and the National Forest Germplasm Resources Platform (2005DKA21003).
Data Availability Statement
Data are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Liu, M. Advances in taxonomy study on the genus Ziziphus. Acta Hortic. 1999, 390, 161–166. [Google Scholar]
- Xu, T.; Zhou, X.; Degen, A.; Yin, J.; Zhang, S.; Chen, N. The inclusion of jujube by-products in animal feed: A review. Sustainability 2022, 14, 7882. [Google Scholar] [CrossRef]
- Dou, J.; Kou, X.; Wu, C.; Fan, G.; Li, T.; Li, X. Recent advances and development of postharvest management research for fresh jujube fruit: A review. Sci. Hortic. 2023, 310, 111769. [Google Scholar] [CrossRef]
- Liu, M.; Wang, J.; Wang, L.; Liu, P.; Zhao, J.; Zhao, Z.; Yao, S.; Stănică, Y.; Liu, Z.; Wang, L.; et al. The historical and current research progress on jujube-a superfruit for the future. Hort. Res. 2020, 7, 119. [Google Scholar] [CrossRef]
- Shen, D.; Kou, X.; Wu, C.; Fan, G.; Li, T.; Dou, J.; Wang, H.; Zhu, J. Cocktail enzyme-assisted alkaline extraction and identification of jujube peel pigments. Food Chem. 2021, 357, 129747. [Google Scholar] [CrossRef]
- Liu, M. Advances of research on germplasm resources of Chinese Jujube. Acta Hortic. 2013, 993, 15–20. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, H.; Jung, S.; Main, D.; Guo, L. Developmental mechanisms of fleshy fruit diversity in rosaceae. Annu. Rev. Plant Biol. 2020, 71, 547–573. [Google Scholar] [CrossRef]
- Chen, P.; Chen, L.; Ye, X.; Tan, B.; Zheng, X.; Cheng, J.; Wang, W.; Yang, Q.; Zhang, Y.; Li, J.; et al. Phytoplasma effector Zaofeng6 induces shoot proliferation by decreasing the expression of ZjTCP7 in Ziziphus jujuba. Hortic. Res. 2022, 9, uhab032. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Lao, F.; Shi, X.; Zhang, D.; Wu, J. Effects of cold plasma, high hydrostatic pressure, ultrasound, and high-pressure carbon dioxide pretreatments on the quality characteristics of vacuum freeze-dried jujube slices. Ultrason. Sonochem. 2022, 90, 106219. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Ahn, J.; Kozukue, N.; Levin, C.; Friedman, M. Distribution of free amino acids, flavonoids, total phenolics, and antioxidative activities of Jujube (Ziziphus jujuba) fruits and seeds harvested from plants grown in Korea. J. Agric. Food Chem. 2011, 59, 6594–6604. [Google Scholar] [CrossRef] [PubMed]
- Bao, T.; Hao, X.; Shishir, M.R.I.; Karim, N.; Chen, W. Cold plasma: An emerging pretreatment technology for the drying of jujube slices. Food Chem. 2021, 337, 127783. [Google Scholar] [CrossRef]
- Arslan, M.; Zareef, M.; Tahir, H. Comparative analyses of phenolic compounds and antioxidant properties of Chinese jujube as affected by geographical region and drying methods (Puff-drying and convective hot air-drying systems). Food Meas. Charact. 2021, 15, 933–943. [Google Scholar] [CrossRef]
- Ahmed, K.; Naymul, K.; Mohammad, R.; Tao, B.; Yang, L.; Wei, C. Jujube fruit: A potential nutritious fruit for the development of functional food products. J. Funct. Foods 2020, 75, 104205. [Google Scholar]
- Ahudoukayoumu, A.; Fan, D.; Yue, W.; Zhan, J.; Hao, Q. Changes of nutrients, endogenous hormones and antioxidant enzymes activities during flower bud differentiation process of Ziziphus jujuba. Acta Bot. Boreali-Occident. Sin. 2021, 41, 142–150. [Google Scholar]
- Abudoukayoumu, A. Observation study of the process of flower bud differentiation and flower development of Jujube. Xinjiang Agric. Sci. 2020, 57, 798–805. [Google Scholar]
- Lu, D.; Wu, Y.; Zhang, J.; Qi, Y.; Zhang, Y.; Pan, Q. Visualizing the distribution of Jujube metabolites at different maturity stages using matrix-assisted laser desorption/ionization mass spectrometry imaging. Foods 2023, 12, 3795. [Google Scholar] [CrossRef]
- Yang, Y.; Saand, M.A.; Huang, L.; Abdelaal, W.B.; Zhang, J.; Wu, Y.; Li, J.; Sirohi, M.H.; Wang, F. Applications of multi-omics technologies for crop improvement. Front. Plant Sci. 2021, 12, 563953. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, J.; Cui, Y.; Cui, R.; Kang, R.; Zhang, Y.; Wang, S.; Zhao, L.; Wang, D.; Lu, X.; et al. Changes in terpene biosynthesis and submergence tolerance in cotton. BMC Plant Biol. 2023, 23, 330. [Google Scholar] [CrossRef]
- Owusu, A.; Lv, Y.; Liu, M.; Wu, Y.; Li, C.; Guo, N.; Li, D.-H.; Gao, J.-S. Transcriptomic and metabolomic analyses reveal the potential mechanism of waterlogging resistance in cotton (Gossypium hirsutum L.). Front. Plant Sci. 2023, 14, 1088537. [Google Scholar] [CrossRef]
- Yan, F.; Luo, Y.; Bao, J.; Pan, Y.; Wang, J.; Wu, C.; Liu, M. Construction of a highly saturated genetic map and identification of quantitative trait loci for leaf traits in jujube. Front. Plant Sci. 2022, 13, 1001850. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhao, J.; Cai, Q.; Liu, G.; Wang, J.; Zhao, Z.; Liu, P.; Li, D.; Yan, G.; Wang, W.; et al. The complex jujube genome provides insights into fruit tree biology. Nat. Commun. 2014, 5, 5315. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, C.; Zhao, X.; Fei, Z.; Wan, K.; Zhang, Z.; Pang, X.; Yin, X.; Bai, Y.; Sun, X.; et al. The jujube genome provides insights into genome evolution and the domestication of sweetness/acidity taste in fruit trees. PLoS Genet. 2016, 12, e1006433. [Google Scholar] [CrossRef]
- Shen, L.; Luo, H.; Wang, X.; Wang, X.; Qiu, X.; Liu, H.; Zhou, S.; Jia, K.; Nie, S.; Bao, Y.; et al. Chromosome-scale genome assembly for chinese sour jujube and insights into its genome evolution and domestication signature. Front. Plant Sci. 2021, 12, 773090. [Google Scholar] [CrossRef]
- Wang, L.; Luo, Z.; Liu, Z.; Liu, P.; Liu, M. Genome size variation within species of chinese jujube (Ziziphus jujuba Mill.) and its wild ancestor sour jujube (Z. acidojujuba Cheng et Liu). Forests 2019, 10, 460. [Google Scholar] [CrossRef]
- Yang, M.; Han, L.; Zhang, S.; Dai, L.; Li, B.; Han, S.; Zhao, J.; Liu, P.; Zhao, Z.; Liu, M. Insights into the evolution and spatial chromosome architecture of jujube from an updated gapless genome assembly. Plant Commun. 2023, 4, 100662. [Google Scholar] [CrossRef]
- Zhao, J.; Jian, J.; Liu, G.; Wang, J.; Lin, M.; Ming, Y.; Liu, Z.; Chen, Y.; Liu, X.; Liu, M. Rapid SNP discovery and a RAD-based high-density linkage map in Jujube (Ziziphus Mill.). PLoS ONE 2014, 9, e109850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wei, T.; Zhong, Y.; Li, X.; Huang, J. Construction of a high-density genetic map of Ziziphus jujuba Mill. using genotyping by sequencing technology. Tree Genet. Genomes 2016, 12, 76. [Google Scholar] [CrossRef]
- Zhang, Z. Optimization of a High-Density Genetic Map for Chinese Jujube and QTL Mapping for Several Important Traits. Master Dissertation, Beijing Forestry University, Beijing, China, 2016. [Google Scholar]
- Wang, Z.; Zhang, Z.; Tang, H.; Zhang, Q.; Zhou, G.; Li, X. High-density genetic map construction and QTL mapping of leaf and needling traits in Ziziphus jujuba Mill. Front. Plant Sci. 2019, 10, 1424. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Zhang, Z.; Li, S.; Lian, Q.; Fu, P.; He, Y.; Qiao, J.; Xu, K.; Liu, L.; Wu, M.; et al. Genomic analyses of diverse wild and cultivated accessions provide insights into the evolutionary history of jujube. Plant Biotechnol. J. 2021, 19, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Zhang, Z.; Cheng, Y.; Li, S.; Shao, P.; Yu, Q.; Wang, J.; Xu, G.; Zhang, X.; Liu, J.; et al. Comparative population genomics dissects the genetic basis of seven domestication traits in jujube. Hortic. Res. 2020, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Fabres, P.; Collins, C.; Cavagnaro, T.; Rodríguez López, C. A concise review on multi-omics data integration for terroir analysis in vitis vinifera. Front. Plant Sci. 2017, 8, 1065. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y. Research progress in chemical constituents of Lycium Ruthenicum Murr. Prog. Pharm. Sci. 2015, 39, 351–356. [Google Scholar]
- Zhang, C. Molecular Mechanism Related to the Metabolism of Sugar, Acid and Domestication for Ziziphus Jujuba Mill. Ph.D. Dissertation, Northwest A&F University, Xianyang, China, 2017. [Google Scholar]
- Zhao, A. Characteristic analysis of sugars and organic acids components and contents of chinese Jujube and wild jujube fruits. J. Tarim. Univ. 2016, 28, 29–36. [Google Scholar]
- Ren, Y.; Liao, S.; Xu, Y. An update on sugar allocation and accumulation in fruits. Plant Physiol. 2023, 193, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, P.; Ma, F.; Dandekar, A.M.; Cheng, L. Sugar metabolism and accumulation in the fruit of transgenic apple trees with decreased sorbitol synthesis. Hortic. Res. 2018, 1, 60. [Google Scholar] [CrossRef]
- Sun, L.; Wang, J.; Lian, L.; Song, J.; Du, X.; Liu, W.; Zhao, W.; Yang, L.; Li, C.; Qin, Y.; et al. Systematic analysis of the sugar accumulation mechanism in sucrose- and hexose- accumulating cherry tomato fruits. BMC Plant Biol. 2022, 22, 303. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, Z.; Li, B.; Qin, G.; Tian, S. Molecular basis for optimizing sugar metabolism and transport during fruit development. aBIOTECH 2021, 2, 330–340. [Google Scholar] [CrossRef]
- Zhang, C.; Bian, Y.; Hou, S.; Li, X. Sugar transport played a more important role than sugar biosynthesis in fruit sugar accumulation during Chinese jujube domestication. Planta 2018, 248, 1187–1199. [Google Scholar] [CrossRef]
- Patrick, J.; Offler, C. Post-sieve element transport of photoassimilates in sink regions. J. Exp. Bot. 1996, 47, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Patrick, J. PHLOEM UNLOADING: Sieve element unloading and post-sieve element transport. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1997, 48, 191–222. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Comparative Proteomics Analysis of the Difference in Fruit Size, Sugar and Acid Content between Jujube and Wild Jujube. Master Dissertation, Northwest A&F University, Xianyang, China, 2019. [Google Scholar]
- Yang, C.; Zhao, X.; Luo, Z.; Wang, L.; Liu, M. Genome-wide identification and expression profile analysis of SWEET genes in Chinese jujube. PeerJ 2023, 11, e14704. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Mao, W.; Jia, M.; Xing, S.; Ali, U.; Zhao, Y.; Chen, Y.; Cao, M.; Dai, Z.; Zhang, K.; et al. FaMYB44.2, a transcriptional repressor, negatively regulates sucrose accumulation in strawberry receptacles through interplay with FaMYB10. J. Exp. Bot. 2018, 69, 4805–4820. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Cheng, M.; Ba, L.; Zeng, R.; Luo, D.; Qin, Y.; Liu, Z.; Kuang, J.; Lu, W.; Chen, J.; et al. Pitaya HpWRKY3 is associated with fruit sugar accumulation by transcriptionally modulating sucrose metabolic genes HpINV2 and HpSuSy1. Int. J. Mol. Sci. 2019, 20, 1890. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Zhang, J.; Ren, Y.; Li, M.; Tian, S.; Yu, Y.; Zuo, Y.; Gong, G.; Zhang, H.; et al. The NAC transcription factor ClNAC68 positively regulates sugar content and seed development in watermelon by repressing ClINV and ClGH3.6. Hortic. Res. 2021, 8, 265. [Google Scholar] [PubMed]
- Fang, H.; Shi, Y.; Liu, S.; Jin, R.; Sun, J.; Grierson, D.; Li, S.; Chen, K. The transcription factor CitZAT5 modifies sugar accumulation and hexose proportion in citrus fruit. Plant Physiol. 2023, 192, 1858–1876. [Google Scholar] [CrossRef]
- Gao, G.; Yang, F.; Wang, C.; Duan, X.; Li, M.; Ma, Y.; Wang, F.; Qi, H. The transcription factor CmERFI-2 represses CmMYB44 expression to increase sucrose levels in oriental melon fruit. Plant Physiol. 2023, 192, 1378–1395. [Google Scholar] [CrossRef]
- Li, B.; Li, H.; Liu, C. Analysis of nutritional quality of different jujube cultivars. Ningxia J. Agri. Fores. Sci. Tech. 2019, 60, 25–27. [Google Scholar]
- Ma, Q. Study on the Changes of Main Organic Acid Content and Acid-Metabolism during the Development of Jujube Fruits. Master Dissertation, Tarim University, Aral, China, 2017. [Google Scholar]
- Tong, P. Acid Accumulation Pattern and Related Gene Mining in Jujube Fruit. Master Dissertation, Tarim University, Aral, China, 2021. [Google Scholar]
- Zhang, C.; Geng, Y.; Liu, H.; Wu, M.; Bi, J.; Wang, Z.; Dong, X.; Li, X. Low-acidity ALUMINUM-DEPENDENT MALATE TRANSPORTER4 genotype determines malate content in cultivated jujube. Plant Physiol. 2023, 191, 414–427. [Google Scholar] [CrossRef]
- Pan, F.; Zhao, X.; Liu, F.; Luo, Z.; Chen, S.; Liu, Z.; Zhao, Z.; Liu, M.; Wang, L. Triterpenoids in jujube: A Review of composition, content diversity, pharmacological effects, synthetic pathway, and variation during domestication. Plants 2023, 12, 1501. [Google Scholar] [CrossRef]
- Guo, S.; Duan, J.; Qian, D.; Tang, Y.; Wu, D.; Su, S.; Wang, H.; Zhao, Y. Content variations of triterpenic acid, nucleoside, nucleobase, and sugar in jujube (Ziziphus jujuba) fruit during ripening. Food Chem. 2015, 167, 468–474. [Google Scholar] [CrossRef]
- Sakna, S.; Yasmin, R.; Mohamed, S.; Mohamed, A. Phytochemical diversity and pharmacological effects of triterpenes from genus Ziziphus: A comprehensive review. Phytochem. Rev. 2022, 22, 1611–1636. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, C. Biosynthesis of plant triterpenoid saponins in microbial cell factories. J. Agric. Food Chem. 2018, 66, 12155–12165. [Google Scholar] [CrossRef]
- Tholl, D. Biosynthesis and biological functions of terpenoids in plants. Biochem. Adv. 2015, 148, 63–106. [Google Scholar]
- Kuzuyama, T. Biosynthetic studies on terpenoids produced by Streptomyces. J. Antibiot. 2017, 70, 811–818. [Google Scholar] [CrossRef]
- Wang, Y.; Li, R.; Chen, B. Cytogenetic characterization and metabolomic differences of full-sib progenies of Saccharum spp. Plants 2023, 12, 810. [Google Scholar] [CrossRef]
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene biosynthesis in plants. Annu. Rev. Plant Biol. 2014, 65, 225–257. [Google Scholar] [CrossRef]
- Wen, C.; Zhang, Z.; Shi, Q.; Yue, R.; Li, X. Metabolite and gene expression analysis underlying temporal and spatial accumulation of pentacyclic triterpenoids in Jujube. Genes 2022, 13, 823. [Google Scholar] [CrossRef]
- Wen, C.; Zhang, Z.; Shi, Q.; Duan, X.; Du, J.; Wu, C.; Li, X. Methyl jasmonate- and salicylic acid-induced transcription factor ZjWRKY18 regulates triterpenoid accumulation and salt stress tolerance in Jujube. Int. J. Mol. Sci. 2023, 24, 3899. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Zhang, Z.; Shi, Q.; Niu, R.; Duan, X.; Shen, B.; Li, X. Transcription factors ZjMYB39 and ZjMYB4 regulate farnesyl diphosphate synthase- and squalene synthase-mediated triterpenoid biosynthesis in Jujube. J. Agric. Food Chem. 2023, 71, 4599–4614. [Google Scholar] [CrossRef] [PubMed]
- Chujo, T.; Miyamoto, K.; Ogawa, S.; Masuda, Y.; Shimizu, T.; Kishi-Kaboshi, M.; Takahashi, A.; Nishizawa, Y.; Minami, E.; Nojiri, H. Overexpression of phosphomimic mutated OsWRKY53 leads to enhanced blast resistance in rice. PLoS ONE 2014, 9, e98737. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hao, X.; Wang, Y.; Maoz, I.; Zhou, W.; Zhou, Z.; Kai, G. Identification of WRKY transcription factors involved in regulating the biosynthesis of the anti-cancer drug camptothecin in Ophiorrhiza pumila. Hortic. Res. 2022, 9, uhac099. [Google Scholar] [CrossRef]
- Zhang, X.; Ge, F.; Deng, B.; Shah, T.; Huang, Z.; Liu, D.; Chen, C. Molecular cloning and characterization of PnbHLH1 transcription factor in Panax notoginseng. Molecules 2017, 22, 1268. [Google Scholar] [CrossRef]
- Aslam, M.; Lin, X.; Li, X.; Yang, N.; Chen, L. Molecular cloning and functional characterization of CpMYC2 and CpBHLH13 transcription factors from Wintersweet (Chimonanthus praecox L.). Plants 2020, 9, 785. [Google Scholar] [CrossRef]
- Yin, J.; Li, X.; Zhan, Y.; Li, Y.; Qu, Z.; Sun, L.; Wang, S.; Yang, J.; Xiao, J. Cloning and expression of BpMYC4 and BpbHLH9 genes and the role of BpbHLH9 in triterpenoid synthesis in birch. BMC Plant Biol. 2017, 17, 214. [Google Scholar] [CrossRef]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 2012, 1819, 86–96. [Google Scholar] [CrossRef]
- Bai, Z.; Wu, J.; Huang, W.; Jiao, J.; Zhang, C.; Hou, Z.; Yan, K.; Zhang, X.; Han, R.; Liang, Z.; et al. The ethylene response factor SmERF8 regulates the expression of SmKSL1 and is involved in tanshinone biosynthesis in Saliva miltiorrhiza hairy roots. J. Plant Physiol. 2020, 244, 153006. [Google Scholar] [CrossRef]
- Wei, C.; Li, M.; Cao, X.; Jin, Z.; Zhang, C.; Xu, M.; Chen, K.; Zhang, B. Linalool synthesis related PpTPS1 and PpTPS3 are activated by transcription factor PpERF61 whose expression is associated with DNA methylation during peach fruit ripening. Plant Sci. 2022, 317, 111200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, L.; Zheng, X.; Zhang, J.; Yang, L.; Tan, R.; Zhao, S. Overexpression of SmMYB9b enhances tanshinone concentration in Salvia miltiorrhiza hairy roots. Plant Cell Rep. 2017, 36, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Han, L.; Zhu, X.; Zhang, C.; Li, Y.; Xue, X.; Wang, Y.; Wang, D.; Niu, J.; Hua, W.; et al. SmMYB4 is a R2R3-MYB transcriptional repressor regulating the Biosynthesis of phenolic acids and tanshinones in Salvia miltiorrhiza. Metabolites 2022, 12, 968. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Pu, Z.; Cao, G.; You, D.; Zhou, Y.; Deng, C.; Shi, M.; Nile, S.; Wang, Y.; Zhou, W.; et al. Tanshinone and salvianolic acid biosynthesis are regulated by SmMYB98 in Salvia miltiorrhiza hairy roots. J. Adv. Res. 2020, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, Y.; Gao, F.; Jin, W.; Li, S.; Kimani, S.; Yang, S.; Bao, T.; Gao, X.; Wang, L. MYB21 interacts with MYC2 to control the expression of terpene synthase genes in flowers of Freesia hybrida and Arabidopsis thaliana. J. Exp. Bot. 2020, 71, 4140–4158. [Google Scholar] [CrossRef]
- Wu, Z.; Li, L.; Liu, H.; Yan, X.; Ma, Y.; Li, Y.; Chen, T.; Wang, C.; Xie, L.; Hao, X.; et al. AaMYB15, an R2R3-MYB TF in Artemisia annua, acts as a negative regulator of artemisinin biosynthesis. Plant Sci. 2021, 308, 110920. [Google Scholar] [CrossRef]
- Ribeiro, B.; Erffelinck, M.; Lacchini, E.; Ceulemans, E.; Colinas, M.; Williams, C.; Hamme Van, E.; Clercq, R.; Perassolo, M.; Goossens, A. Interference between ER stress-related bZIP-type and jasmonate-inducible bHLH-type transcription factors in the regulation of triterpene saponin biosynthesis in Medicago truncatula. Front. Plant Sci. 2022, 13, 903793. [Google Scholar] [CrossRef]
- Hao, X.; Zhong, Y.; Nï Tzmann, H.; Fu, X.; Yan, T.; Shen, Q.; Chen, M.; Ma, Y.; Zhao, J.; Osbourn, A.; et al. Light-induced artemisinin biosynthesis is regulated by the bZIP transcription factor AaHY5 in Artemisia annua. Plant Cell Physiol. 2019, 60, 1747–1760. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Z.; Ji, A.; Luo, H.; Song, J. Genomic survey of bZIP transcription factor genes related to tanshinone biosynthesis in Salvia miltiorrhiza. Acta Pharm. Sin. B 2018, 8, 295–305. [Google Scholar] [CrossRef]
- Cárdenas, P.; Sonawane, P.; Pollier, J.; Vanden Bossche, R.; Dewangan, V.; Weithorn, E.; Tal, L.; Meir, S.; Rogachev, I.; Malitsky, S.; et al. GAME9 regulates the biosynthesis of steroidal alkaloids and upstream isoprenoids in the plant mevalonate pathway. Nat. Commun. 2016, 7, 10654. [Google Scholar] [CrossRef] [PubMed]
- Thagun, C.; Imanishi, S.; Kudo, T.; Nakabayashi, R.; Ohyama, K.; Mori, T.; Kawamoto, K.; Nakamura, Y.; Katayama, M.; Nonaka, S.; et al. Jasmonate-responsive ERF transcription factors regulate steroidal glycoalkaloid biosynthesis in Tomato. Plant Cell Physiol. 2016, 57, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Sun, M.; Yuan, T.; Wang, Y.; Shi, M.; Lu, S.; Tang, B.; Pan, J.; Wang, Y.; Kai, G. The AP2/ERF transcription factor SmERF1L1 regulates the biosynthesis of tanshinones and phenolic acids in Salvia miltiorrhiza. Food Chem. 2019, 274, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Pollier, J.; Vanden Bossche, R.; Lopez-Vidriero, I.; Franco-Zorrilla, J.; Goossens, A. The bHLH transcription factors TSAR1 and TSAR2 regulate triterpene saponin biosynthesis in medicago truncatula. Plant Physiol. 2016, 170, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Sun, L.; Li, Y.; Xiao, J.; Wang, S.; Yang, J.; Qu, Z.; Zhan, Y. Functional identification of BpMYB21 and BpMYB61 transcription factors responding to MeJA and SA in birch triterpenoid synthesis. BMC Plant Biol. 2020, 20, 374. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, A. Flavonoids of Zizyphus jujuba L. and Zizyphus spina-christi (L.) Willd (Rhamnaceae) fruits. Food Chem. 2009, 112, 858–862. [Google Scholar] [CrossRef]
- Wang, C.; Cheng, D.; Cao, J.; Jiang, W. Antioxidant capacity and chemical constituents of Chinese jujube (Ziziphus jujuba Mill.) at different ripening stages. Food Sci. Biotechnol. 2013, 22, 639–644. [Google Scholar] [CrossRef]
- Li, X. The patterns of flavonoids accumulation and the expression of biosynthesis related genes during the course of maturation of the Chinese jujube fruit. J. Fruit. Sci. 2020, 37, 1464–1474. [Google Scholar]
- Shi, Q.; Du, J.; Zhu, D.; Li, X.; Li, X. Metabolomic and transcriptomic analyses of anthocyanin biosynthesis mechanisms in the color mutant Ziziphus jujuba cv. Tailihong. J. Agric. Food Chem. 2020, 68, 15186–15198. [Google Scholar] [CrossRef]
- Li, S.; Shen, Y.; Zheng, S.; Zhu, Q.; Cai, L.; Wang, Y.; Zhao, X. ZjFAS2 is involved in the fruit coloration in Ziziphus jujuba Mill. by regulating anthocyanin accumulation. Front. Plant Sci. 2023, 14, 1142757. [Google Scholar] [CrossRef]
- Muhammad, N.; Luo, Z.; Zhao, X.; Yang, M.; Liu, Z.; Liu, M. Transcriptome-wide expression analysis of MYB gene family leads to functional characterization of flavonoid biosynthesis in fruit coloration of Ziziphus Mill. Front. Plant Sci. 2023, 14, 1171288. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pu, Y.; Wen, H.; Lu, D.; Yan, M.; Liu, M.; Wu, M.; Bai, H.; Shen, L.; Wu, C. Transcriptome and weighted gene co-expression network analysis of jujube (Ziziphus jujuba Mill.) fruit reveal putative genes involved in proanthocyanin biosynthesis and regulation. FSHW 2023, 12, 1557–1570. [Google Scholar] [CrossRef]
- Wang, L.; Li, M.; Liu, Z.; Dai, L.; Zhang, M.; Wang, L.; Zhao, J.; Liu, M. Genome-wide identification of CNGC genes in Chinese jujube (Ziziphus jujuba Mill.) and ZjCNGC2 mediated signalling cascades in response to cold stress. BMC Genom. 2020, 21, 191. [Google Scholar] [CrossRef]
- Ma, Y.; Han, Y.; Feng, X.; Gao, H.; Cao, B.; Song, L. Genome-wide identification of BAM (β-amylase) gene family in jujube (Ziziphus jujuba Mill.) and expression in response to abiotic stress. BMC Genom. 2022, 23, 438. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, L.; Luo, Z.; Zhao, R.; Liu, Z.; Liu, P.; Liu, M.; Wang, L. Molecular characteristics of CML genes in Chinese jujube and their expression patterns in resistance to cold stress. J. Beijing For. Univ. 2023, 45, 58–67. [Google Scholar]
- Zhou, H. Comparative Transcriptome Analysis of Jujube under Freezing Stress and Functional Studies of Related Genes. Ph.D. Dissertation, Beijing Forestry University, Beijing, China, 2020. [Google Scholar]
- Chen, X.; Chen, R.; Wang, Y.; Wu, C.; Huang, J. Genome-wide identification of WRKY transcription factors in Chinese jujube (Ziziphus jujuba Mill.) and their involvement in fruit developing, ripening, and abiotic stress. Genes 2019, 10, 360. [Google Scholar] [CrossRef]
- He, A.; Ma, Z.; Li, Y.; Huang, C.; Yong, J.; Huang, J. Spatiotemporal, physiological and transcriptomic dynamics of wild jujube seedlings under saline conditions. Tree Physiol. 2023, 43, 832–850. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hou, L.; Liu, S.; Zhang, C.; Yang, W.; Pang, X.; Li, Y. Genome-wide identification and expression analysis of NAC transcription factors in Ziziphus jujuba Mill. reveal their putative regulatory effects on tissue senescence and abiotic stress responses. Ind. Crops Prod. 2021, 173, 114093. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, S.; Liu, B. Research progress on pangenome and its application in plant functional genetics. J. Plant Genet. Resour. 2021, 22, 7–15. [Google Scholar]
- Wang, J.; Yang, W.; Zhang, S.; Hu, H.; Yuan, Y.; Dong, J.; Chen, L.; Ma, Y.; Yang, T.; Zhou, L.; et al. A pangenome ana1ysis pipe1ine provides insights into functiona1 gene identification in rice. Genome Biol. 2023, 24, 19. [Google Scholar] [CrossRef]
- Wang, B.; Hou, M.; Shi, J.; Ku, L.; Song, W.; Li, C.; Ning, Q.; Li, X.; Li, C.; Zhao, B.; et al. De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat. Genet. 2023, 55, 312–323. [Google Scholar] [CrossRef]
- Wang, M.; Li, J.; Qi, Z.; Long, Y.; Pei, L.; Huang, X.; Grover, C.; Du, X.; Xia, C.; Wang, P.; et al. Genomic innovation and regulatory rewiring during evolution of the cotton genus Gossypium. Nat. Genet. 2022, 54, 1959–1971. [Google Scholar] [CrossRef]
- Xia, R.; Chen, C.; Pokhrel, S.; Ma, W.; Huang, K.; Patel, P.; Wang, F.; Xu, J.; Liu, Z.; Li, J.; et al. 24-nt reproductive phasiRNAs are broadly present in angiosperms. Nat. Commun. 2019, 10, 627. [Google Scholar] [CrossRef]
- Zhan, J.; Meyers, B. Plant small RNAs: Their biogenesis, regulatory roles, and functions. Annu. Rev. Plant Biol. 2023, 74, 21–51. [Google Scholar] [CrossRef]
- Kong, X.; Yang, M.; Le, B.H.; He, W.; Hou, Y. The master role of siRNAs in plant immunity. Mol. Plant Pathol. 2022, 23, 1565–1574. [Google Scholar] [CrossRef]
- Meers, C.; Le, H.; Pesari, S.; Hoffmann, F.; Walker, M.; Gezelle, J.; Tang, S.; Sternberg, S. Transposon-encoded nucleases use guide RNAs to promote their selfish spread. Nature 2023, 622, 863–871. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).