
Genome-Wide Identification of WRKY in Suaeda australis against Salt Stress


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, cDNA Synthesis, and Transcriptome Sequencing
2.2. Identification of SaWRKY Genes in S. australis
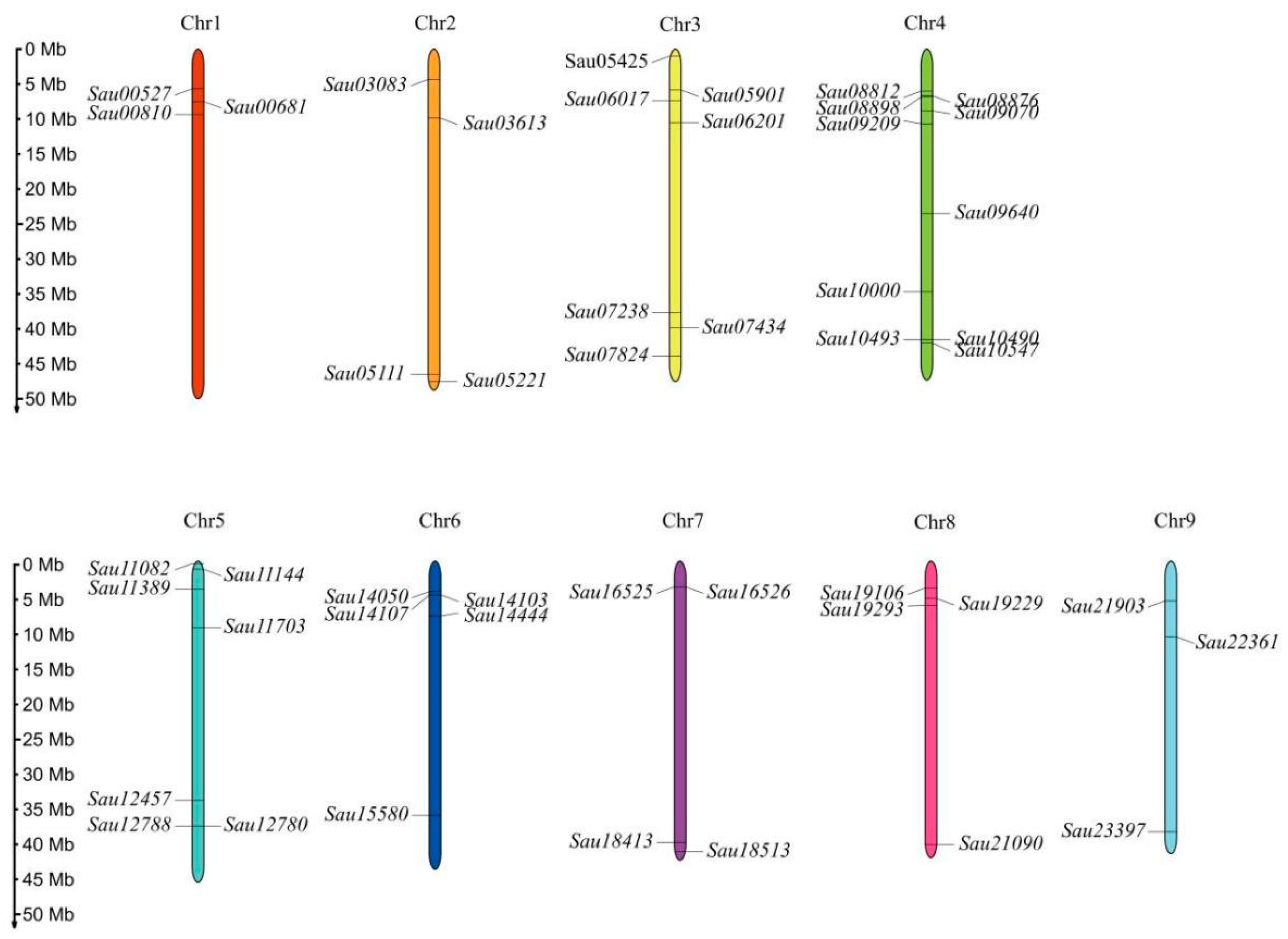
2.3. Chromosomal Location and Synteny Analysis of SaWRKY Genes
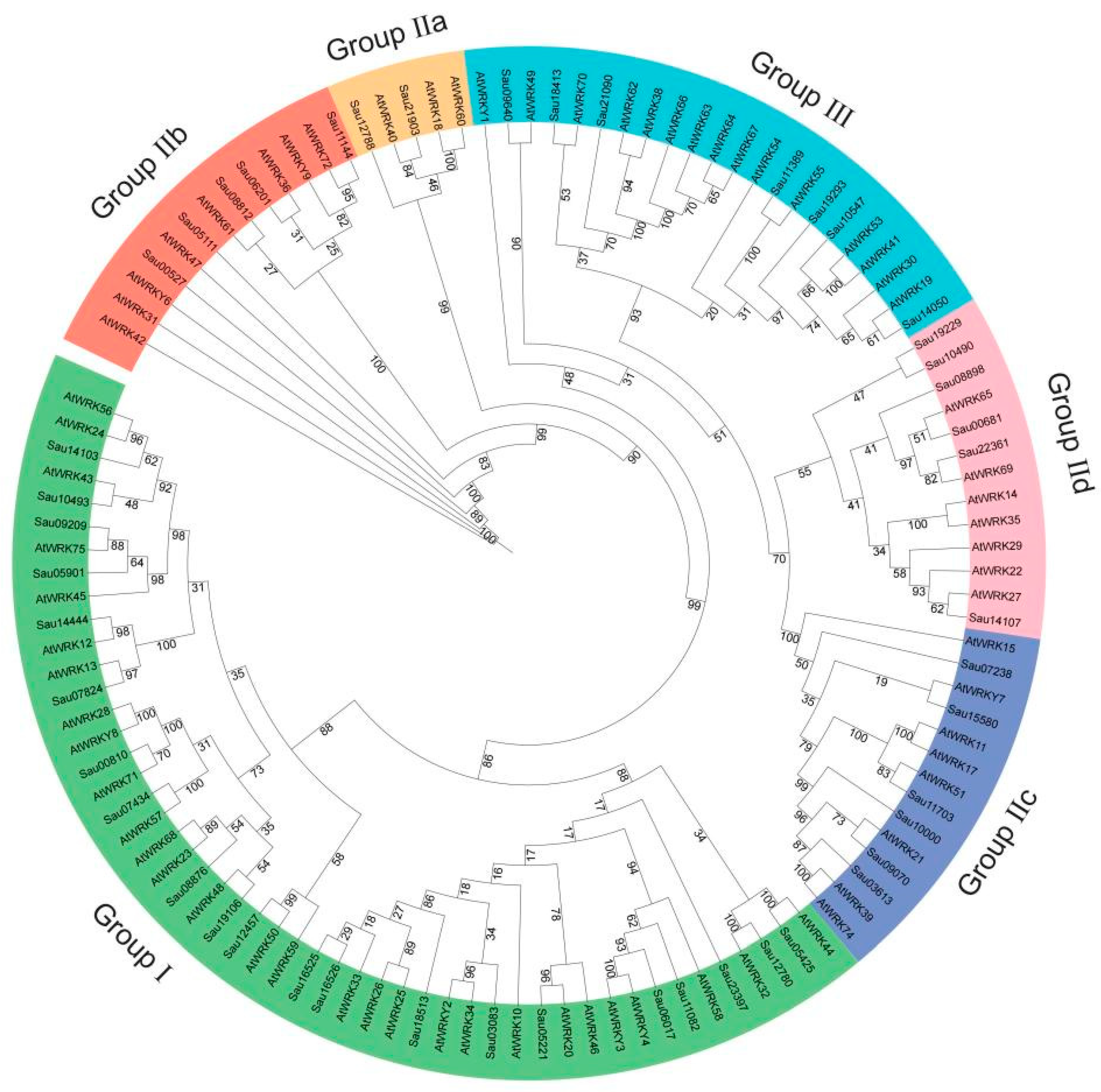
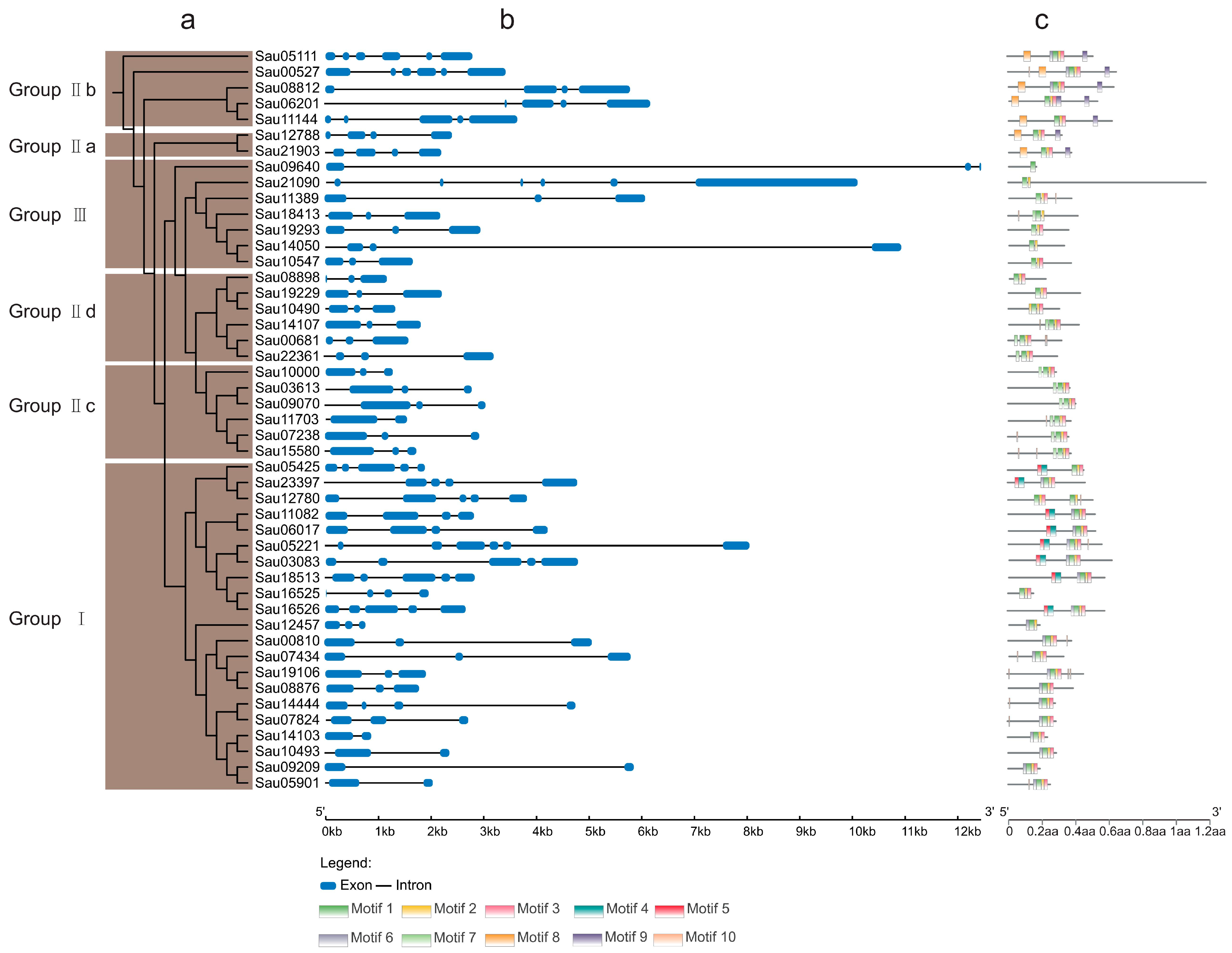
2.4. Phylogeny, Structural, and Motif Analyses of SaWRKY Genes
2.5. Prediction of Regulatory Elements of Promoter Region
2.6. Estimation of Ka/Ks Values
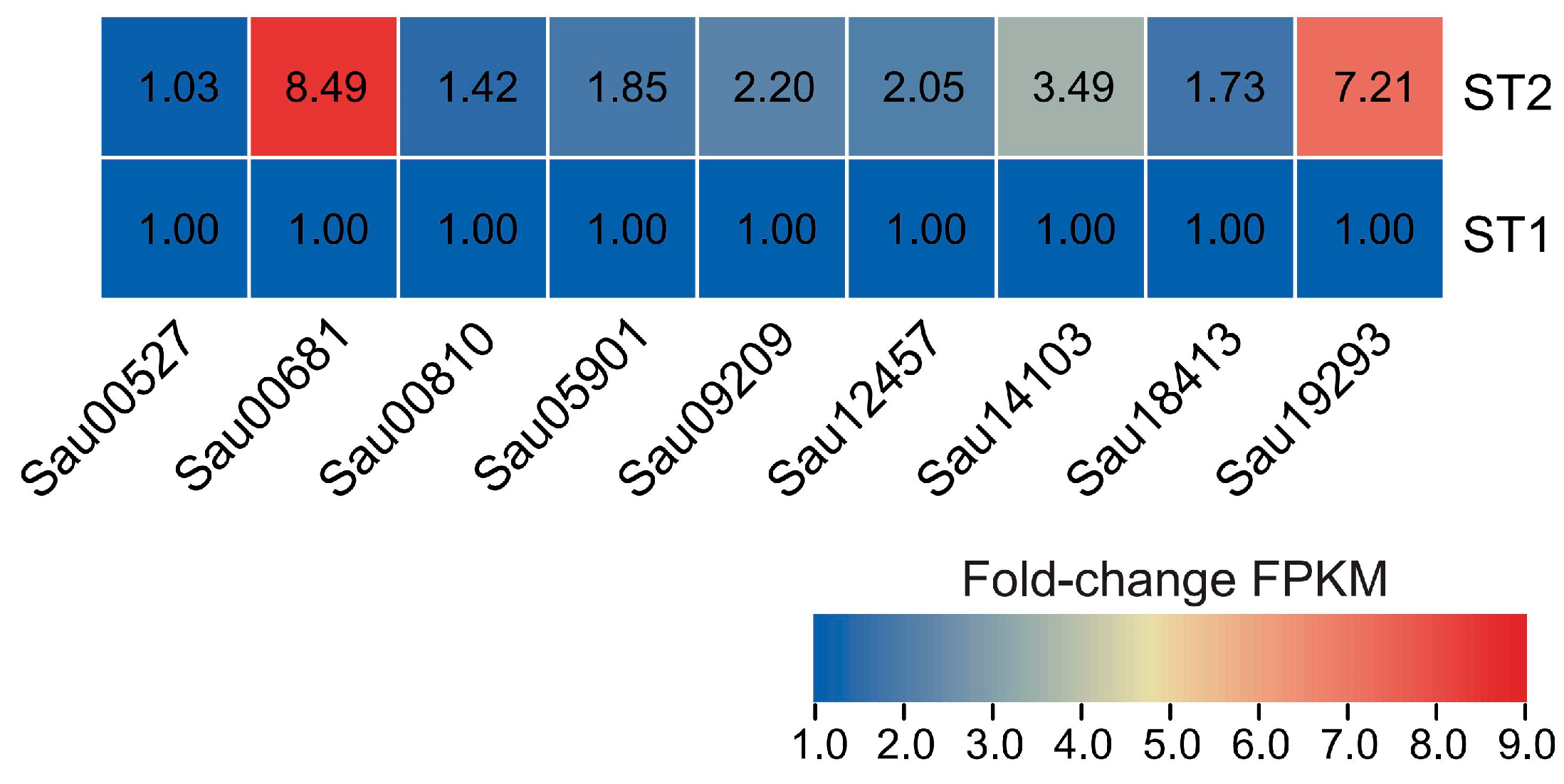
2.7. Expression Analysis of Selected SaWRKY Genes
2.8. Measurements of Physiological and Biochemical Traits
2.9. Statistical Analysis
3. Results
3.1. Genome-Wide Identification of the SaWRKY Gene Family in S. australis
3.2. Phylogeny, Structural, and Conserved Motif Analyses of SaWRKY Genes
3.3. Estimation of Ka and Ks Substitution Rates and Ka/Ks Values
3.4. Elements in the Promoters of Suaeda australis WRKY Genes
3.5. Expression Profiles of SaWRKY Genes in Relation to Salt Stress
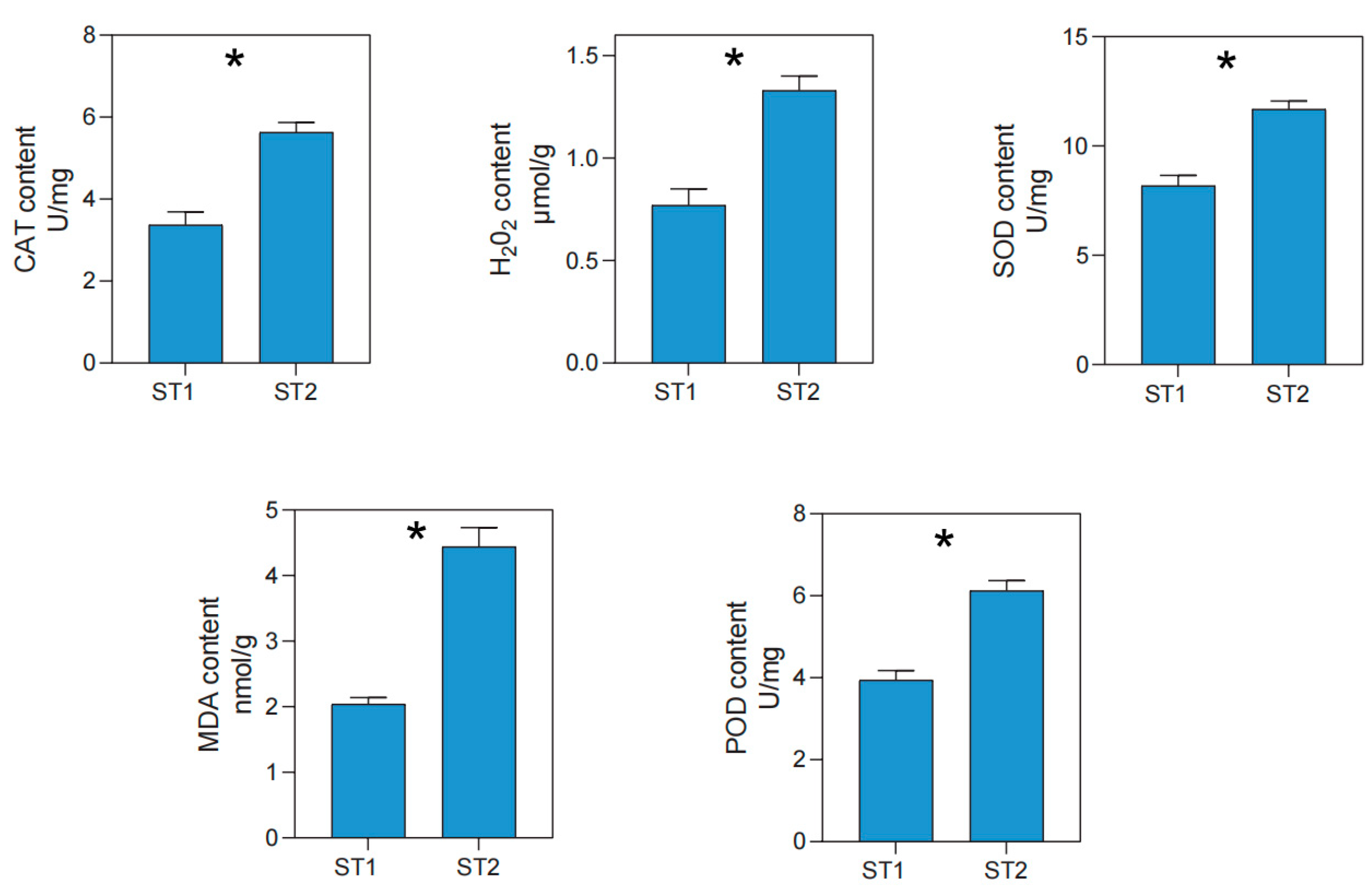
3.6. Variations in Physiological and Biochemical Traits of S. australis under Salt Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Serrano, O.; Lovelock, C.E.; Atwood, T.B.; Macreadie, P.I.; Canto, R.; Phinn, S.; Arias-Ortiz, A.; Bai, L.; Baldock, J.; Bedulli, C.; et al. Australian vegetated coastal ecosystems as global hotspots for climate change mitigation. Nat. Commun. 2019, 10, 4313. [Google Scholar] [CrossRef] [PubMed]
- Cresta, E.; Battisti, C. Anthropogenic litter along a coastal-wetland gradient: Reed-bed vegetation in the backdunes may act as a sink for expanded polystyrene. Mar. Pollut. Bull. 2021, 172, 112829. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.F.; Wang, W.J.; Li, H.; Wu, H.T.; Yan, B.X. Soil organic matter and salinity as critical factors affecting the bacterial community and function of Phragmites australis dominated riparian and coastal wetlands. Sci. Total Environ. 2021, 762, 143156. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.P.; Downton, W.J.S. Potassium, sodium and chloride ion concentrations in leaves and isolated chloroplasts of the halophyte Suaeda australis R. Br. Funct. Plant Biol. 1985, 12, 471–479. [Google Scholar] [CrossRef]
- Alam, M.R.; Tran, T.K.A.; Stein, T.J.; Rahman, M.M.; Griffin, A.S.; Yu, R.M.K.; MacFarlane, G.R. Accumulation and distribution of metal (loid) s in the halophytic saltmarsh shrub, Austral seablite, Suaeda australis in New South Wales, Australia. Mar. Pollut. Bull. 2021, 169, 112475. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.Q.; Wu, M.; Wang, Q.; Jiang, K.Y.; Shao, X.X. Correlation of seed germination capacities under salt stress with four plant species distribution in the Hangzhou Bay Wetlands. J. Zhejiang AF Univ. 2012, 29, 739–743. [Google Scholar]
- Mu, Y.N.; Ding, L.X.; Li, N.; Lu, L.Y.; Wu, M. Classification of coastal wetland vegetation in Hangzhou Bay with an object-oriented, random forest model. J. Zhejiang AF Univ. 2018, 35, 1088–1097. [Google Scholar]
- Eulgem, T.; Rushton, P.J.; Robatzek, S. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, R.; Jiang, S.Y.; Kumar, N.; Venkatesh, P.N.; Ramachandran, S. A comprehensive transcriptional profiling of the WRKY gene family in rice under various abiotic and phytohormone treatments. Plant Cell Physiol. 2008, 49, 865–879. [Google Scholar] [CrossRef]
- Dong, J.X.; Chen, C.H.; Chen, Z.X. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol. Biol. 2003, 51, 21–37. [Google Scholar] [CrossRef]
- Hu, W.; Ren, Q.; Chen, Y.; Xu, G.; Qian, Y. Genome-wide identification and analysis of WRKY gene family in maize provide insights into regulatory network in response to abiotic stresses. BMC Plant Biol. 2021, 21, 427. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, W.; Fang, L.; Sun, X.; Su, L.; Liang, Z.; Wang, N.; Londo, J.P.; Li, H.; Xin, H.P. Genome-wide identification of WRKY family genes and their response to cold stress in Vitis vinifera. BMC Plant Biol. 2014, 14, 103. [Google Scholar] [CrossRef]
- Huang, S.; Gao, Y.; Liu, J.; Peng, X.; Niu, X.; Fei, Z.; Liu, Y. Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mo. Genet. Genomics. 2012, 287, 495–513. [Google Scholar] [CrossRef]
- Ma, Z.B.; Li, W.; Wang, H.P.; Yu, D.Q. WRKY transcription factors WRKY12 and WRKY13 interact with SPL10 to modulate age-mediated flowering. J. Integr. Plant Biol. 2020, 62, 1659–1673. [Google Scholar] [CrossRef]
- Gao, Y.F.; Liu, J.K.; Yang, F.M.; Zhang, G.Y.; Wang, D.; Zhang, L.; Ou, Y.B.; Yao, Y.A. The WRKY transcription factor WRKY8 promotes resistance to pathogen infection and mediates drought and salt stress tolerance in Solanum lycopersicum. Physiol. Plantarum. 2020, 168, 98–117. [Google Scholar] [CrossRef]
- Niu, C.F.; Wei, W.; Zhou, Q.Y.; Tian, A.G.; Hao, Y.J.; Zhang, W.K.; Ma, B.; Lin, Q.; Zhang, Z.B.; Zhang, J.S.; et al. Wheat WRKY genes TaWRKY2 and TaWRKY19 regulate abiotic stress tolerance in transgenic Arabidopsis plants. Plant Cell Environ. 2012, 35, 1156–1170. [Google Scholar] [CrossRef]
- Zhu, D.; Hou, L.X.; Xiao, P.L.; Guo, Y.; Deyholos, M.K.; Liu, X. VvWRKY30, a grape WRKY transcription factor, plays a positive regulatory role under salinity stress. Plant Sci. 2019, 280, 132–142. [Google Scholar] [CrossRef]
- Ye, H.; Qiao, L.Y.; Guo, H.Y.; Guo, L.P.; Ren, F.; Bai, J.F.; Wang, Y.K. Genome-wide identification of wheat WRKY gene family reveals that TaWRKY75-A is referred to drought and salt resistances. Front. Plant Sci. 2021, 12, 663118. [Google Scholar] [CrossRef]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Prakash, A.; Jeffryes, M.; Bateman, A.; Finn, R.D. The HMMER web server for protein sequence similarity search. Curr. Protoc. Bioinform. 2017, 60, 3–15. [Google Scholar] [CrossRef]
- Wang, Y.P.; Tang, H.B.; Debarry, J.D.; Tan, X.; Li, J.P.; Wang, X.Y.; Lee, T.H.; Jin, H.Z.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Alexandros, S. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Peer, Y.V.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Zhang, Z. KaKs_Calculator 3.0: Calculating selective pressure on coding and non-coding sequences. Genom. Proteom. Bioinf. 2022, 20, 536–540. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Shumate, A.; Wong, B.; Pertea, G.; Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 2022, 18, e1009730. [Google Scholar] [CrossRef]
- Chen, X.L.; Mao, X.; Huang, P.; Fang, S.Z. Morphological characterization of flower buds development and related gene expression profiling at bud break stage in heterodichogamous Cyclocarya paliurus (Batal.) lljinskaja. Genes 2019, 10, 818. [Google Scholar] [CrossRef]
- Goyal, P.; Manzoor, M.M.; Ram, A.V.; Deepak, S.; Manoj, K.D.; Suphla, G. A Comprehensive transcriptome-wide identification and screening of WRKY gene family engaged in abiotic stress in Glycyrrhiza glabra. Sci. Rep. 2020, 10, 373. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, S.; Huang, X.; Lyu, L.; Li, W.; Wu, W. Genome-wide identification of WRKY gene family members in black raspberry and their response to abiotic stresses. Sci. Hortic. 2022, 304, 111338. [Google Scholar] [CrossRef]
- Wei, Y.L.; Jin, J.P.; Liang, D.; Gao, J.; Li, J.; Xie, Q.; Lu, C.Q.; Zhu, G.F. Genome-wide identification of Cymbidium sinense WRKY gene family and the importance of its Group III members in response to abiotic stress. Front. Plant Sci. 2022, 13, 969010. [Google Scholar] [CrossRef]
- Wang, C.; Ye, D.; Li, Y.; Hu, P.L.; Xu, R.; Wang, X.J. Genome-wide identification and bioinformatics analysis of the WRKY transcription factors and screening of candidate genes for anthocyanin biosynthesis in azalea (Rhododendron simsii). Front. Genet. 2023, 14, 1172321. [Google Scholar] [CrossRef]
- Muhammad, A.K.; Kang, D.R.; Wu, Y.F.; Wang, Y.; Ai, P.H.; Wang, Z.C. Characterization of WRKY gene family in whole-genome and exploration of flowering improvement genes in Chrysanthemum lavandulifolium. Front. Plant Sci. 2022, 13, 861193. [Google Scholar]
- Wen, F.; Wu, X.Z.; Li, T.J.; Jia, M.L.; Liao, L. Characterization of the WRKY gene family in Akebia trifoliata and their response to Colletotrichum acutatum. BMC Plant Bio. 2022, 22, 115. [Google Scholar] [CrossRef]
- Wu, W.H.; Zhu, S.; Xu, L.; Zhu, L.M.; Wang, D.D.; Liu, Y.; Liu, S.Q.; Hao, Z.D.; Lu, Y.; Yang, L.M.; et al. Genome-wide identification of the Liriodendron chinense WRKY gene family and its diverse roles in response to multiple abiotic stress. BMC Plant Biol. 2022, 22, 25. [Google Scholar] [CrossRef]
- Hou, X.; Wang, D.P.; Cheng, Z.K.; Wang, Y.; Jiao, Y.L. A near-complete assembly of an Arabidopsis thaliana genome. Mol. Plant. 2022, 15, 1247–1250. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Wei, J.T.; Cai, K.W.; Zhang, H.Z.; Ge, L.L.; Ren, Z.J.; Zhao, C.L.; Zhao, X.Y. Genome-wide identification and analysis of the WRKY gene family and cold stress response in Acer truncatum. Genes 2021, 12, 1867. [Google Scholar] [CrossRef]
- Jing, C.J.; Wang, D.; Liu, Z.K.; Chen, X.F.; Dong, H.; Wu, X.H. Identification of the WRKY gene family in apricot and its response to drought stress. Hortic. Environ. Biotechnol. 2023, 64, 269–282. [Google Scholar] [CrossRef]
- Xiong, R.Q.; Peng, Z.H.; Zhou, H.; Xue, G.X.; He, A.L.; Yao, X.; Weng, W.F.; Wu, W.J.; Ma, C.; Bai, Q.; et al. Genome-wide identification, structural characterization and gene expression analysis of the WRKY transcription factor family in pea (Pisum sativum L.). BMC Plant Biol. 2024, 24, 113. [Google Scholar]
- Kong, W.L.; Gong, Z.Y.; Zhong, H.; Zhang, Y.; Zhao, G.Q.; Gautam, M.; Deng, X.X.; Liu, C.; Zhang, C.H.; Li, Y.S. Expansion and evolutionary patterns of glycosyltransferase family 8 in gramineae crop genomes and their expression under salt and cold stresses in Oryza sativa ssp. japonica. Biomolecules 2019, 9, 188. [Google Scholar] [CrossRef]
- Wu, G.Q.; Li, Z.Q.; Cao, H.; Wang, J.L. Genome-wide identification and expression analysis of the WRKY genes in sugar beet (Beta vulgaris L.) under alkaline stress. PeerJ 2019, 7, e7817. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, S.; Yu, H.; Li, R.; Lin, Z.; Cai, M.; Ming, R. Expression profiling of WRKY transcription factors in Spinach. Euphytica 2021, 217, 217. [Google Scholar] [CrossRef]
- Lu, K.K.; Song, R.F.; Guo, J.X.; Zhang, Y.; Zuo, J.X.; Chen, H.H.; Liao, C.Y.; Hu, X.Y.; Ren, F.; Lu, Y.T.; et al. CycC1; 1–WRKY75 complex-mediated transcriptional regulation of SOS1 controls salt stress tolerance in Arabidopsis. Plant Cell 2023, 35, 2570–2591. [Google Scholar] [CrossRef]
- Wang, J.H.; Gu, K.D.; Zhang, Q.Y.; Yu, Q.J.; Wang, C.K.; You, C.X.; Cheng, L.G.; Hu, D.G. Ethylene inhibits malate accumulation in apple by transcriptional repression of aluminum-activated malate transporter 9 via the WRKY31-ERF72 network. New Phytol. 2023, 239, 1014–1034. [Google Scholar] [CrossRef]
- Chen, J.N.; Nolan, T.M.; Ye, H.X.; Zhang, M.C.; Tong, H.N.; Xin, P.Y.; Chu, J.F.; Chu, C.C.; Li, Z.H.; Yin, Y.H. Arabidopsis WRKY46, WRKY54, and WRKY70 transcription factors are involved in brassinosteroid-regulated plant growth and drought responses. Plant Cell 2017, 29, 1425–1439. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Exon Count | Chromosome Localization | Genomic Sequence (bp) | CDS (bp) | Amino Acid (aa) | PI | MW (kDa) |
|---|---|---|---|---|---|---|---|---|
| SaWRKY01 | Sau00527 | 6 | Chr01:5,596,347–5,600,163 | 3817 | 1938 | 645 | 6.72 | 70.13 |
| SaWRKY02 | Sau00681 | 3 | Chr01:7,485,495–7,487,452 | 1958 | 960 | 319 | 6.23 | 35.79 |
| SaWRKY03 | Sau00810 | 3 | Chr01:9,356,913–9,362,321 | 5409 | 1134 | 377 | 6.34 | 41.65 |
| SaWRKY04 | Sau03083 | 5 | Chr02:4,315,820–4,320,608 | 4789 | 1839 | 612 | 6.56 | 68.49 |
| SaWRKY05 | Sau03613 | 3 | Chr02:9,765,700–9,768,801 | 3102 | 1107 | 368 | 9.98 | 41.17 |
| SaWRKY06 | Sau05111 | 6 | Chr02:46,391,992–46,394,779 | 2788 | 1536 | 511 | 7.21 | 57.16 |
| SaWRKY07 | Sau05221 | 6 | Chr02:47,318,618–47,326,924 | 8307 | 1671 | 556 | 6.39 | 60.15 |
| SaWRKY08 | Sau05425 | 5 | Chr03:968,760–970,650 | 1891 | 1362 | 453 | 9.09 | 50.29 |
| SaWRKY09 | Sau05901 | 2 | Chr03:5,737,788–5,739,946 | 2159 | 753 | 250 | 6.51 | 29.02 |
| SaWRKY10 | Sau06017 | 4 | Chr03:7,255,251–7,259,940 | 4690 | 1554 | 517 | 6.52 | 56.85 |
| SaWRKY11 | Sau06201 | 5 | Chr03:10,485,361–10,491,762 | 6402 | 1581 | 526 | 6.39 | 57.05 |
| SaWRKY12 | Sau07238 | 3 | Chr03:37,516,523–37,520,160 | 3636 | 1086 | 361 | 9.51 | 39.30 |
| SaWRKY13 | Sau07434 | 3 | Chr03:39,659,365–39,665,672 | 6278 | 975 | 324 | 7.10 | 35.95 |
| SaWRKY14 | Sau07824 | 3 | Chr03:43,631,812–43,634,859 | 3048 | 867 | 288 | 8.49 | 32.67 |
| SaWRKY15 | Sau08812 | 4 | Chr04:5,969,824–5,975,609 | 5786 | 1884 | 627 | 6.16 | 68.84 |
| SaWRKY16 | Sau08876 | 3 | Chr04:6,616,826–6,619,034 | 2209 | 1155 | 384 | 5.70 | 41.69 |
| SaWRKY17 | Sau08898 | 3 | Chr04:6,857,924–6,859,392 | 1469 | 651 | 216 | 9.39 | 24.74 |
| SaWRKY18 | Sau09070 | 3 | Chr04:8,790,234–8,793,597 | 3364 | 1215 | 404 | 9.48 | 44.04 |
| SaWRKY19 | Sau09209 | 2 | Chr04:10,564,046–10,570,220 | 6175 | 573 | 190 | 9.27 | 22.04 |
| SaWRKY20 | Sau09640 | 3 | Chr04:23,324,654–23,337,094 | 12,441 | 495 | 164 | 7.06 | 18.52 |
| SaWRKY21 | Sau10000 | 3 | Chr04:34,579,825–34,581,100 | 1276 | 858 | 285 | 8.96 | 32.03 |
| SaWRKY22 | Sau10490 | 3 | Chr04:41,258,580–41,259,996 | 1417 | 915 | 304 | 5.89 | 34.50 |
| SaWRKY23 | Sau10493 | 2 | Chr04:41,275,640–41,278,332 | 2693 | 864 | 287 | 5.89 | 34.50 |
| SaWRKY24 | Sau10547 | 3 | Chr04:41,804,302–41,806,554 | 2253 | 1119 | 372 | 5.89 | 41.47 |
| SaWRKY25 | Sau11082 | 4 | Chr05:300,843–303,661 | 2819 | 1554 | 517 | 8.44 | 56.64 |
| SaWRKY26 | Sau11144 | 5 | Chr05:1,086,695–1,090,392 | 3698 | 1854 | 617 | 6.11 | 61.85 |
| SaWRKY27 | Sau11389 | 3 | Chr05:4,026,282–4,032,409 | 6128 | 1116 | 371 | 5.96 | 41.63 |
| SaWRKY28 | Sau11703 | 2 | Chr05:9,412,532–9,414,487 | 1956 | 1116 | 371 | 9.78 | 40.79 |
| SaWRKY29 | Sau12457 | 3 | Chr05:34,048,180–34,049,567 | 1388 | 552 | 183 | 4.91 | 21.04 |
| SaWRKY30 | Sau12780 | 5 | Chr05:37,640,971–37,645,313 | 4343 | 1518 | 505 | 7.29 | 55.33 |
| SaWRKY31 | Sau12788 | 4 | Chr05:37,723,284–37,726,031 | 2748 | 945 | 314 | 8.37 | 34.83 |
| SaWRKY32 | Sau14050 | 3 | Chr06:4,300,250–4,311,580 | 11,331 | 987 | 328 | 6.02 | 36.73 |
| SaWRKY33 | Sau14103 | 2 | Chr06:4,804,678–4,805,924 | 1247 | 711 | 236 | 5.21 | 26.87 |
| SaWRKY34 | Sau14107 | 3 | Chr06:4,845,544–4,847,964 | 2421 | 1251 | 416 | 6.51 | 45.15 |
| SaWRKY35 | Sau14444 | 4 | Chr06:7,846,244–7,851,364 | 5121 | 837 | 278 | 7.35 | 32.01 |
| SaWRKY36 | Sau15580 | 3 | Chr06:36,258,891–36,261,065 | 2175 | 1128 | 375 | 9.68 | 40.97 |
| SaWRKY37 | Sau16525 | 4 | Chr07:3,604,255–3,606,636 | 2382 | 456 | 151 | 9.16 | 16.77 |
| SaWRKY38 | Sau16526 | 5 | Chr07:3,610,119–3,613,215 | 3097 | 1737 | 578 | 6.73 | 64.35 |
| SaWRKY39 | Sau18413 | 3 | Chr07:40,047,343–40,049,893 | 2551 | 1251 | 416 | 6.09 | 47.16 |
| SaWRKY40 | Sau18513 | 5 | Chr07:41,275,001–41,278,109 | 3109 | 1716 | 571 | 6.17 | 63.19 |
| SaWRKY41 | Sau19106 | 3 | Chr08:3,857,398–3,859,865 | 2468 | 1359 | 452 | 6.67 | 49.49 |
| SaWRKY42 | Sau19229 | 3 | Chr08:5,383,303–5,385,512 | 2210 | 1284 | 427 | 5.49 | 47.84 |
| SaWRKY43 | Sau19293 | 3 | Chr08:6,244,515–6,248,341 | 3827 | 1083 | 360 | 5.67 | 40.75 |
| SaWRKY44 | Sau21090 | 7 | Chr08:40,259,570–40,269,879 | 10,310 | 3537 | 1178 | 7.70 | 133.24 |
| SaWRKY45 | Sau21903 | 4 | Chr09:5,673,708–5,676,152 | 2445 | 1119 | 372 | 6.77 | 40.86 |
| SaWRKY46 | Sau22361 | 3 | Chr09:10,741,005–10,744,503 | 3499 | 876 | 291 | 5.25 | 31.49 |
| SaWRKY47 | Sau23397 | 5 | Chr09:38,591,545–38,596,704 | 5160 | 1383 | 460 | 5.62 | 50.61 |
| Tandem Duplicated Genes | Ka | Ks | Ka/Ks | Purifying Selection |
|---|---|---|---|---|
| Sau16525 and Sau16526 | 0.9588 | 1.1405 | 0.8407 | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, Y.; Wang, J.; Qu, C.; Mo, X.; Zhang, X. Genome-Wide Identification of WRKY in Suaeda australis against Salt Stress. Forests 2024, 15, 1297. https://doi.org/10.3390/f15081297
Qu Y, Wang J, Qu C, Mo X, Zhang X. Genome-Wide Identification of WRKY in Suaeda australis against Salt Stress. Forests. 2024; 15(8):1297. https://doi.org/10.3390/f15081297
Chicago/Turabian StyleQu, Yinquan, Ji Wang, Caihui Qu, Xiaoyun Mo, and Xiumei Zhang. 2024. "Genome-Wide Identification of WRKY in Suaeda australis against Salt Stress" Forests 15, no. 8: 1297. https://doi.org/10.3390/f15081297