
Comparative and Phylogenetic Analysis of Six New Complete Chloroplast Genomes of Rubus (Rosaceae)

Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction, and Sequencing
2.2. Chloroplast Genome Assembly and Annotation
2.3. The Characteristics Analysis of the Cp Genome
2.4. Comparison of Diversity in Cp Genomes
2.5. Repeat Sequences Identification
2.6. A Gene Selection Pressure Analysis of PCGs in Rubus
2.7. Codon Usage Bias Analysis
2.8. Phylogenetic Analysis
3. Results
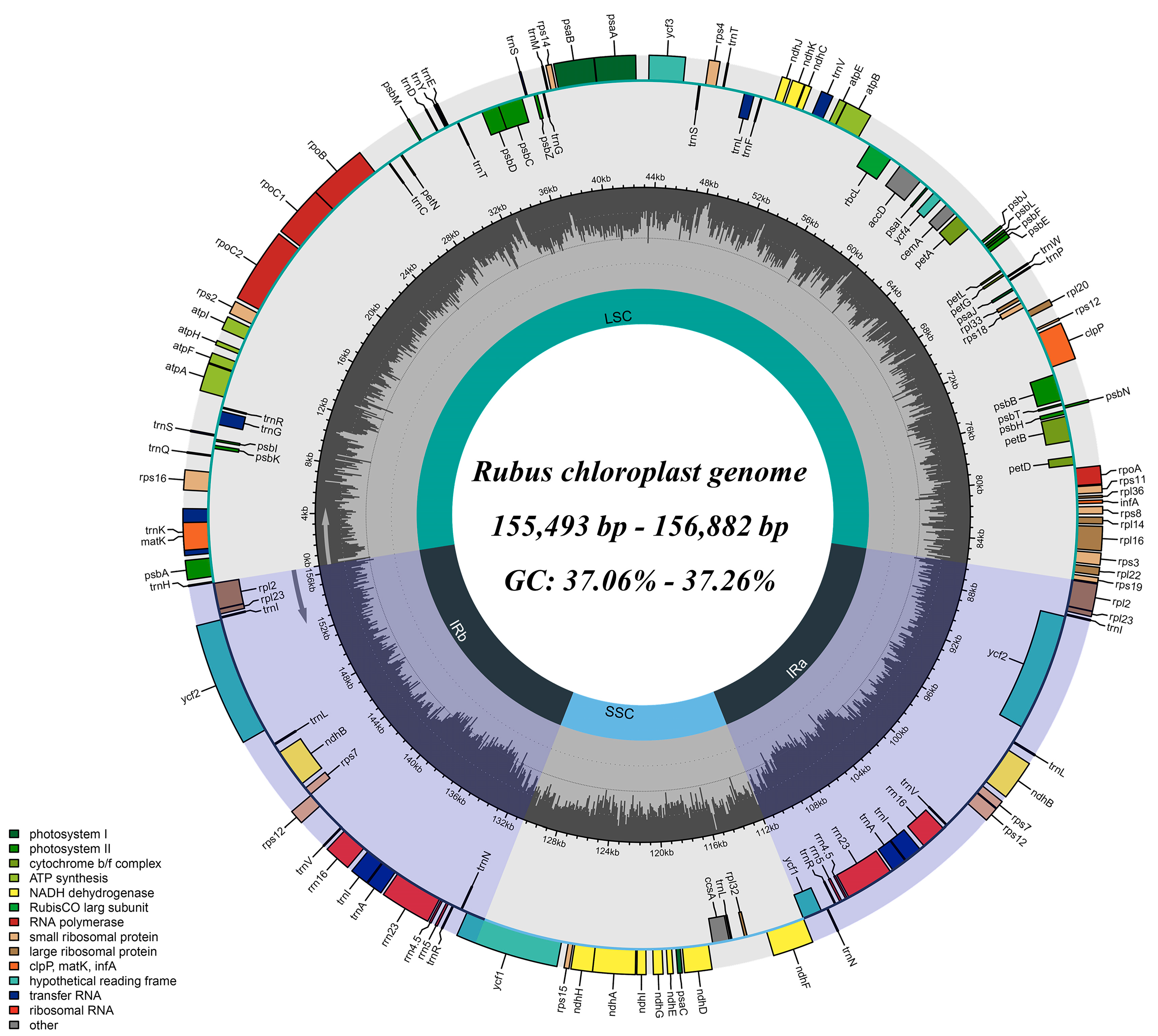
3.1. Basic Characteristics of Rubus Chloroplast Genomes
3.2. Comparison of the IR Region Expansion/Contraction among Different Species
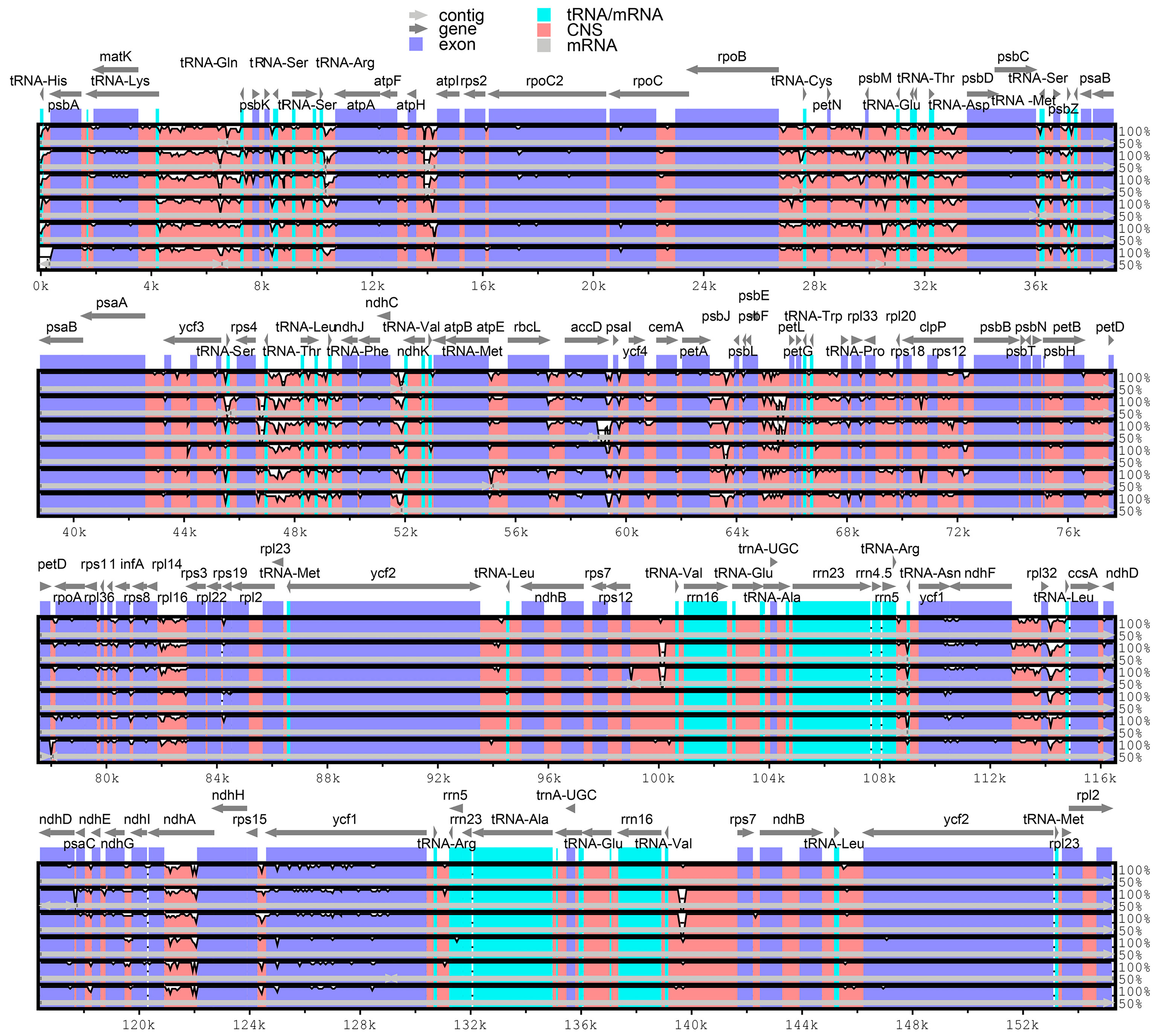
3.3. Comparative Cp Genome Sequences Diversities and Hotspots Regions
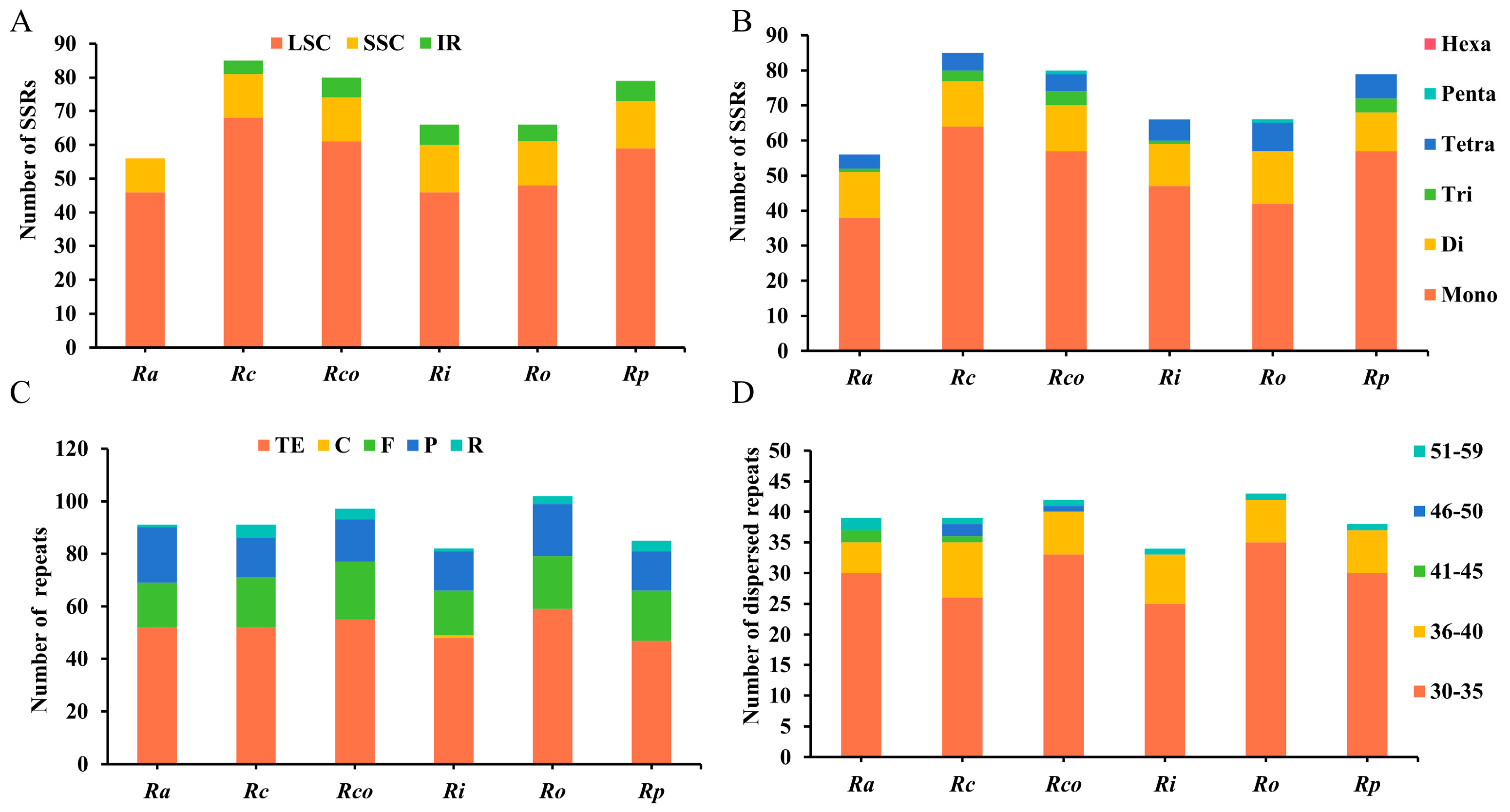
3.4. Repeat Sequences Analysis
3.5. Synonymous and Non-Synonymous Substitution Rate Analysis
3.6. Codon Usage Bias Statistics
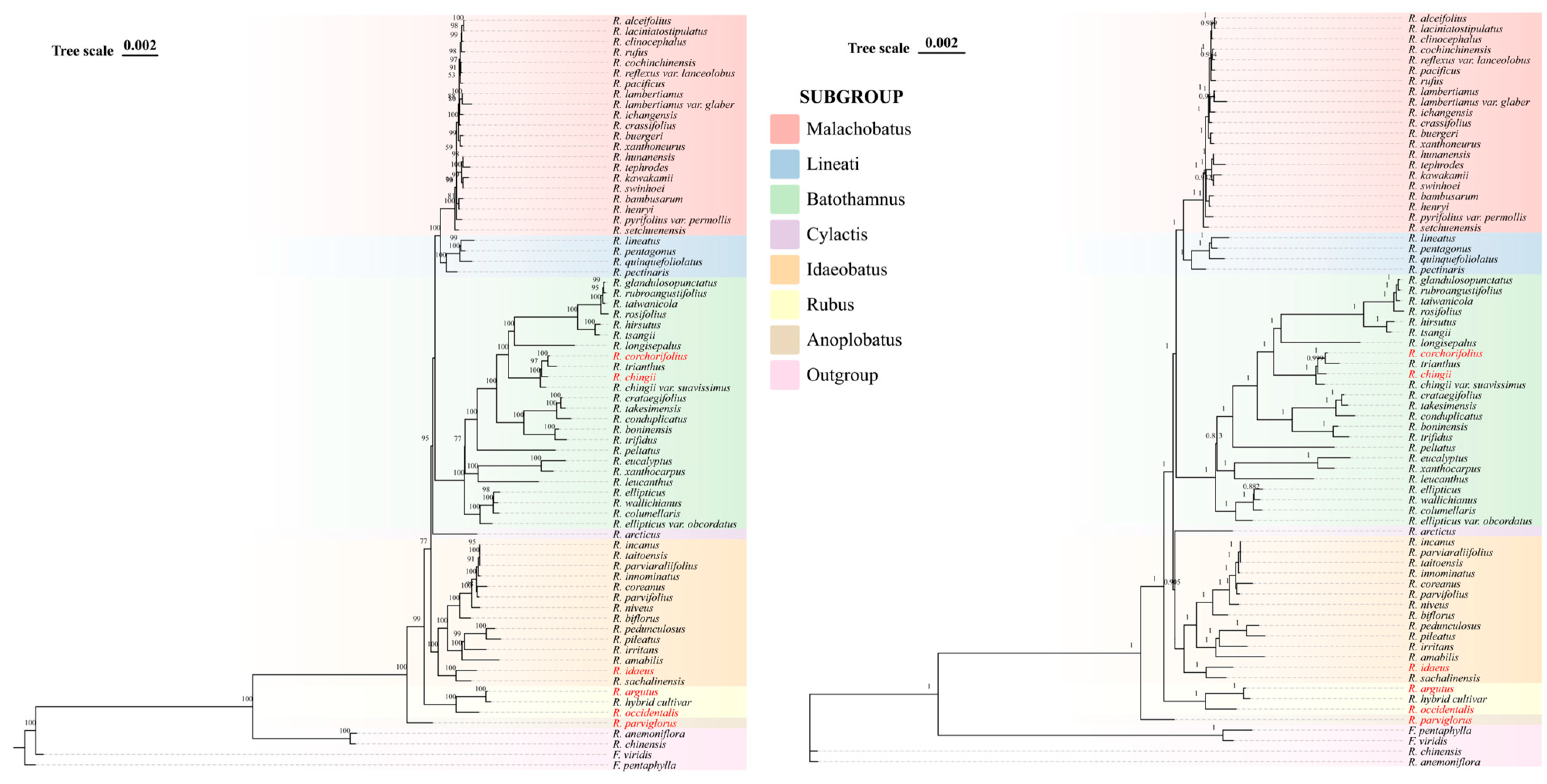
3.7. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Moreno-Medina, B.L.; Casierra-Posada, F.; Cutler, J. Phytochemical composition and potential use of Rubus species. Gesunde Pflanz. 2018, 70, 65–74. [Google Scholar] [CrossRef]
- Yu, J.; Fu, J.; Fang, Y.; Xiang, J.; Dong, H. Complete chloroplast genomes of Rubus species (Rosaceae) and comparative analysis within the genus. BMC Genom. 2022, 23, 32. [Google Scholar] [CrossRef] [PubMed]
- Focke, W.O. Species Ruborum: Monographiae Generis Rubi Prodromus; E. Schweizerbart: Stuttgart, Germany, 1914. [Google Scholar]
- Lu, L.; Boufford, D.E. Rubus Linnaeus, Sp. P1.1: 492.1753. In Flora of China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2003; Volume 9, pp. 192–285. [Google Scholar]
- Foster, T.M.; Bassil, N.V.; Dossett, M.; Leigh Worthington, M.; Graham, J. Genetic and genomic resources for Rubus breeding: A roadmap for the future. Hortic. Res. 2019, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Moyer, R.A.; Hummer, K.E.; Finn, C.E.; Frei, B.; Wrolstad, R.E. Anthocyanins, phenolics, and antioxidant capacity in diverse small fruits: Vaccinium, Rubus, and Ribes. J. Agric. Food Chem. 2002, 50, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Kaume, L.; Howard, L.R.; Devareddy, L. The blackberry fruit: A review on its composition and chemistry, metabolism and bioavailability, and health benefits. J. Agric. Food Chem. 2012, 60, 5716–5727. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Luo, Z.; Wang, W.; Li, Y.; Zhou, Y.; Shi, Y. Rubus chingii Hu: A review of the phytochemistry and pharmacology. Front. Pharmacol. 2019, 10, 799. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.P.; Moreira, H.; Rangel, A.O.; Castro, P.M. Arsenic, lead and nickel accumulation in Rubus ulmifolius growing in contaminated soil in Portugal. J. Hazard. Mater. 2009, 165, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, H.; Zhang, T.; Sen, L.; Ni, W. Classification and identification of metal-accumulating plant species by cluster analysis. Environ. Sci. Pollut. Res. 2014, 21, 10626–10637. [Google Scholar] [CrossRef]
- He, B.; Dai, L.; Jin, L.; Liu, Y.; Li, X.; Luo, M.; Wang, Z.; Kai, G. Bioactive components, pharmacological effects, and drug development of traditional herbal medicine Rubus chingii Hu (Fu-Pen-Zi). Front. Nutr. 2023, 9, 1052504. [Google Scholar] [CrossRef] [PubMed]
- Alice, L. Evolutionary relationships in Rubus (Rosaceae) based on molecular data. Acta Hortic. 2002, 585, 79–83. [Google Scholar] [CrossRef]
- Alice, L.A.; Campbell, C.S. Phylogeny of Rubus (Rosaceae) based on nuclear ribosomal DNA internal transcribed spacer region sequences. Am. J. Bot. 1999, 86, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.M. Survey of chromosome numbers in Rubus (Rosaceae: Rosoideae). Ann. Mo. Bot. Gard. 1997, 84, 128–164. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Q.; Chen, T.; Tang, H.; Liu, L.; Wang, X. Phylogenetic insights into Chinese Rubus (Rosaceae) from multiple chloroplast and nuclear DNAs. Front. Plant Sci. 2016, 7, 968. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Q.; Chen, T.; Zhang, J.; He, W.; Liu, L.; Luo, Y.; Sun, B.; Zhang, Y.; Tang, H.-R. Allopolyploid origin in Rubus (Rosaceae) inferred from nuclear granule-bound starch synthase I (GBSS I) sequences. BMC Plant Biol. 2019, 19, 303. [Google Scholar]
- Yang, J.Y.; Pak, J.-H. Phylogeny of Korean Rubus (Rosaceae) based on ITS (nrDNA) and trnL/F intergenic region (cpDNA). J. Plant Biol. 2006, 49, 44–54. [Google Scholar] [CrossRef]
- Yang, J.; Yoon, H.-S.; Pak, J.-H. Phylogeny of Korean Rubus (Rosaceae) based on the second intron of the LEAFY gene. Can. J. Plant Sci. 2012, 92, 461–472. [Google Scholar] [CrossRef]
- Yang, J.; Chiang, Y.-C.; Hsu, T.-W.; Kim, S.-H.; Pak, J.-H.; Kim, S.-C. Characterization and comparative analysis among plastome sequences of eight endemic Rubus (Rosaceae) species in Taiwan. Sci. Rep. 2021, 11, 1152. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Tian, Q.; Gu, W.; Yang, C.-X.; Wang, D.-J.; Yi, T.-S. Comparative genomics on chloroplasts of Rubus (Rosaceae). Genomics 2024, 116, 110845. [Google Scholar] [CrossRef]
- Brůna, T.; Aryal, R.; Dudchenko, O.; Sargent, D.J.; Mead, D.; Buti, M.; Cavallini, A.; Hytönen, T.; Andrés, J.; Pham, M.; et al. A chromosome-length genome assembly and annotation of blackberry (Rubus argutus, cv. “Hillquist”). G3 Genes Genomes Genet. 2022, 13, jkac289. [Google Scholar] [CrossRef]
- Wang, L.; Lei, T.; Han, G.; Yue, J.; Zhang, X.; Yang, Q.; Ruan, H.; Gu, C.; Zhang, Q.; Qian, T. The chromosome-scale reference genome of Rubus chingii Hu provides insight into the biosynthetic pathway of hydrolyzable tannins. Plant J. 2021, 107, 1466–1477. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, K.; Xiao, Y.; Zhang, L.; Huang, Y.; Li, X.; Chen, S.; Peng, Y.; Yang, S.; Liu, Y. Genome assembly and population resequencing reveal the geographical divergence of shanmei (Rubus corchorifolius). Genom. Proteom. Bioinform. 2022, 20, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Davik, J.; Røen, D.; Lysøe, E.; Buti, M.; Rossman, S.; Alsheikh, M.; Aiden, E.L.; Dudchenko, O.; Sargent, D.J. A chromosome-level genome sequence assembly of the red raspberry (Rubus idaeus L.). PLoS ONE 2022, 17, e0265096. [Google Scholar] [CrossRef] [PubMed]
- Price, R.J.; Davik, J.; Fernandéz Fernandéz, F.; Bates, H.J.; Lynn, S.; Nellist, C.F.; Buti, M.; Røen, D.; Šurbanovski, N.; Alsheikh, M. Chromosome-scale genome sequence assemblies of the ‘Autumn Bliss’ and ‘Malling Jewel’ cultivars of the highly heterozygous red raspberry (Rubus idaeus L.) derived from long-read Oxford Nanopore sequence data. PLoS ONE 2023, 18, e0285756. [Google Scholar] [CrossRef] [PubMed]
- VanBuren, R.; Bryant, D.; Bushakra, J.M.; Vining, K.J.; Edger, P.P.; Rowley, E.R.; Priest, H.D.; Michael, T.P.; Lyons, E.; Filichkin, S.A. The genome of black raspberry (Rubus occidentalis). Plant J. 2016, 87, 535–547. [Google Scholar] [CrossRef] [PubMed]
- VanBuren, R.; Wai, C.M.; Colle, M.; Wang, J.; Sullivan, S.; Bushakra, J.M.; Liachko, I.; Vining, K.J.; Dossett, M.; Finn, C.E.; et al. A near complete, chromosome-scale assembly of the black raspberry (Rubus occidentalis) genome. GigaScience 2018, 7, giy094. [Google Scholar] [CrossRef] [PubMed]
- Caplan, J.S.; Yeakley, J.A. Functional morphology underlies performance differences among invasive and non-invasive ruderal Rubus species. Oecologia 2013, 173, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Maliga, P. Plastid transformation in higher plants. Annu. Rev. Plant Biol. 2004, 55, 289–313. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Wingett, S.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control [version 2; peer review: 4 approved]. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Jian, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ni, Y.; Li, J.; Zhang, X.; Yang, H.; Chen, H.; Liu, C. CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 2023, 23, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. S2), W273–W279. [Google Scholar] [CrossRef]
- Rozas, J.; Sánchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chen, Z.; Shen, M.; Li, Q.; Wang, S.; Jiang, J.; Zeng, W. Identification and Functional Verification of the Glycosyltransferase Gene Family Involved in Flavonoid Synthesis in Rubus chingii Hu. Plants 2024, 13, 1390. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization by One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, gkad359. [Google Scholar] [CrossRef]
- Huang, T.-R.; Chen, J.-H.; Hummer, K.E.; Alice, L.A.; Wang, W.-H.; He, Y.; Yu, S.-X.; Yang, M.-F.; Chai, T.-Y.; Zhu, X.-Y.; et al. Phylogeny of Rubus (Rosaceae): Integrating molecular and morphological evidence into an infrageneric revision. TAXON 2023, 72, 278–306. [Google Scholar] [CrossRef]
- Ravi, V.; Khurana, J.; Tyagi, A.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Luo, C.; Huang, W.; Sun, H.; Yer, H.; Li, X.; Li, Y.; Yan, B.; Wang, Q.; Wen, Y.; Huang, M. Comparative chloroplast genome analysis of Impatiens species (Balsaminaceae) in the karst area of China: Insights into genome evolution and phylogenomic implications. BMC Genom. 2021, 22, 571. [Google Scholar] [CrossRef]
- Downie, S.R.; Palmer, J.D. Use of chloroplast DNA rearrangements in reconstructing plant phylogeny. In Molecular Systematics of Plants; Springer: Boston, MA, USA, 1992; pp. 14–35. [Google Scholar]
- Xiao, S.; Xu, P.; Deng, Y.; Dai, X.; Zhao, L.; Heider, B.; Zhang, A.; Zhou, Z.; Cao, Q. Correction to: Comparative analysis of chloroplast genomes of cultivars and wild species of sweetpotato (Ipomoea batatas [L.] Lam). BMC Genom. 2021, 22, 368. [Google Scholar] [CrossRef]
- Li, D.-M.; Li, J.; Wang, D.-R.; Xu, Y.-C.; Zhu, G.-F. Molecular evolution of chloroplast genomes in subfamily Zingiberoideae (Zingiberaceae). BMC Plant Biol. 2021, 21, 558. [Google Scholar] [CrossRef]
- Li, B.; Liu, T.; Ali, A.; Xiao, Y.; Shan, N.; Sun, J.; Huang, Y.; Zhou, Q.; Zhu, Q. Complete chloroplast genome sequences of three aroideae species (Araceae): Lights into selective pressure, marker development and phylogenetic relationships. BMC Genom. 2022, 23, 218. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Kim, K.-A.; Yoo, K.-O. The complete chloroplast genome sequences of three Adenophora species and comparative analysis with Campanuloid species (Campanulaceae). PLoS ONE 2017, 12, e0183652. [Google Scholar] [CrossRef]
- Sun, Y.; Moore, M.J.; Zhang, S.; Soltis, P.S.; Soltis, D.E.; Zhao, T.; Meng, A.; Li, X.; Li, J.; Wang, H. Phylogenomic and structural analyses of 18 complete plastomes across nearly all families of early-diverging eudicots, including an angiosperm-wide analysis of IR gene content evolution. Mol. Phylogenet. Evol. 2016, 96, 93–101. [Google Scholar] [CrossRef]
- Downie, S.R.; Jansen, R.K. A comparative analysis of whole plastid genomes from the Apiales: Expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 2015, 40, 336–351. [Google Scholar] [CrossRef]
- Wang, R.-J.; Cheng, C.-L.; Chang, C.-C.; Wu, C.-L.; Su, T.-M.; Chaw, S.-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, Z.; Gao, C.; Ge, Y.; Cheng, R. The complete chloroplast genome sequence of Rubus hirsutus Thunb. and a comparative analysis within Rubus species. Genetica 2021, 149, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- McNeal, J.R.; Kuehl, J.V.; Boore, J.L.; De Pamphilis, C.W. Complete plastid genome sequences suggest strong selection for retention of photosynthetic genes in the parasitic plant genus Cuscuta. BMC Plant Biol. 2007, 7, 57. [Google Scholar] [CrossRef]
- Funk, H.T.; Berg, S.; Krupinska, K.; Maier, U.G.; Krause, K. Complete DNA sequences of the plastid genomes of two parasitic flowering plant species, Cuscuta reflexa and Cuscuta gronovii. BMC Plant Biol. 2007, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-S.; Fu, P.-C.; Zhou, X.-J.; Cheng, Y.-W.; Zhang, F.-Q.; Chen, S.-L.; Gao, Q.-B. The complete plastome sequences of seven species in Gentiana sect. Kudoa (Gentianaceae): Insights into plastid gene loss and molecular evolution. Front. Plant Sci. 2018, 9, 493. [Google Scholar] [PubMed]
- Yao, X.; Tang, P.; Li, Z.; Li, D.; Liu, Y.; Huang, H. The first complete chloroplast genome sequences in Actinidiaceae: Genome structure and comparative analysis. PLoS ONE 2015, 10, e0129347. [Google Scholar] [CrossRef]
- Park, I.; Kim, W.J.; Yeo, S.-M.; Choi, G.; Kang, Y.-M.; Piao, R.; Moon, B.C. The complete chloroplast genome sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and comparative analysis with other Fritillaria species. Molecules 2017, 22, 982. [Google Scholar] [CrossRef]
- Wu, Z.; Gui, S.; Quan, Z.; Pan, L.; Wang, S.; Ke, W.; Liang, D.; Ding, Y. A precise chloroplast genome of Nelumbo nucifera (Nelumbonaceae) evaluated with Sanger, Illumina MiSeq, and PacBio RS II sequencing platforms: Insight into the plastid evolution of basal eudicots. BMC Plant Biol. 2014, 14, 289. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, N.; Wu, H. Analyzing and characterizing the chloroplast genome of Salix wilsonii. BioMed Res. Int. 2019, 2019, 5190425. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.-L.; Wang, R.-N.; Zhang, N.-Y.; Fan, W.-B.; Fang, M.-F.; Li, Z.-H. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [PubMed]
- Drescher, A.; Ruf, S.; Calsa, T., Jr.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Akhunov, E.D.; Akhunova, A.R.; Anderson, O.D.; Anderson, J.A.; Blake, N.; Clegg, M.T.; Coleman-Derr, D.; Conley, E.J.; Crossman, C.C.; Deal, K.R. Nucleotide diversity maps reveal variation in diversity among wheat genomes and chromosomes. BMC Genom. 2010, 11, 702. [Google Scholar] [CrossRef]
- Alice, L.; Dodson, T.; Sutherland, B. Diversity and relationships of Bhutanese Rubus. Acta Hortic. 2008, 777, 63–70. [Google Scholar] [CrossRef]
- Li, Z.; Yan, W.; Qing, C.; Ya, L.; Yong, Z.; Tang, H.-R.; Wang, X.-R. Phylogenetic utility of Chinese Rubus (Rosaceae) based on ndhF sequence. Acta Hortic. Sin. 2015, 42, 19. [Google Scholar]
- Morden, C.W.; Gardner, D.E.; Weniger, D.A. Phylogeny and biogeography of Pacific Rubus subgenus Idaeobatus (Rosaceae) species: Investigating the origin of the endemic Hawaiian raspberry R. macraei. Pac. Sci. 2003, 57, 181–197. [Google Scholar] [CrossRef]
- Imanishi, H.; Nakahara, K.; Tsuyuzaki, H. Genetic relationships among native and introduced Rubus species in Japan based on rbcL sequence. Acta Hortic. 2008, 918, 195–199. [Google Scholar] [CrossRef]
- Milligan, B.G.; Hampton, J.N.; Palmer, J.D. Dispersed repeats and structural reorganization in subclover chloroplast DNA. Mol. Biol. Evol. 1989, 6, 355–368. [Google Scholar]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.; Rafalski, J. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Shi, T.; Luo, W.; Ni, X.; Iqbal, S.; Ni, Z.; Huang, X.; Yao, D.; Shen, Z.; Gao, Z. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic. Res. 2019, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Somaratne, Y.; Guan, D.-L.; Wang, W.-Q.; Zhao, L.; Xu, S.-Q. Complete chloroplast genome sequence of Xanthium sibiricum provides useful DNA barcodes for future species identification and phylogeny. Plant Syst. Evol. 2019, 305, 949–960. [Google Scholar] [CrossRef]
- Li, X.; Zuo, Y.; Zhu, X.; Liao, S.; Ma, J. Complete chloroplast genomes and comparative analysis of sequences evolution among seven Aristolochia (Aristolochiaceae) medicinal species. Int. J. Mol. Sci. 2019, 20, 1045. [Google Scholar] [CrossRef] [PubMed]
- Yanfei, N.; Tai, S.; Chunhua, W.; Jia, D.; Fazhong, Y. Complete chloroplast genome sequences of the medicinal plant Aconitum transsectum (Ranunculaceae): Comparative analysis and phylogenetic relationships. BMC Genom. 2023, 24, 90. [Google Scholar] [CrossRef] [PubMed]
- Nekrutenko, A.; Makova, K.D.; Li, W.-H. The KA/KS ratio test for assessing the protein-coding potential of genomic regions: An empirical and simulation study. Genome Res. 2002, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Ding, X.; Guan, W.; Zhang, D.; Zhang, J.; Bai, J.; Xu, W.; Huang, J.; Qiu, X.; Zheng, X. Comparative chloroplast genome analyses of Amomum: Insights into evolutionary history and species identification. BMC Plant Biol. 2022, 22, 520. [Google Scholar] [CrossRef] [PubMed]
- Rensing, S.A.; Fritzowsky, D.; Lang, D.; Reski, R. Protein encoding genes in an ancient plant: Analysis of codon usage, retained genes and splice sites in a moss, Physcomitrella patens. BMC Genom. 2005, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Quax, T.E.; Claassens, N.J.; Söll, D.; van der Oost, J. Codon bias as a means to fine-tune gene expression. Mol. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef]
- Mehmood, F.; Shahzadi, I.; Ahmed, I.; Waheed, M.T.; Mirza, B. Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics 2020, 112, 1522–1530. [Google Scholar] [CrossRef]
- Li, H.; Wu, M.; Lai, Q.; Zhou, W.; Song, C. Complete chloroplast of four Sanicula taxa (Apiaceae) endemic to China: Lights into genome structure, comparative analysis, and phylogenetic relationships. BMC Plant Biol. 2023, 23, 444. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Duan, H.; Tao, K.; Luo, Y.; Li, Q.; Li, L. Complete chloroplast genome structural characterization of two Phalaenopsis (Orchidaceae) species and comparative analysis with their alliance. BMC Genom. 2023, 24, 359. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | R. argutus | R. chingii | R. corchorifolius | R. idaeus | R. occidentalis | R. parviflorus | |
|---|---|---|---|---|---|---|---|
| Length (bp) | Total | 156,630 | 155,563 | 155,493 | 155,702 | 156,712 | 156,882 |
| LSC | 85,962 | 85,322 | 85,271 | 85,026 | 85,929 | 86,069 | |
| SSC | 18,754 | 18,743 | 18,702 | 18,706 | 18,843 | 18,795 | |
| IR | 25,957 | 25,749 | 25,760 | 25,985 | 25,970 | 26,009 | |
| Region size (%) | CDS | 49.90 | 50.91 | 50.58 | 51.49 | 49.61 | 50.13 |
| Cis-spliced intron | 12.19 | 11.88 | 12.37 | 12.55 | 11.74 | 12.68 | |
| tRNA | 1.74 | 1.79 | 1.75 | 1.80 | 1.69 | 1.78 | |
| rRNA | 5.78 | 5.82 | 5.82 | 6.01 | 5.77 | 5.76 | |
| Non-coding region | 31.46 | 30.69 | 30.56 | 29.24 | 32.25 | 31.00 | |
| GC content (%) | Total | 37.13 | 37.06 | 37.06 | 37.26 | 37.11 | 37.18 |
| LSC | 35.01 | 34.94 | 34.91 | 35.18 | 34.97 | 35.08 | |
| SSC | 31.23 | 30.83 | 30.91 | 31.36 | 31.1 | 31.25 | |
| IR | 42.79 | 42.84 | 42.83 | 42.80 | 42.81 | 42.78 | |
| CDS | 37.95 | 37.75 | 37.84 | 37.95 | 37.85 | 38.01 | |
| Cis-spliced intron | 37 | 37.35 | 36.73 | 37.01 | 37.17 | 36.97 | |
| tRNA | 53.71 | 53.48 | 53.78 | 53.45 | 53.68 | 53.44 | |
| rRNA | 55.44 | 55.42 | 55.39 | 55.00 | 55.44 | 55.46 | |
| Non-coding region | 31.52 | 31.23 | 31.32 | 31.44 | 31.68 | 31.62 | |
| Gene numbers | Total (unique) | 131(111) | 131(111) | 131(111) | 131(111) | 131(111) | 131(111) |
| PCG (unique) | 86(79) | 86(79) | 86(79) | 86(79) | 86(79) | 86(79) | |
| tRNA (unique) | 37(28) | 37(28) | 37(28) | 37(28) | 37(28) | 37(28) | |
| rRNA (unique) | 8(4) | 8(4) | 8(4) | 8(4) | 8(4) | 8(4) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Chen, Z.; Jiang, J.; Li, X.; Zeng, W. Comparative and Phylogenetic Analysis of Six New Complete Chloroplast Genomes of Rubus (Rosaceae). Forests 2024, 15, 1167. https://doi.org/10.3390/f15071167
Shi Y, Chen Z, Jiang J, Li X, Zeng W. Comparative and Phylogenetic Analysis of Six New Complete Chloroplast Genomes of Rubus (Rosaceae). Forests. 2024; 15(7):1167. https://doi.org/10.3390/f15071167
Chicago/Turabian StyleShi, Yujie, Zhen Chen, Jingyong Jiang, Xiaobai Li, and Wei Zeng. 2024. "Comparative and Phylogenetic Analysis of Six New Complete Chloroplast Genomes of Rubus (Rosaceae)" Forests 15, no. 7: 1167. https://doi.org/10.3390/f15071167
APA StyleShi, Y., Chen, Z., Jiang, J., Li, X., & Zeng, W. (2024). Comparative and Phylogenetic Analysis of Six New Complete Chloroplast Genomes of Rubus (Rosaceae). Forests, 15(7), 1167. https://doi.org/10.3390/f15071167