
Full-Length Transcriptome Assembly of Platycladus orientalis Root Integrated with RNA-Seq to Identify Genes in Response to Root Pruning

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Experimental Materials
2.2. RNA Extraction, RNA-Seq, and PacBio Full-Length ISO-Seq Library Preparation and Sequencing
2.3. SSR Analysis
2.4. Prediction of lncRNAs
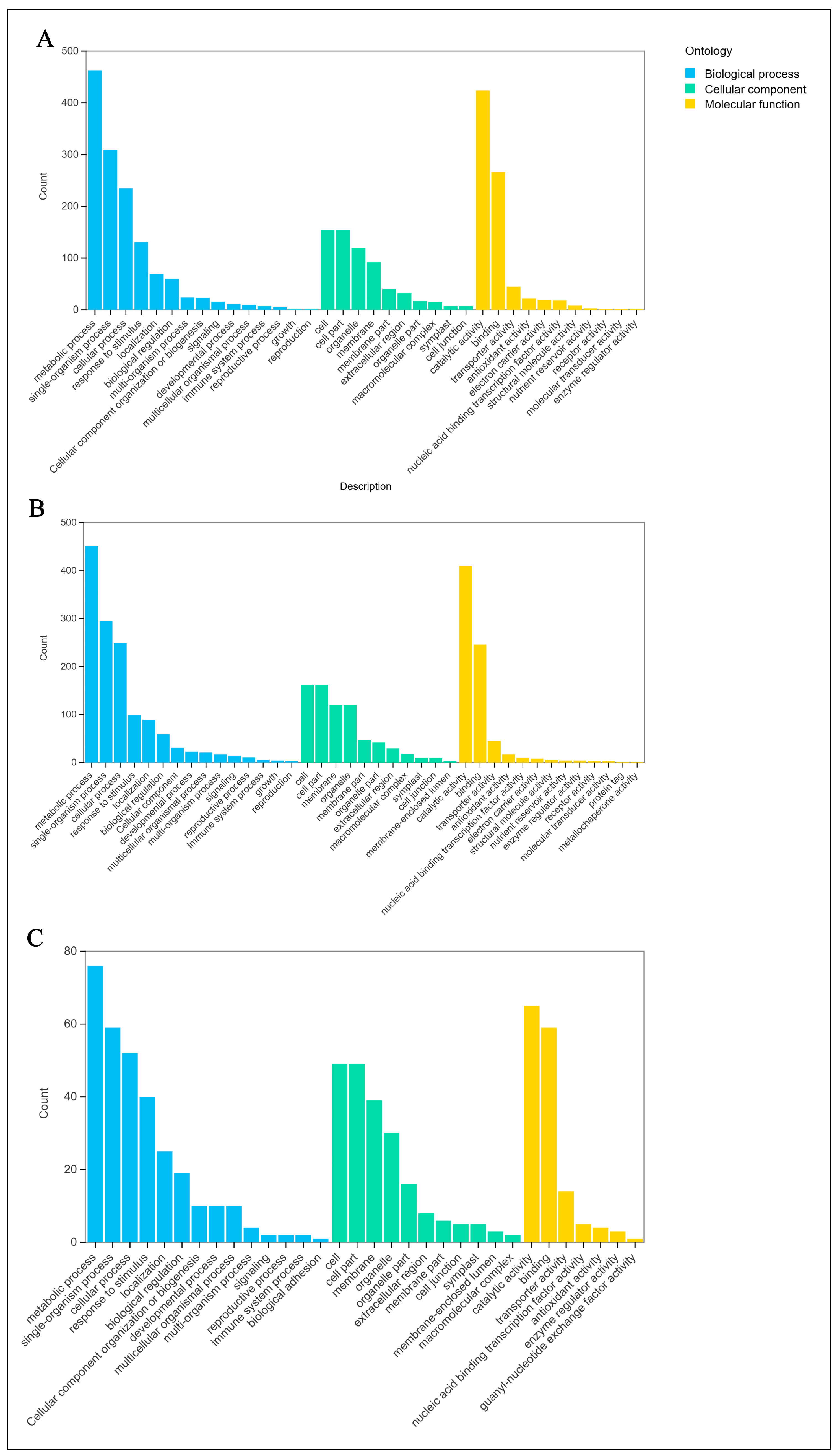
2.5. Gene Function Annotation
2.6. Transcript Expression Analysis
2.7. Differential Gene Expression Analysis and Enrichment Evaluation
2.8. Screening of Key Genes
2.9. Validation of DEGs with Real-Time Fluorescence Quantitative PCR (RT-qPCR)
3. Results
3.1. Root Development of P. orientalis in Different Periods
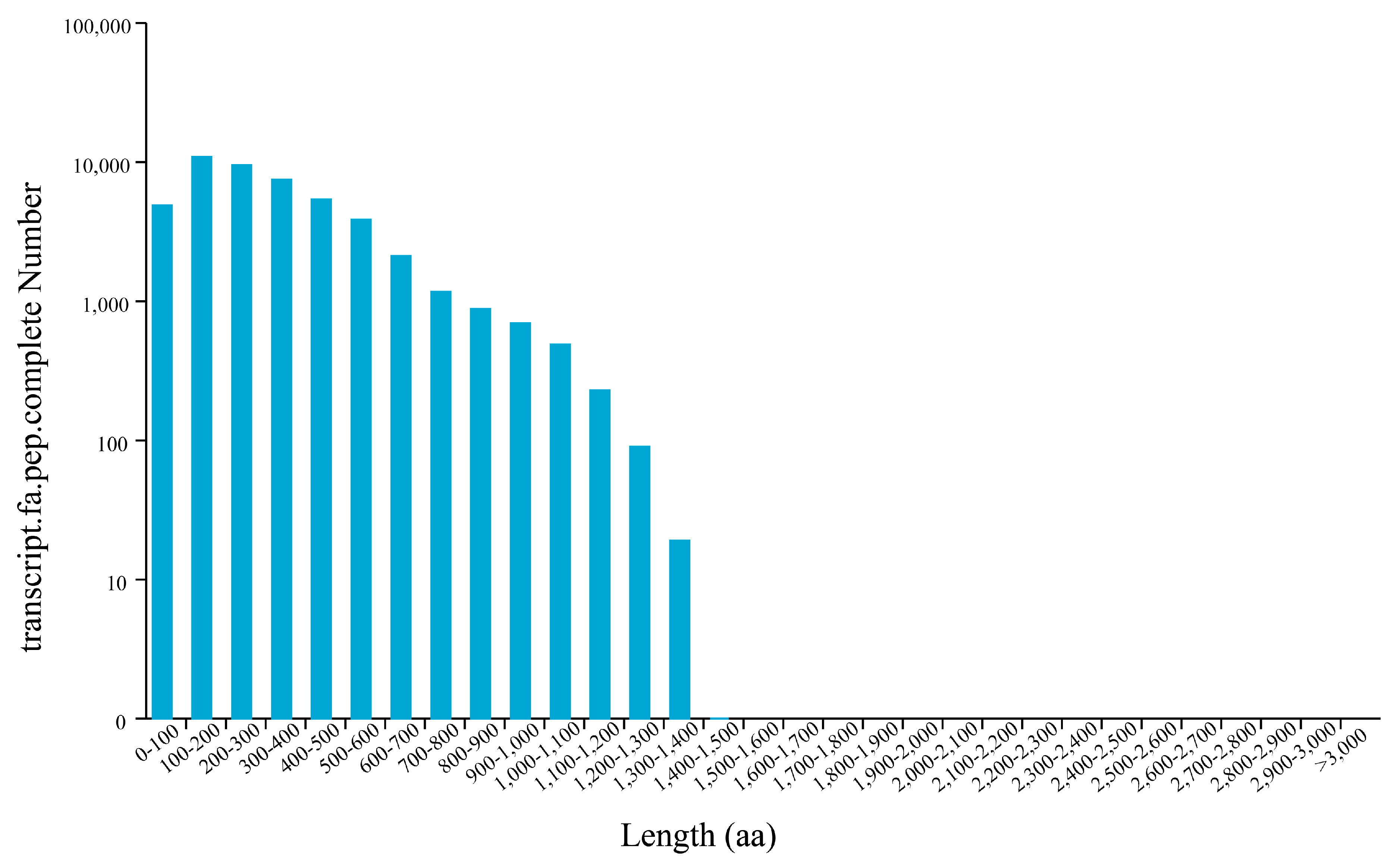
3.2. Construction of P. orientalis Full-Length Transcriptome Database
3.3. SSR Analysis
3.4. Prediction of lncRNAs
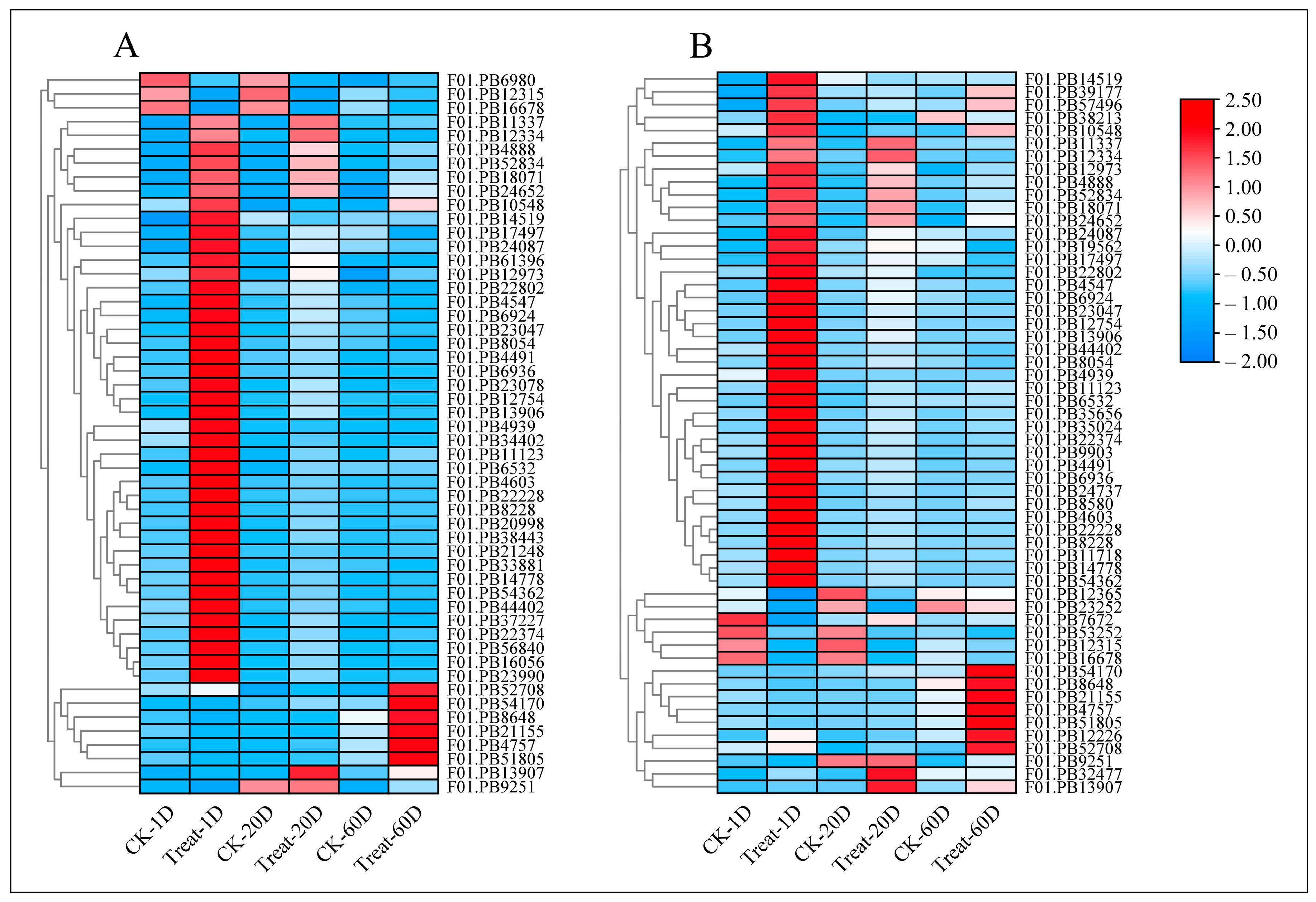
3.5. Identification of DEGs
3.6. Identification of DEGs
3.7. Expression Profiles and Functional Analysis of Genes Related to 2 Key Pathways
3.8. Identification of Hub Genes from Candidate Genes
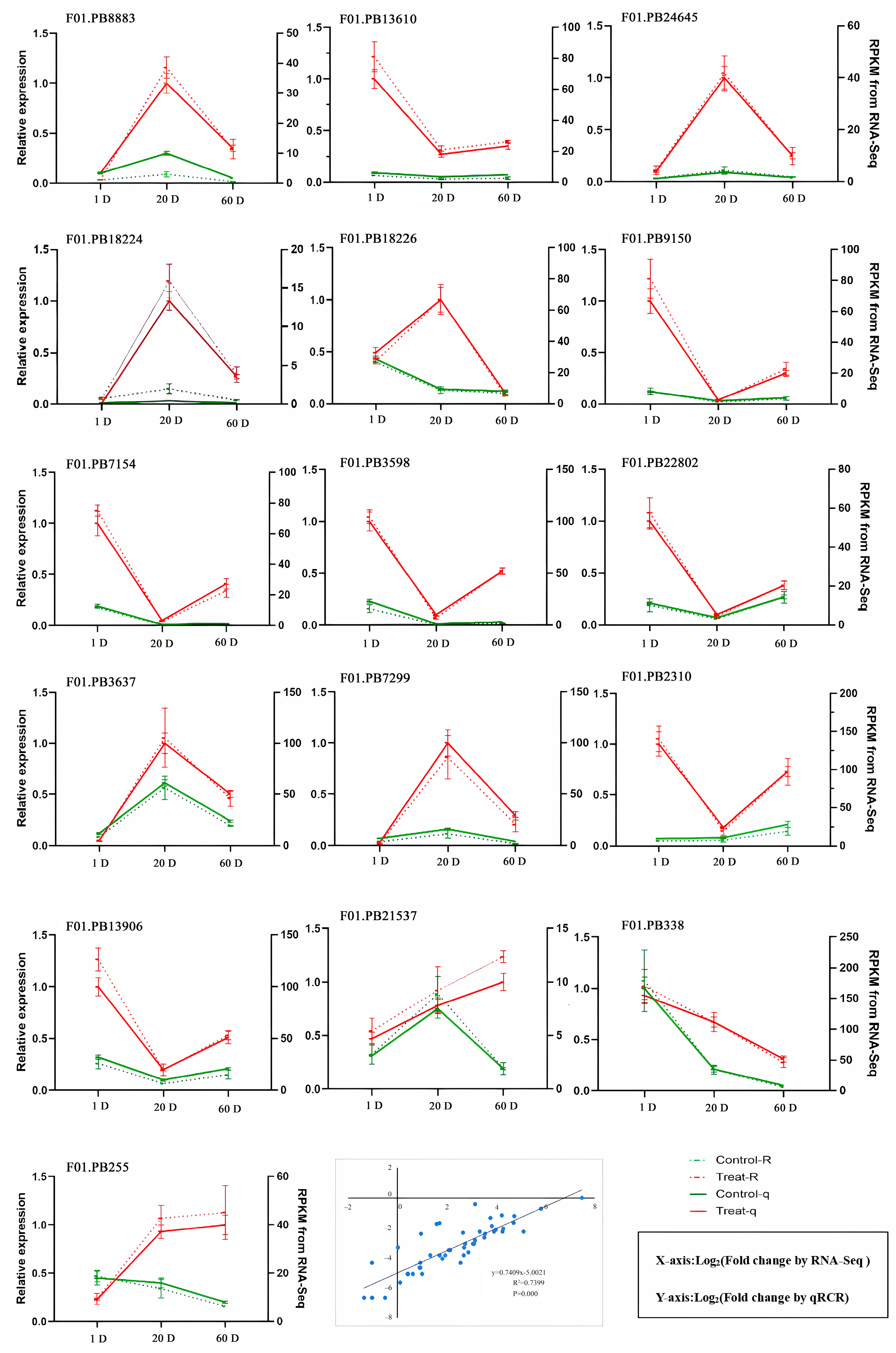
3.9. RT-qPCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Toca, A.; Oliet, J.A.; Villar-Salvador, P.; Martínez Catalán, R.A.; Jacobs, D.F. Ecologically distinct pine species show differential root development after outplanting in response to nursery nutrient cultivation. For. Ecol. Manag. 2019, 451, 117562. [Google Scholar] [CrossRef]
- Liangpeng, Y.; Jian, M.; Yan, L. Impact of salt stress on the features and activities of root system for three desert halophyte species in their seedling stage. Sci. China Ser. D 2007, 50, 97–106. [Google Scholar]
- Yang, X.T.; Yang, X.B.; Zeng, L.L.; Fan, Z.W. Ecological functions of root system of forest trees and factors affecting root distribution. J. Henan Agric. Univ. 2009, 43, 681–690. [Google Scholar]
- Lu, W.; Wang, X.; Wang, F. Adaptive minirhizotron for pepper roots observation and its installation based on root system architecture traits. Plant Methods 2019, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Zhang, Y.; Krehbiel, P.R. Analysis of the relationship between the morphological characteristics of lightning channels and turbulent dynamics based on the localization of VHF radiation sources. Geophys. Res. Lett. 2024, 51, e2023GL106024. [Google Scholar] [CrossRef]
- Zhou, B.Z.; Zhang, S.G.; Fu, M.Y. The origin, development and application of Minirhizotron, a new technology for plant root. J. Ecol. 2007, 2, 253–256. [Google Scholar]
- Cai, T.; Xu, H.; Peng, D.; Yin, Y.; Yang, W.; Ni, Y.; Chen, X.; Xu, C.; Yang, D.; Cui, Z.; et al. Exogenous hormonal application improves grain yield of wheat by optimizing tiller productivity. Field Crop Res. 2014, 155, 172–183. [Google Scholar] [CrossRef]
- Yu, H.; Hu, Y.; Qi, L.; Zhang, K.; Jiang, J.; Li, H.; Zhang, X.; Zhang, Z. Hyperspectral Detection of Moisture Content in Rice Straw Nutrient Bowl Trays Based on PSO-SVR. Sustainability. 2023, 15, 8703. [Google Scholar] [CrossRef]
- Löf, M.; Thomsen, A.; Madsen, P. Sowing and transplanting of broadleaves (Fagus sylvatica L., Quercus robur L., Prunus avium L. and Crataegus monogyna Jacq.) for afforestation of farmland. For. Ecol. Manag. 2004, 188, 113–123. [Google Scholar] [CrossRef]
- Agibalova, A.V. On the completeness of systems of root functions of differential operators of fractional order with matrix coefficients. Math. Notes 2010, 88, 287–290. [Google Scholar] [CrossRef]
- Ganin, H.; Kemper, N.; Meir, S.; Rogachev, I.; Ely, S.; Massalha, H.; Mandaby, A.; Shanzer, A.; Keren-Paz, A.; Meijler, M.M.; et al. Indole Derivatives Maintain the Status Quo between Beneficial Biofilms and Their Plant Hosts. Mol. Plant Microbe Interact. 2019, 32, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.D.; Luo, F.Q.; Qian, G.Q. Study on the cultivation of Pinus massoniana seedlings in containers by transplanting mycorrhizal roots. J. Fujian For. Coll. 1994, 2, 128–132. [Google Scholar]
- Yang, S.M.; Yang, B.C.; Jiang, L.Y.; Yang, P.; Lu, Y. Technology of cultivation and cultivation management of green Zanthoxylum zanthoxylum root transplanting seedlings. Hunan For. Sci. Technol. 2020, 47, 112–115. [Google Scholar]
- Du, Y.W.; Deng, X.Z.; Cheng, J.Y. Effect of root cutting on quality of thin shelled hickory stock. For. Sci. West. China 2021, 50, 92–98. [Google Scholar]
- Thomas, P.; Ravindra, M.B. Effect of pruning or removal of in vitro formed roots on ex vitro root regeneration and growth in micropropagated grapes. Plant Cell 1997, 51, 177–180. [Google Scholar] [CrossRef]
- Ma, S.C.; Xu, B.C.; Li, F.M.; Huang, Z.B. Effects of root pruning on root efficiency, water use and yield of winter wheat. J. Appl. Environ. Biol. 2009, 15, 606–609. [Google Scholar] [CrossRef]
- Wang, S.Y.; Wang, P.; Tu, G.Q.; Li, B.M.; Chen, Q.; Zhang, M.; Tao, H.H.; Chen, D.Y.; Jin, L.L. Effects of root cutting treatment on growth potential and fruit quality of kiwi fruit. Chin. Fruit Tree 2022, 12, 32–37. [Google Scholar]
- Hou, D.P.; Xu, Y.X.; Li, M.J.; Liu, H.S.; Xu, S.Z.; He, C.S. Experiment on the cultivation of Keeleria phyllosa seedlings by cutting root and matching matrix. Green Technol. 2018, 15, 87–89. [Google Scholar]
- Fan, G.Y.; Hu, L.D.; Zhou, Z.R. Study on the technology of osmanthus seedling cutting and transplanting. Mod. Hortic. 2017, 21, 78. [Google Scholar]
- Yan, M.; Cui, F.; Liu, Y.; Zhang, Z.; Zhang, J.; Ren, H.; Li, Z. Vegetation type and plant diversity affected soil carbon accumulation in a postmining area in Shanxi Province, China. Land Degrad. Dev. 2019, 31, 181–189. [Google Scholar] [CrossRef]
- Wang, R.X.; Kong, Q.Y.; Yu, H.; Guo, J.; Wu, D.; Xin, X.B. Study on multifunctional evaluation of Platycladus orientalis plantation in Mentougou district, Beijing. J. Cent. South Univ. For. Technol. 2016, 36, 58–62. [Google Scholar]
- Hu, Y.Z. The medicine of dark hair and hair—Platycypress leaf. All Things Rural 2017, 14, 54. [Google Scholar]
- Yang, X.; Yan, D.; Liu, C. Natural regeneration of trees in three types of afforested stands in the Taihang Mountains, China. PLoS ONE 2014, 9, e108744. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.T.; Wang, G.L.; Zhao, N.; Fan, Z.W. Effects of different root cutting treatments on lateral root growth of tree seedlings. J. Henan Agric. Univ. 2010, 44, 155–159. [Google Scholar]
- Ayadi, L.; Motorin, Y.; Marchand, V. Quantification of 2′-O-Me Residues in RNA Using Next-Generation Sequencing (Illumina RiboMethSeq Protocol). Methods Mol. Biol. 2018, 1649, 29–48. [Google Scholar] [CrossRef]
- Sun, J.; Xu, Y.; Ye, S.; Jiang, H.; Chen, Q.; Liu, F.; Zhou, W.; Chen, R.; Li, X.; Tietz, O.; et al. Arabidopsis ASA1 is important for jasmonate-mediated regulation of auxin biosynthesis and transport during lateral root formation. Plant cell. 2019, 21, 1495–1511. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Mo, X.; Shou, H.; Wu, P. Cytokinin-mediated cell cycling arrest of pericycle founder cells in lateral root initiation of Arabidopsis. Plant Cell Physiol. 2006, 47, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Eyun, S.I.; Arora, K.; Tan, S.Y.; Gandra, P.; Moriyama, E.; Khajuria, C.; Jurzenski, J.; Li, H.; Donahue, M.; et al. Patterns of Gene Expression in Western Corn Rootworm (Diabrotica virgifera virgifera) Neonates, Challenged with Cry34Ab1, Cry35Ab1 and Cry34/35Ab1, Based on Next-Generation Sequencing. Toxins 2017, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Ichihashi, Y.; Kimura, S.; Chitwood, D.H.; Headland, L.R.; Peng, J.; Maloof, J.N.; Sinha, N.R. A High-Throughput Method for Illumina RNA-Seq Library Preparation. Frontiers in plant science. Front. Plant Sci. 2012, 3, 202. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, M.O.; Russ, C.; Ross, M.G.; Gabriel, S.B.; Nusbaum, C.; DePristo, M.A. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genom. 2012, 13, 375. [Google Scholar] [CrossRef]
- Pan, L.; Dinh, H.Q.; Pawitan, Y.; Vu, T.N. Isoform-level quantification for single-cell RNA sequencing. Bioinformatics 2022, 38, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Kainth, A.S.; Haddad, G.A.; Hall, J.M.; Ruthenburg, A.J. Merging short and stranded long reads improves transcript assembly. PLoS Comput. Biol. 2023, 19, e1011576. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Feng, S.; Liu, Y.; Zhao, L.; Tian, L.; Hu, Y.; Yang, T.; Wei, A. Single-Molecule Long-Read Sequencing of Zanthoxylum bungeanum Maxim. Transcriptome: Identification of Aroma-Related Genes. Forests 2018, 9, 765. [Google Scholar] [CrossRef]
- Zhou, Y.; Tang, Q.; Wu, M.; Mou, D.; Liu, H.; Wang, S.; Zhang, C.; Ding, L.; Luo, J. Comparative transcriptomics provides novel insights into the mechanisms of selenium tolerance in the hyperaccumulator plant Cardamine hupingshanensis. Sci. Rep. 2018, 8, 2789. [Google Scholar] [CrossRef]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. B Biol Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef]
- Rhoads, D.M.; Subbaiah, C.C. Mitochondrial retrograde regulation in plants. Mitochondrion 2007, 7, 177–194. [Google Scholar] [CrossRef]
- Mhamdi, A.; Noctor, G.; Baker, A. Plant catalases: Peroxisomal redox guardians. Arch. Biochem. Biophys. 2012, 525, 181–194. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, H.; Ruan, W.; Deng, M.; Wang, F.; Peng, J.; Luo, J.; Chen, Z.; Yi, K. ABNORMAL INFLORESCENCE MERISTEM1 Functions in Salicylic Acid Biosynthesis to Maintain Proper Reactive Oxygen Species Levels for Root Meristem Activity in Rice. Plant Cell 2017, 29, 560–574. [Google Scholar] [CrossRef]
- Zhu, X.F.; Lu, Y.N.; Wang, J.D.; Xu, Q. Investigation of the interactions between indole-3-acetic acid and catalase: A spectroscopic study in combination with second-order calibration and molecular docking methods. Anal. Methods 2013, 5, 6037–6044. [Google Scholar] [CrossRef]
- Anjum, N.A.; Sharma, P.; Gill, S.S.; Hasanuzzaman, M.; Khan, E.A.; Kachhap, K.; Mohamed, A.A.; Thangavel, P.; Devi, G.D.; Vasudhevan, P.; et al. Catalase and ascorbate peroxidase-representative H2O2-detoxifying heme enzymes in plants. Environ. Sci. Pollut. Res. Int. 2016, 23, 19002–19029. [Google Scholar] [CrossRef]
- Liu, C.; Dong, L.A.; Lin, J.Z.; Liu, X.M. Research Advances on Regulation Mechanism of Reactive Oxygen Species Metabolism under Stresses. Life Sci. Res. 2019, 23, 253–258. [Google Scholar]
- Del Río, L.A. ROS and RNS in plant physiology: An overview. J. Exp. Bot. 2015, 66, 2827–2837. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; He, Y.; Yin, X.; Zhong, X.; Yan, B.; Wu, Y.; Chen, J.; Li, X.; Zhai, K.; Huang, Y.; et al. Ca2+ sensor-mediated ROS scavenging suppresses rice immunity and is exploited by a fungal effector. Cell 2021, 184, 5391–5404.e17. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hu, W.Z.; Bi, Y.; Jiang, A.L. Effect of exogenous ethylene and methyl jasmonate (MeJA) on reactive oxygen metabolism in tissue of fresh-cut cabbage. Sci. Technol. Food Ind. 2013, 34, 336–339+343. [Google Scholar] [CrossRef]
- Saidi, I.; Nawel, N.; Djebali, W. Role of selenium in preventing manganese toxicity in sunflower (Helianthus annuus) seedling. S. Afr. J. Bot. 2014, 94, 88–94. [Google Scholar] [CrossRef]
- Zhang, M.; Li, Q.; Liu, T.; Liu, L.; Shen, D.; Zhu, Y.; Liu, P.; Zhou, J.M.; Dou, D. Two cytoplasmic effectors of Phytophthora sojae regulate plant cell death via interactions with plant catalases. Plant Physiol. 2015, 167, 164–175. [Google Scholar] [CrossRef]
- Mathioudakis, M.M.; Veiga, R.S.; Canto, T.; Medina, V.; Mossialos, D.; Makris, A.M.; Livieratos, I. Pepino mosaic virus triple gene block protein 1 (TGBp1) interacts with and increases tomato catalase 1 activity to enhance virus accumulation. Mol. Plant Pathol. 2013, 14, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, Y.; Miao, L.F. Comparative physiological and proteomic responses to drought stress in two poplar species originating from different altitudes. Physiol. Plant 2010, 139, 388–400. [Google Scholar] [CrossRef]
- Rizhsky, L.; Hallak-Herr, E.; Van Breusegem, F.; Rachmilevitch, S.; Barr, J.E.; Rodermel, S.; Inzé, D.; Mittler, R. Double antisense plants lacking ascorbate peroxidase and catalase are less sensitive to oxidative stress than single antisense plants lacking ascorbate peroxidase or catalase. Plant J. 2002, 32, 329–342. [Google Scholar] [CrossRef]

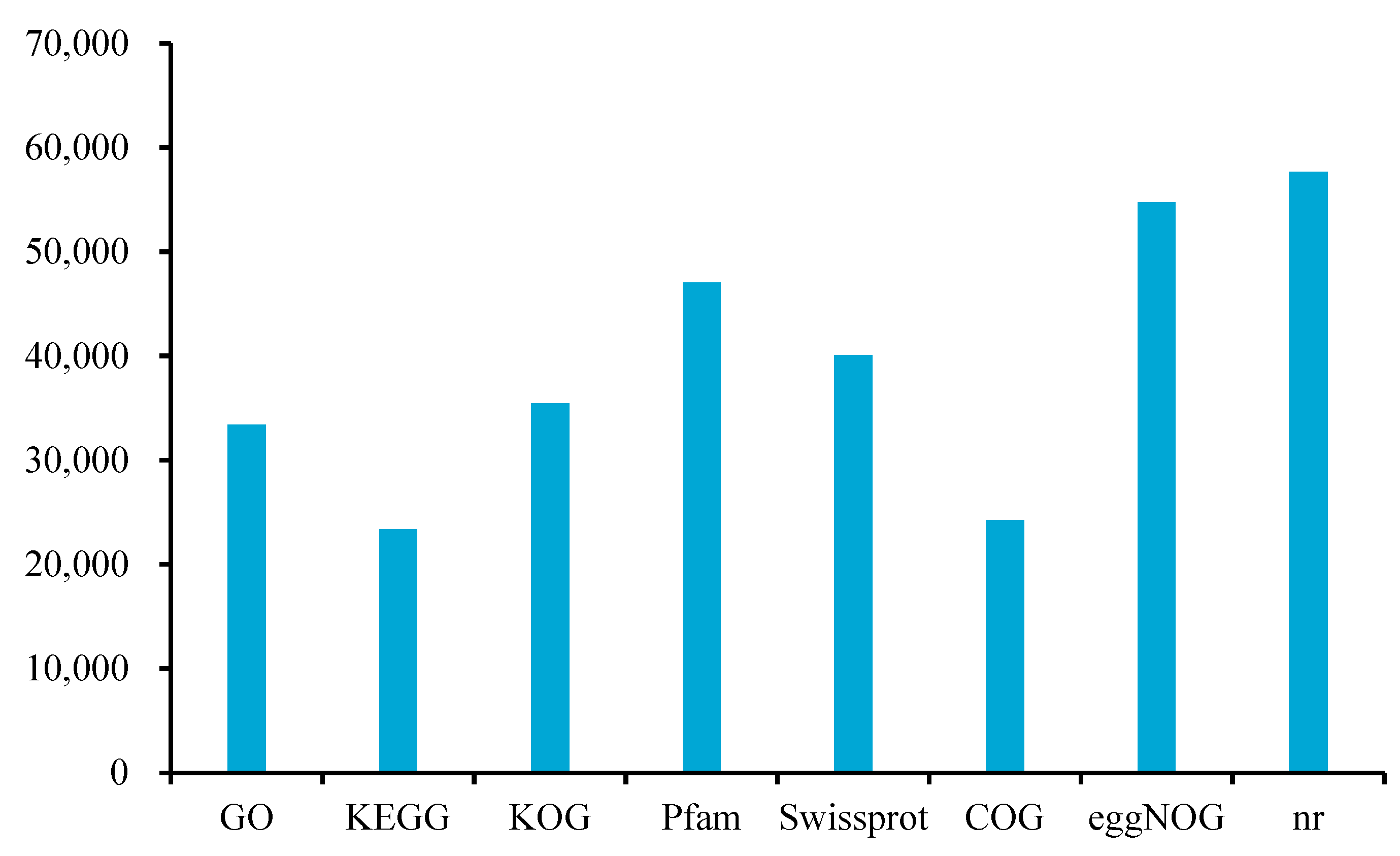





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dou, H.; Sun, H.; Feng, X.; Wang, T.; Wang, Y.; Quan, J.; Yang, X. Full-Length Transcriptome Assembly of Platycladus orientalis Root Integrated with RNA-Seq to Identify Genes in Response to Root Pruning. Forests 2024, 15, 1232. https://doi.org/10.3390/f15071232
Dou H, Sun H, Feng X, Wang T, Wang Y, Quan J, Yang X. Full-Length Transcriptome Assembly of Platycladus orientalis Root Integrated with RNA-Seq to Identify Genes in Response to Root Pruning. Forests. 2024; 15(7):1232. https://doi.org/10.3390/f15071232
Chicago/Turabian StyleDou, Hao, Huijuan Sun, Xi Feng, Tiantian Wang, Yilin Wang, Jin’e Quan, and Xitian Yang. 2024. "Full-Length Transcriptome Assembly of Platycladus orientalis Root Integrated with RNA-Seq to Identify Genes in Response to Root Pruning" Forests 15, no. 7: 1232. https://doi.org/10.3390/f15071232
APA StyleDou, H., Sun, H., Feng, X., Wang, T., Wang, Y., Quan, J., & Yang, X. (2024). Full-Length Transcriptome Assembly of Platycladus orientalis Root Integrated with RNA-Seq to Identify Genes in Response to Root Pruning. Forests, 15(7), 1232. https://doi.org/10.3390/f15071232