
Integration of Illumina and PacBio HiFi Sequencing Reveals a Three-Linear-Molecule Mitogenome with RNA-Editing Sites and Phylogeny in Arrow Bamboo (Fargesia qinlingensis)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, Sequencing, and Library Construction
2.2. De Novo Assembly of the Mitogenome and Gene Annotation
2.3. Analysis of Codon Usage Preference and Repetitive Sequences
2.4. Analysis of the Migration of MTPTs
2.5. RNA-Editing Site Prediction and Experimental Evaluation
2.6. Collinearity and Phylogenetic Tree Construction
3. Results
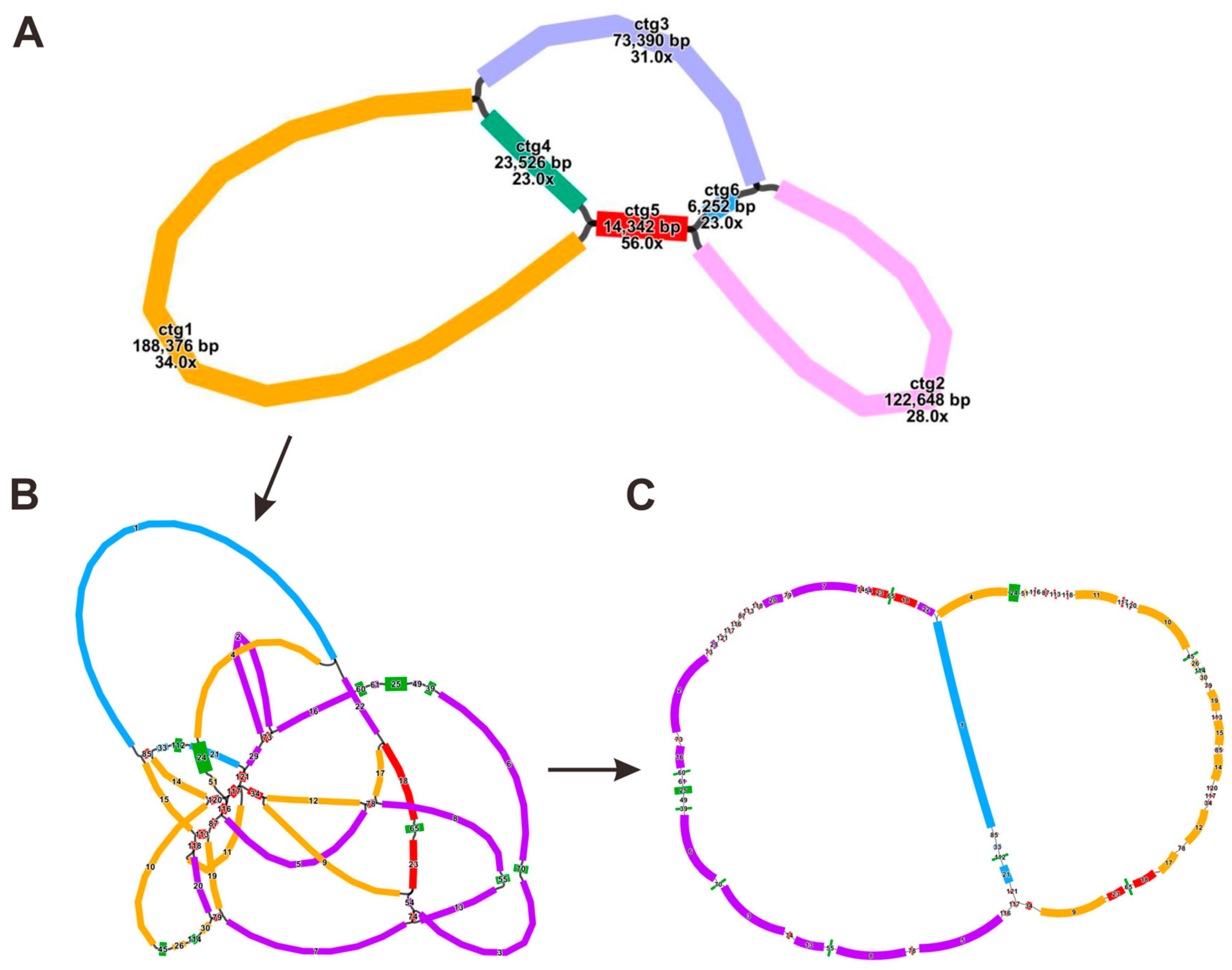
3.1. Mitogenome Assembly and Configuration of F. qinlingensis
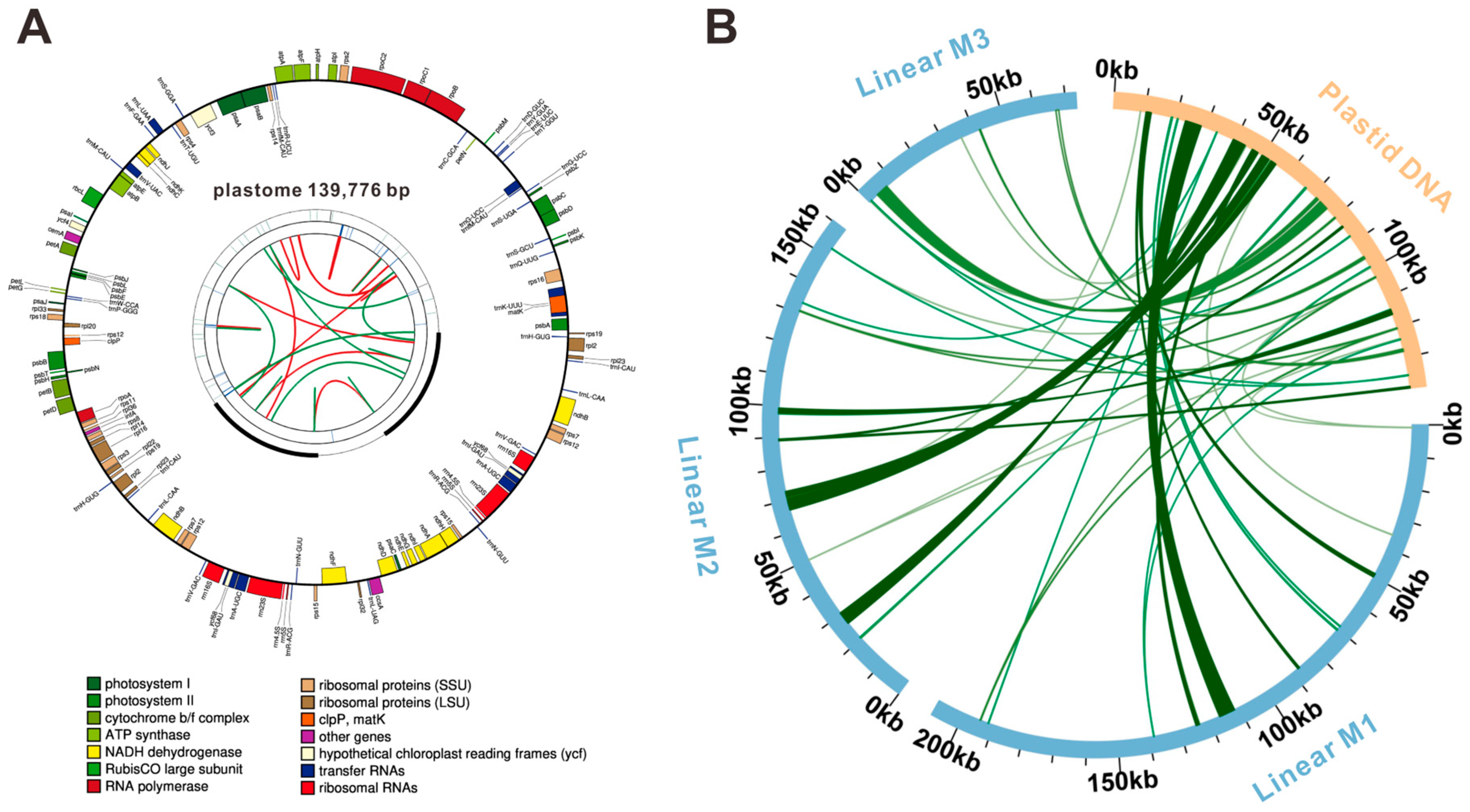
3.2. Mitogenome Annotation of F. qinlingensis
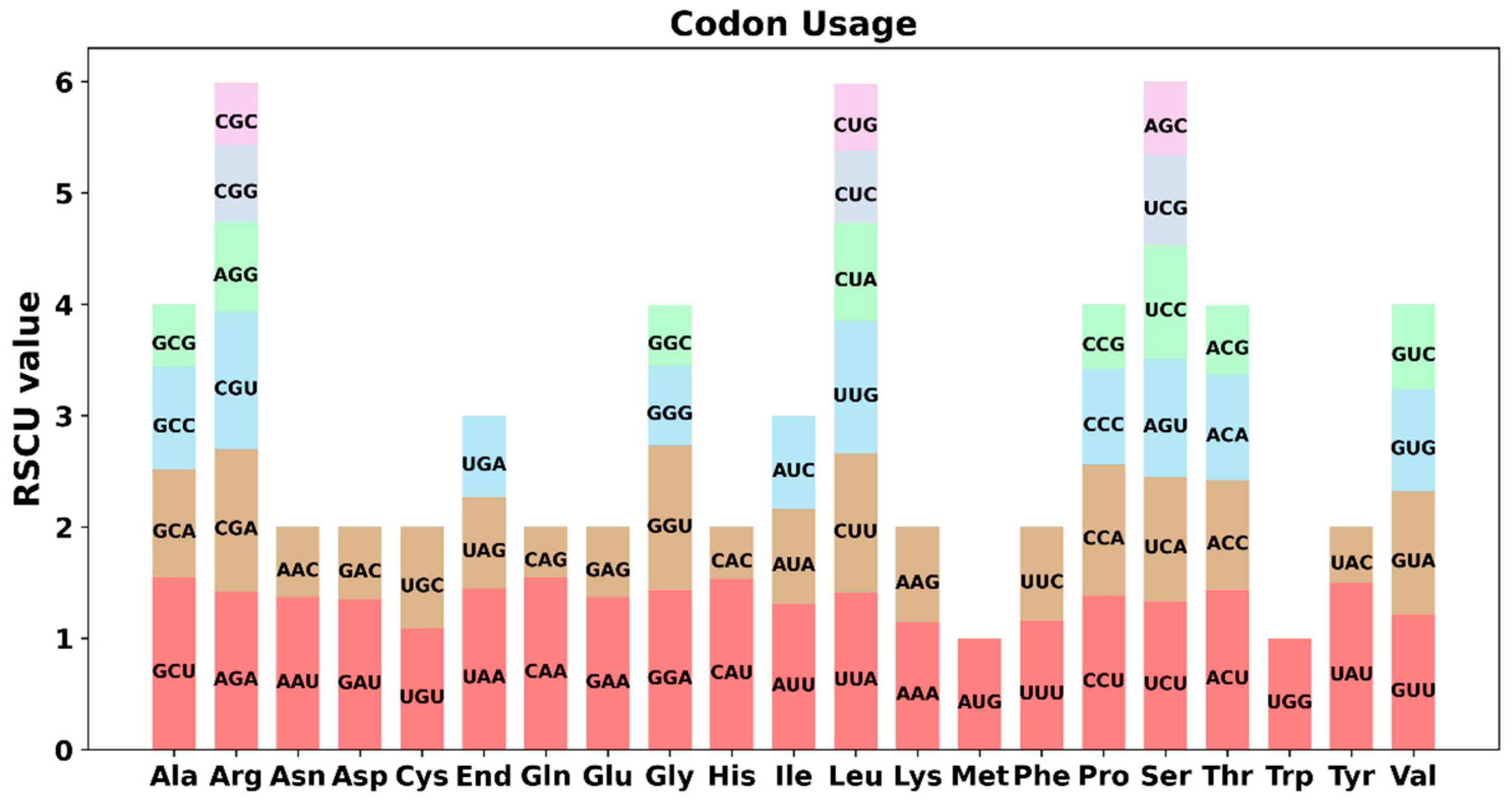
3.3. Detection of Codon Usage Preference in PCGs
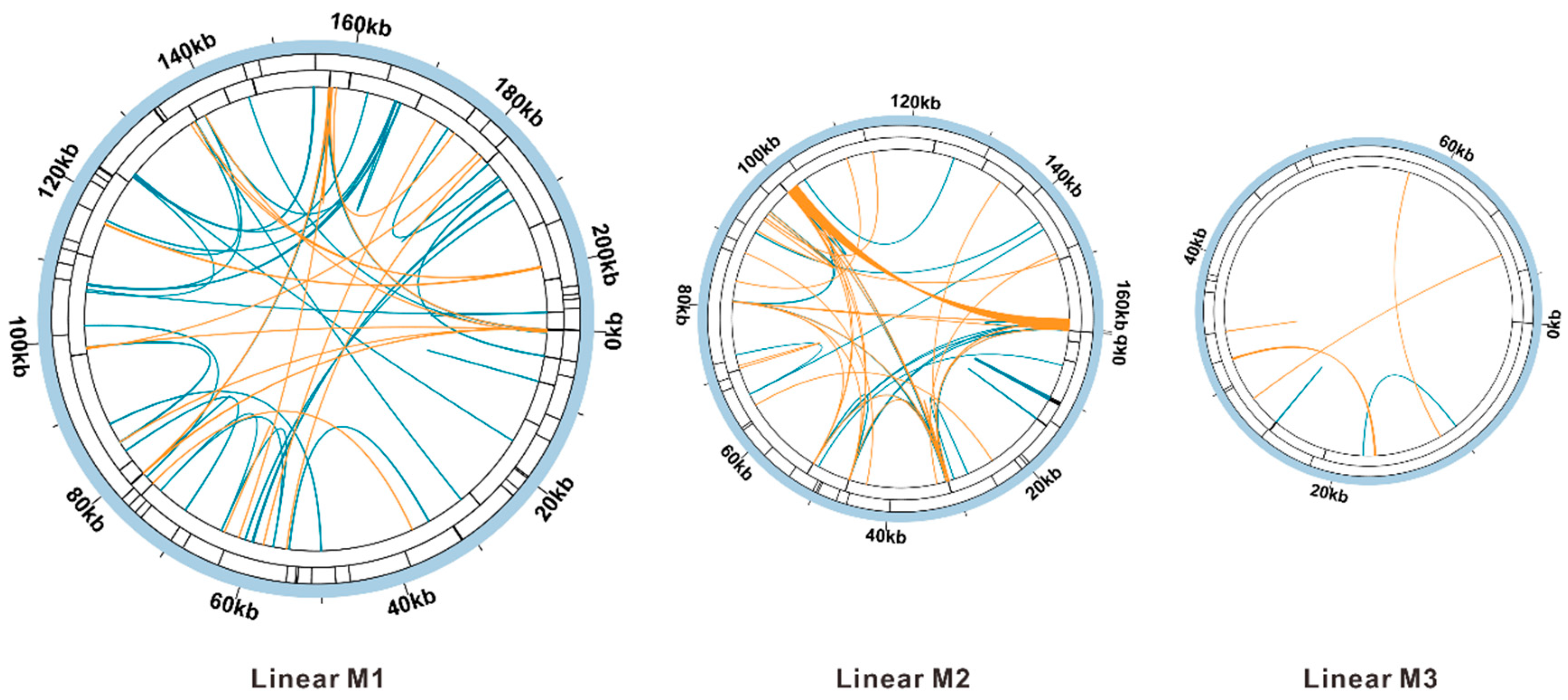
3.4. Analyses of Various Types of Repetitive Sequences
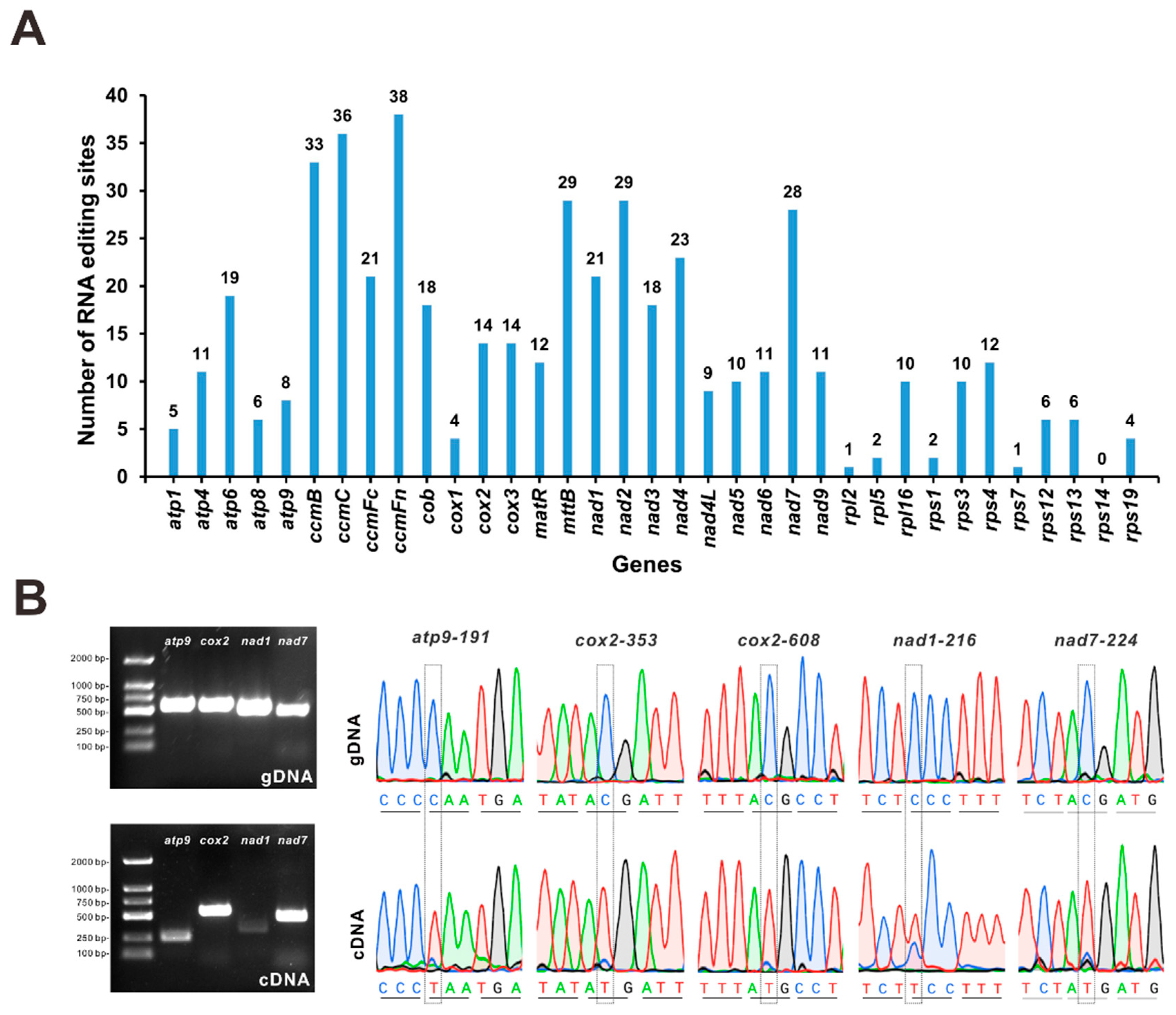
3.5. Prediction of RNA-Editing Sites and Experimental Evaluation
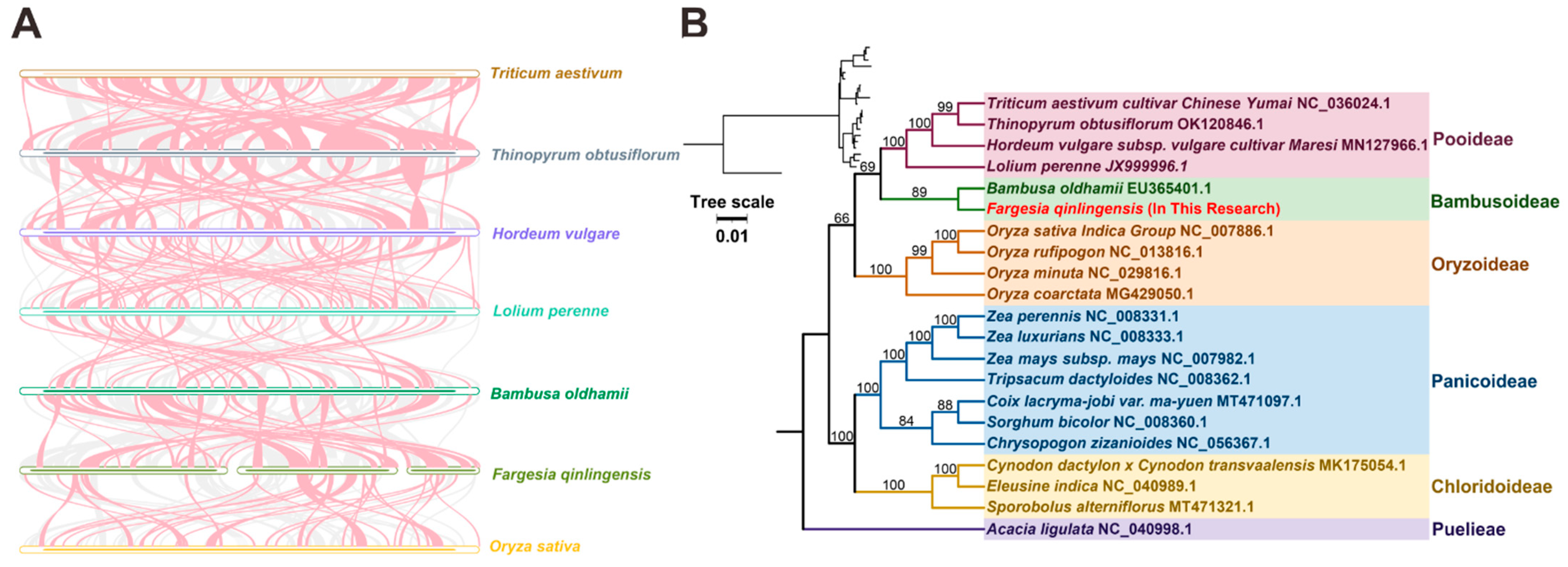
3.6. Sequence Collinearity and Phylogenetic Evolution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turmel, M.; Lopes dos Santos, A.; Otis, C.; Sergerie, R.; Lemieux, C. Tracing the Evolution of the Plastome and Mitogenome in the Chloropicophyceae Uncovered Convergent tRNA Gene Losses and a Variant Plastid Genetic Code. Genome Biol. Evol. 2019, 11, 1275–1292. [Google Scholar] [CrossRef]
- Kummer, E.; Ban, N. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Biol. 2021, 22, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Chai, P.; Zhu, J.; Zhang, K. High-resolution in situ structures of mammalian respiratory supercomplexes. Nature 2024, 631, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Møller, I.M.; Rasmusson, A.G.; Van Aken, O. Plant mitochondria—Past, present and future. Plant J. 2021, 108, 912–959. [Google Scholar] [CrossRef] [PubMed]
- Ghifari, A.S.; Saha, S.; Murcha, M.W. The biogenesis and regulation of the plant oxidative phosphorylation system. Plant Physiol. 2023, 192, 728–747. [Google Scholar] [CrossRef] [PubMed]
- Munasinghe, M.; Ågren, J.A. When and why are mitochondria paternally inherited? Curr. Opin. Genet. Dev. 2023, 80, 102053. [Google Scholar] [CrossRef] [PubMed]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Eric Schranz, M.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liao, X.; Zhang, X.; Tembrock, L.R.; Broz, A. Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 2022, 60, 160–168. [Google Scholar] [CrossRef]
- Lei, B.; Li, S.; Liu, G.; Chen, Z.; Su, A.; Li, P.; Li, Z.; Hua, J. Evolution of mitochondrial gene content: Loss of genes, tRNAs and introns between Gossypium harknessii and other plants. Plant Syst. Evol. 2013, 299, 1889–1897. [Google Scholar] [CrossRef]
- Grewe, F.; Viehoever, P.; Weisshaar, B.; Knoop, V. A trans-splicing group I intron and tRNA-hyperediting in the mitochondrial genome of the lycophyte Isoetes engelmannii. Nucleic Acids Res. 2009, 37, 5093–5104. [Google Scholar] [CrossRef]
- Tong, W.; Kim, T.-S.; Park, Y.-J. Rice Chloroplast Genome Variation Architecture and Phylogenetic Dissection in Diverse Oryza Species Assessed by Whole-Genome Resequencing. Rice 2016, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Soreng, R.J.; Peterson, P.M.; Zuloaga, F.O.; Romaschenko, K.; Clark, L.G.; Teisher, J.K.; Gillespie, L.J.; Barberá, P.; Welker, C.A.D.; Kellogg, E.A.; et al. A worldwide phylogenetic classification of the Poaceae (Gramineae) III: An update. J. Syst. Evol. 2022, 60, 476–521. [Google Scholar] [CrossRef]
- Guo, Z.-H.; Ma, P.-F.; Yang, G.-Q.; Hu, J.-Y.; Liu, Y.-L.; Xia, E.-H.; Zhong, M.-C.; Zhao, L.; Sun, G.-L.; Xu, Y.-X.; et al. Genome Sequences Provide Insights into the Reticulate Origin and Unique Traits of Woody Bamboos. Mol. Plant 2019, 12, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Sanchez, E.; Tyrrell, C.D.; Londoño, X.; Oliveira, R.P.; Clark, L.G. Diversity, distribution, and classification of neotropical woody bamboos (Poaceae: Bambusoideae) in the 21st century. Bot. Sci. 2021, 99, 198–228. [Google Scholar] [CrossRef]
- Lv, S.-Y.; Ye, X.-Y.; Li, Z.-H.; Ma, P.-F.; Li, D.-Z. Testing complete plastomes and nuclear ribosomal DNA sequences for species identification in a taxonomically difficult bamboo genus Fargesia. Plant Divers. 2023, 45, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.-Q.; Xing, X.-C.; Zhang, J.-Q.; Ren, Y. Straight From the Plastome: Molecular Phylogeny and Morphological Evolution of Fargesia (Bambusoideae: Poaceae). Front. Plant Sci. 2019, 10, 981. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yuan, S.; Nie, Y.; Zhao, J.; Cao, X.; Dai, Y.; Zhang, Z.; Wei, F. Dietary flavonoids and the altitudinal preference of wild giant pandas in Foping National Nature Reserve, China. Glob. Ecol. Conserv. 2020, 22, e00981. [Google Scholar] [CrossRef]
- Du, X.C.; Ren, Y.; Di Dang, G.; Lundholm, J. Distribution and plant community associations of the understory bamboo Fargesia qinlingensis in the Foping National Nature Reserve, China. Ann. For. Sci. 2011, 68, 1197–1206. [Google Scholar] [CrossRef]
- Wang, W.; Franklin, S.B.; Lu, Z.; Rude, B.J. Delayed Flowering in Bamboo: Evidence from Fargesia qinlingensis in the Qinling Mountains of China. Front. Plant Sci. 2016, 7, 151. [Google Scholar] [CrossRef]
- Ye, X.; Ma, P.; Yang, G.; Guo, C.; Zhang, Y.; Chen, Y.; Guo, Z.; Li, D. Rapid diversification of alpine bamboos associated with the uplift of the Hengduan Mountains. J. Biogeogr. 2019, 46, 2678–2689. [Google Scholar] [CrossRef]
- Porcher, A.; Kangasjärvi, S. Plant biology: Unlocking mitochondrial stress signals. Curr. Biol. 2024, 34, R59–R61. [Google Scholar] [CrossRef]
- Guo, S.; Li, Z.; Li, C.; Liu, Y.; Liang, X.; Qin, Y. Assembly and characterization of the complete mitochondrial genome of Ventilago leiocarpa. Plant Cell Rep. 2024, 43, 77. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.-F.; Guo, Z.-H.; Li, D.-Z. Rapid Sequencing of the Bamboo Mitochondrial Genome Using Illumina Technology and Parallel Episodic Evolution of Organelle Genomes in Grasses. PLoS ONE 2012, 7, e30297. [Google Scholar] [CrossRef] [PubMed]
- Arseneau, J.; Steeves, R.; Laflamme, M. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 2017, 17, 686–693. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Xia, H.; Zhao, W.; Shi, Y.; Wang, X.-R.; Wang, B. Microhomologies Are Associated with Tandem Duplications and Structural Variation in Plant Mitochondrial Genomes. Genome Biol. Evol. 2020, 12, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Gerke, P.; Szövényi, P.; Neubauer, A.; Lenz, H.; Gutmann, B.; McDowell, R.; Small, I.; Schallenberg-Rüdinger, M.; Knoop, V. Towards a plant model for enigmatic U-to-C RNA editing: The organelle genomes, transcriptomes, editomes and candidate RNA editing factors in the hornwort Anthoceros agrestis. New Phytol. 2020, 225, 1974–1992. [Google Scholar] [CrossRef]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant organellar RNA editing: What 30 years of research has revealed. Plant J. 2020, 101, 1040–1056. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhi, X.; Yu, R.; Liu, Y.; Zhou, R. Factors contributing to mitogenome size variation and a recurrent intracellular DNA transfer in Melastoma. BMC Genom. 2023, 24, 370. [Google Scholar] [CrossRef]
- Chase, M.W.; Christenhusz, M.J.M.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; Stevens, P.F.; et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Jiang, M.; Ni, Y.; Zhang, J.; Li, J.; Liu, C. Complete mitochondrial genome of Mentha spicata L. reveals multiple chromosomal configurations and RNA editing events. Int. J. Biol. Macromol. 2023, 251, 126257. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef]
- Wang, W.; Franklin, S.B.; Ouellette, J.R. Clonal regeneration of an arrow bamboo, Fargesia qinlingensis, following giant panda herbivory. Plant Ecol. 2007, 192, 97–106. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Zhou, Y.; Hou, X.-Q.; Huang, L.; Kang, J.-Q.; Zhang, J.-Q.; Ren, Y. Phylogeny of Fargesia (Poaceae: Bambusoideae) and infrageneric adaptive divergence inferred from three cpDNA and nrITS sequence data. Plant Syst. Evol. 2019, 305, 61–75. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, C.B. Socialized mitochondria: Mitonuclear crosstalk in stress. Exp. Mol. Med. 2024, 56, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Van Aken, O. Mitochondrial redox systems as central hubs in plant metabolism and signaling. Plant Physiol. 2021, 186, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Qu, Y.; Hou, J.; Wu, K.; Ye, N.; Yin, T. Deciphering the Multi-Chromosomal Mitochondrial Genome of Populus simonii. Front. Plant Sci. 2022, 13, 914635. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, H.; Ni, Y.; Li, J.; Cai, Y.; Ma, B.; Yu, J.; Wang, J.; Liu, C. De Novo Hybrid Assembly of the Salvia miltiorrhiza Mitochondrial Genome Provides the First Evidence of the Multi-Chromosomal Mitochondrial DNA Structure of Salvia Species. Int. J. Mol. Sci. 2022, 23, 14267. [Google Scholar] [CrossRef]
- Yang, L.; Liu, J.; Guo, W.; Zheng, Z.; Xu, Y.; Xia, H.; Xiao, T. Insights into the multi-chromosomal mitochondrial genome structure of the xero-halophytic plant Haloxylon Ammodendron (C.A.Mey.) Bunge ex Fenzl. BMC Genom. 2024, 25, 123. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. Variation in protein gene and intron content among land plant mitogenomes. Mitochondrion 2020, 53, 203–213. [Google Scholar] [CrossRef]
- Darracq, A.; Varré, J.-S.; Touzet, P. A scenario of mitochondrial genome evolution in maize based on rearrangement events. BMC Genom. 2010, 11, 233. [Google Scholar] [CrossRef]
- Knoop, V. The mitochondrial DNA of land plants: Peculiarities in phylogenetic perspective. Curr. Genet. 2004, 46, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Newton, K.J. Plant Mitochondrial Genomes: Dynamics and Mechanisms of Mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Bi, C.; Zhao, Y.; Gao, M.; Wang, Y.; Chen, Y. Unraveling the complex evolutionary features of the Cinnamomum camphora mitochondrial genome. Plant Cell Rep. 2024, 43, 183. [Google Scholar] [CrossRef] [PubMed]
- McCauley, D.E. Paternal leakage, heteroplasmy, and the evolution of plant mitochondrial genomes. New Phytol. 2013, 200, 966–977. [Google Scholar] [CrossRef]
- Hia, F.; Takeuchi, O. The effects of codon bias and optimality on mRNA and protein regulation. Cell. Mol. Life Sci. 2021, 78, 1909–1928. [Google Scholar] [CrossRef]
- Barik, S. The Uniqueness of Tryptophan in Biology: Properties, Metabolism, Interactions and Localization in Proteins. Int. J. Mol. Sci. 2020, 21, 8776. [Google Scholar] [CrossRef] [PubMed]
- Radrizzani, S.; Kudla, G.; Izsvák, Z.; Hurst, L.D. Selection on synonymous sites: The unwanted transcript hypothesis. Nat. Rev. Genet. 2024, 25, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.; Liu, Y.; Wang, C.; Li, L.; Chao, Y. Complete mitochondrial genome of Agrostis stolonifera: Insights into structure, Codon usage, repeats, and RNA editing. BMC Genom. 2023, 24, 466. [Google Scholar] [CrossRef]
- Zardoya, R. Recent advances in understanding mitochondrial genome diversity. F1000Research 2020, 9, 270. [Google Scholar] [CrossRef]
- Zou, Y.; Zhu, W.; Sloan, D.B.; Wu, Z. Long-read sequencing characterizes mitochondrial and plastid genome variants in Arabidopsis msh1 mutants. Plant J. 2022, 112, 738–755. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.-F.; Zhang, Y.-X.; Guo, Z.-H.; Li, D.-Z. Evidence for horizontal transfer of mitochondrial DNA to the plastid genome in a bamboo genus. Sci. Rep. 2015, 5, 11608. [Google Scholar] [CrossRef]
- Ala, K.G.; Zhao, Z.; Ni, L.; Wang, Z. Comparative analysis of mitochondrial genomes of two alpine medicinal plants of Gentiana (Gentianaceae). PLoS ONE 2023, 18, e0281134. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Wu, Z. History of Plastid DNA Insertions Reveals Weak Deletion and AT Mutation Biases in Angiosperm Mitochondrial Genomes. Genome Biol. Evol. 2014, 6, 3210–3221. [Google Scholar] [CrossRef] [PubMed]
- Masutani, B.; Arimura, S.; Morishita, S. Investigating the mitochondrial genomic landscape of Arabidopsis thaliana by long-read sequencing. PLoS Comput. Biol. 2021, 17, e1008597. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Xiao, G.; Liu, H.; Zhang, L.; Zhao, L.; Tang, M.; Huang, S.; An, Y.; Yu, J. Two pivotal RNA editing sites in the mitochondrial atp1 mRNA are required for ATP synthase to produce sufficient ATP for cotton fiber cell elongation. New Phytol. 2018, 218, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, X.; Priyadarshani, S.V.G.N.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.; Zhou, Q.; Luo, Z.; Deng, F.; et al. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef]
- Bi, R.; Zhang, W.; Zhang, D.-F.; Xu, M.; Fan, Y.; Hu, Q.-X.; Jiang, H.-Y.; Tan, L.; Li, T.; Fang, Y.; et al. Genetic association of the cytochrome c oxidase-related genes with Alzheimer’s disease in Han Chinese. Neuropsychopharmacology 2018, 43, 2264–2276. [Google Scholar] [CrossRef]
- Analin, B.; Mohanan, A.; Bakka, K.; Challabathula, D. Cytochrome oxidase and alternative oxidase pathways of mitochondrial electron transport chain are important for the photosynthetic performance of pea plants under salinity stress conditions. Plant Physiol. Biochem. 2020, 154, 248–259. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Type | Path |
|---|---|---|
| M1 | linear | 117-116-5-78-8-55-13-74-3-70-6-39-49-25-61-60-16-73-2-73-29-121-117-116-87-113-118-20-79-7-74-54-23-65-18-22 |
| M2 | linear | 4-24-51-116-87-113-118-11-117-120-10-45-26-114-30-79-19-113-15-85-14-120-117-34-12-78-17-18-65-23-9-34 |
| M3 | linear | 1-85-33-112-21-121 |
| Gene Group | Gene Name |
|---|---|
| ATP synthase | atp1 (×2 *), atp4, atp6, atp8, atp9 |
| NADH dehydrogenase | nad1, nad2, nad3 (×2), nad4, nad4L, nad5, nad6, nad7, nad9 |
| Cytochrome b reductase | cob |
| Ubiquinol cytochrome c reductase | ccmB, ccmC, ccmFC, ccmFN |
| Cytochrome c oxidase | cox1, cox2, cox3 |
| Maturase | matR |
| Membrane transport protein | mttB |
| Ribosomal large subunits | rpl2, rpl5 (×2), rpl16 |
| Ribosomal small subunits | rps1, rps3, rps4, rps7, rps12, rps13, rps14 (×2), rps19 |
| Ribosome RNA | rrn5, rrn18, rrn26 |
| Transfer RNA | trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnH-GUG, trnK-UUU, trnM-CAU (×2), trnN-GUU, trnP-UGG (×2), trnQ-UUG, trnR-ACG (×2), trnS-GCU (×2), trnS-UGA, trnT-UGU, trnW-CCA, trnY-GUA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.; Li, X.; Qu, K.; Yang, L.; Su, T.; Yong, L.; Han, M.; Cao, F. Integration of Illumina and PacBio HiFi Sequencing Reveals a Three-Linear-Molecule Mitogenome with RNA-Editing Sites and Phylogeny in Arrow Bamboo (Fargesia qinlingensis). Forests 2024, 15, 1267. https://doi.org/10.3390/f15071267
Wu H, Li X, Qu K, Yang L, Su T, Yong L, Han M, Cao F. Integration of Illumina and PacBio HiFi Sequencing Reveals a Three-Linear-Molecule Mitogenome with RNA-Editing Sites and Phylogeny in Arrow Bamboo (Fargesia qinlingensis). Forests. 2024; 15(7):1267. https://doi.org/10.3390/f15071267
Chicago/Turabian StyleWu, Hao, Xue Li, Ke Qu, Lele Yang, Tao Su, Lijun Yong, Mei Han, and Fuliang Cao. 2024. "Integration of Illumina and PacBio HiFi Sequencing Reveals a Three-Linear-Molecule Mitogenome with RNA-Editing Sites and Phylogeny in Arrow Bamboo (Fargesia qinlingensis)" Forests 15, no. 7: 1267. https://doi.org/10.3390/f15071267