
Genome-Wide Identification of Phenylacetaldehyde Reductase Genes and Molecular Docking Simulation Study of OePAR1 in Olives


Abstract
1. Introduction
2. Materials and Methods
2.1. Identification and Physicochemical Property Analysis of Gene Family Members
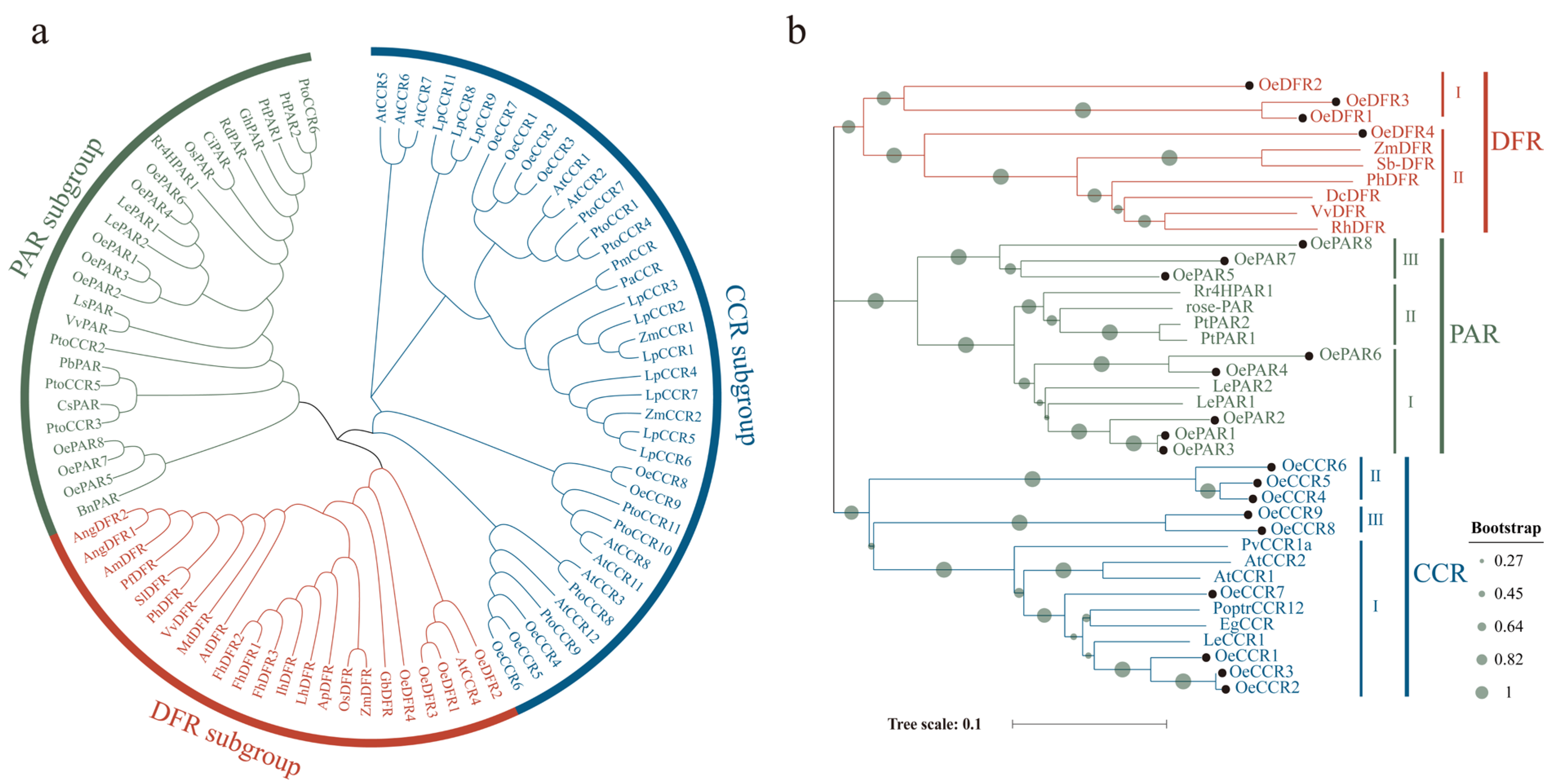
2.2. Phylogenetic Analysis
2.3. Conserved Motif and Gene Structure Analysis
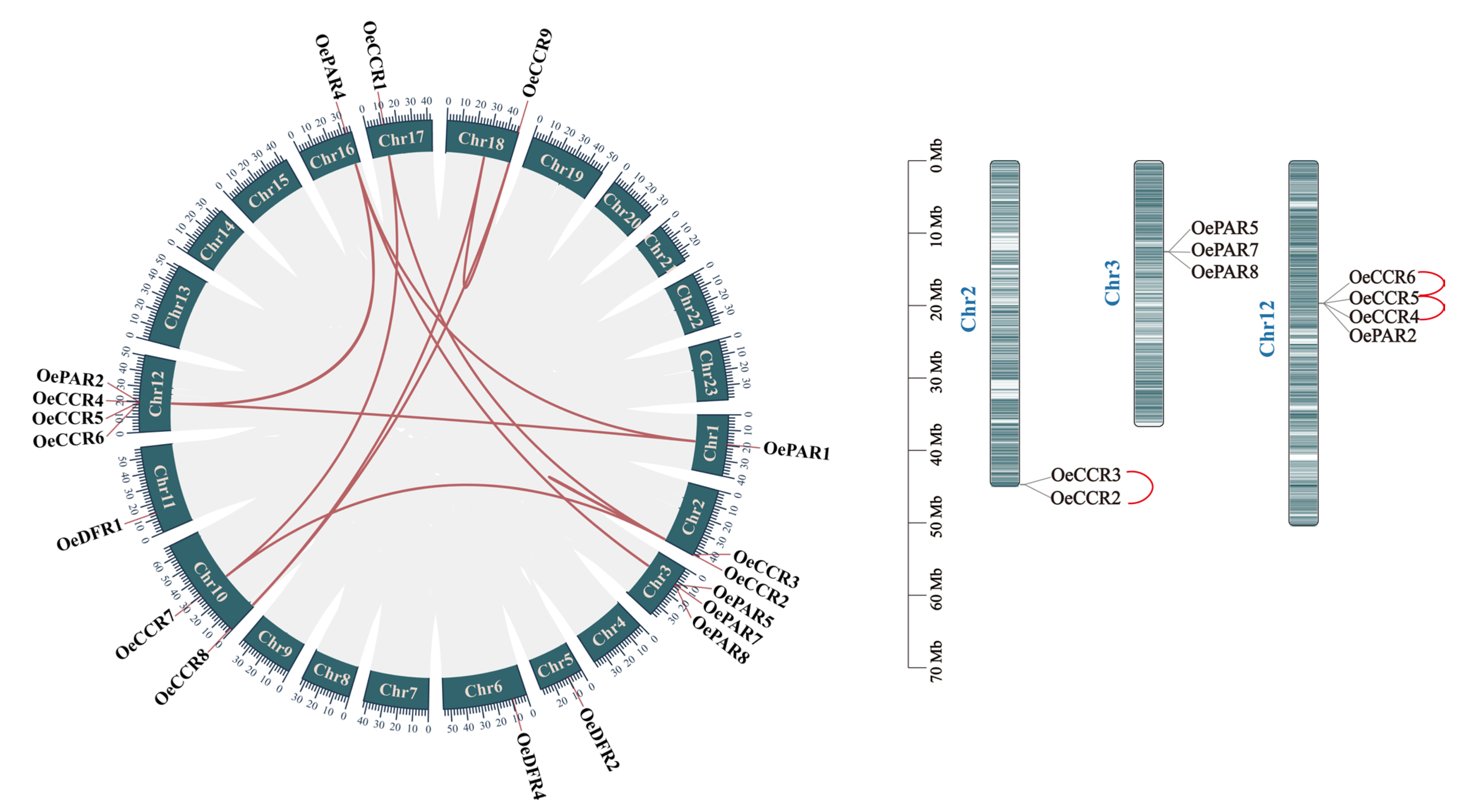
2.4. Chromosomal Localization and Duplication Event Analysis
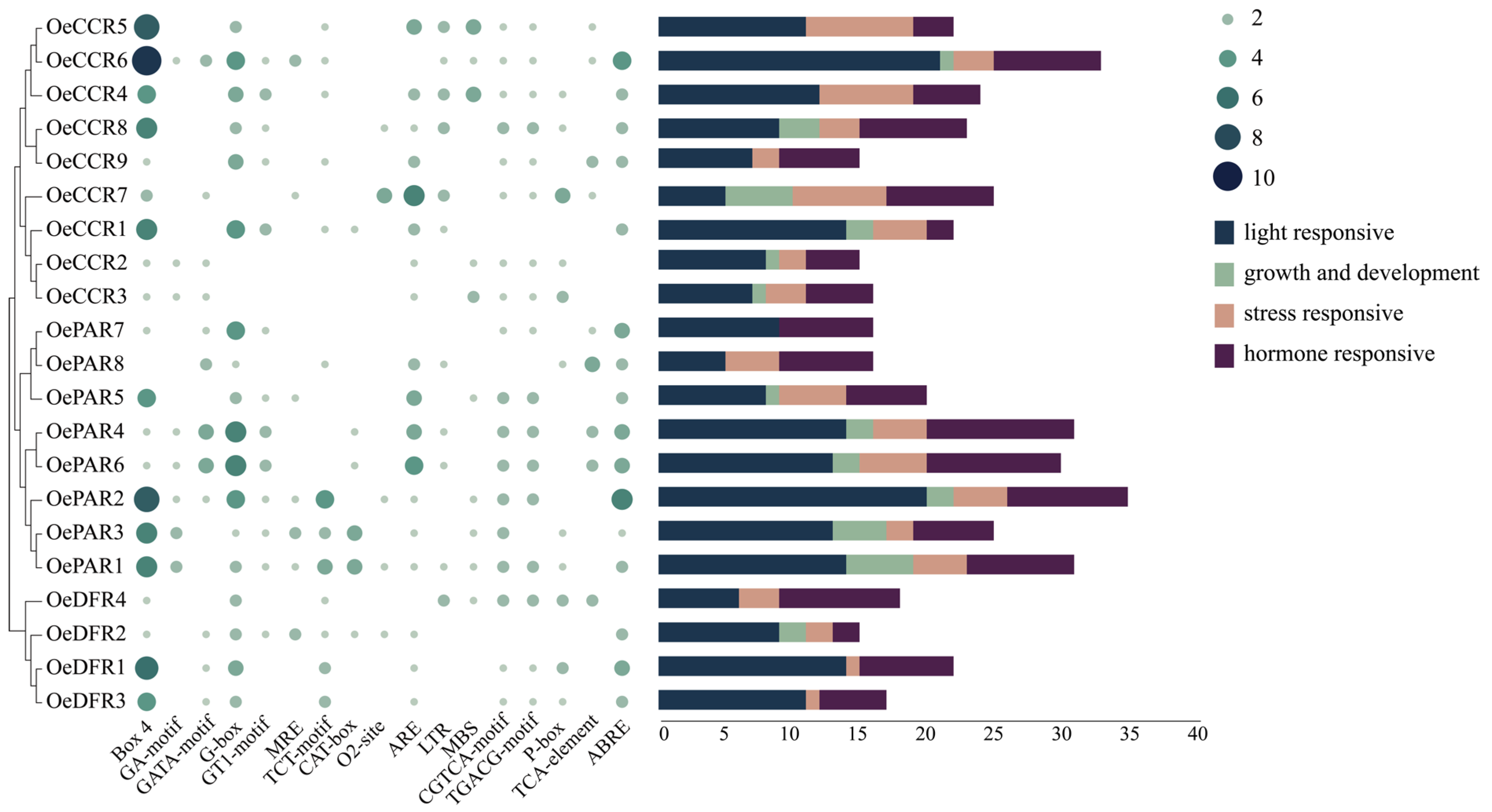
2.5. Promoter cis-Acting Elements Analysis
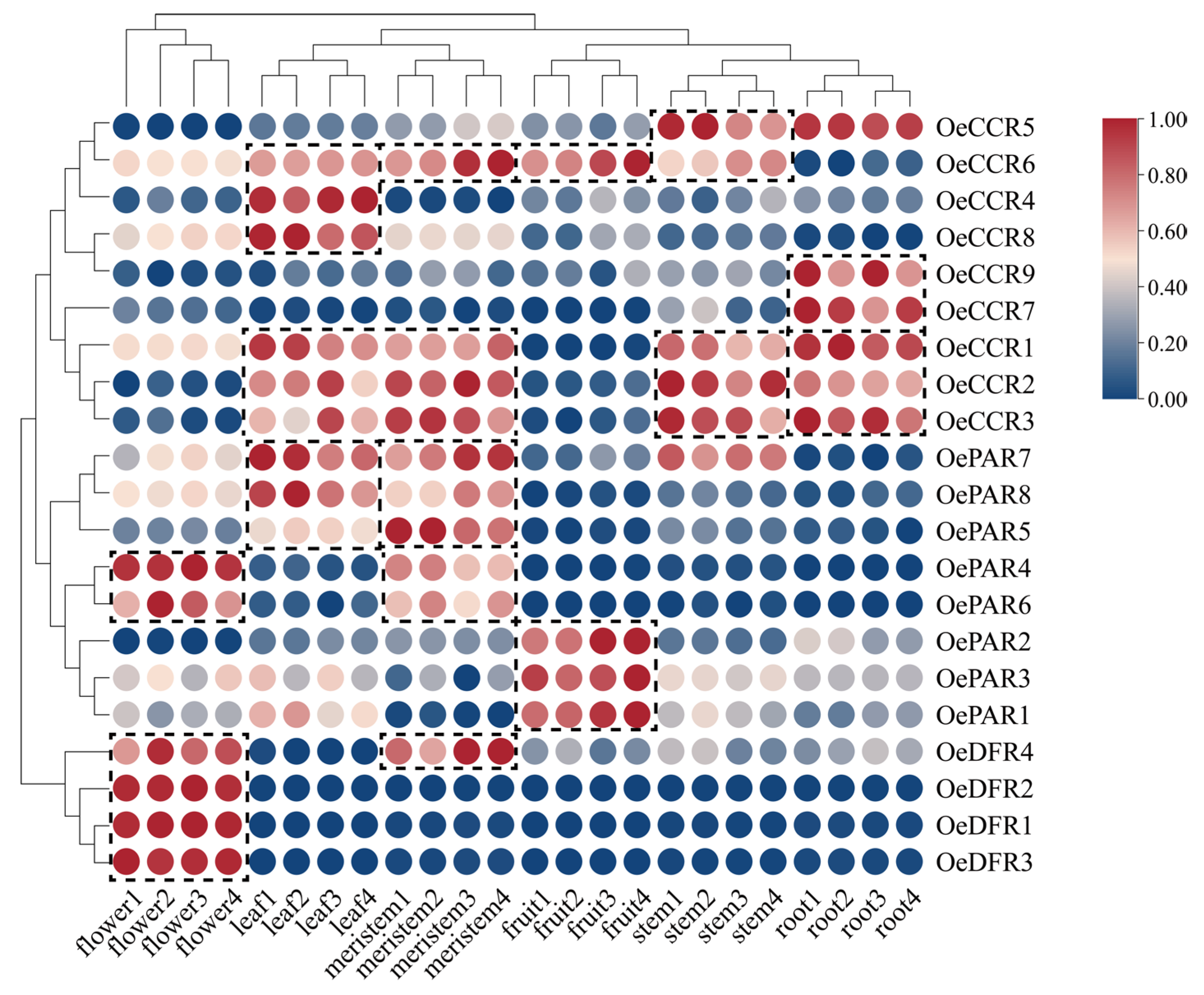
2.6. Expression Pattern Analysis
2.7. Three-Dimensional Structure Modeling and Molecular Docking of OePAR1
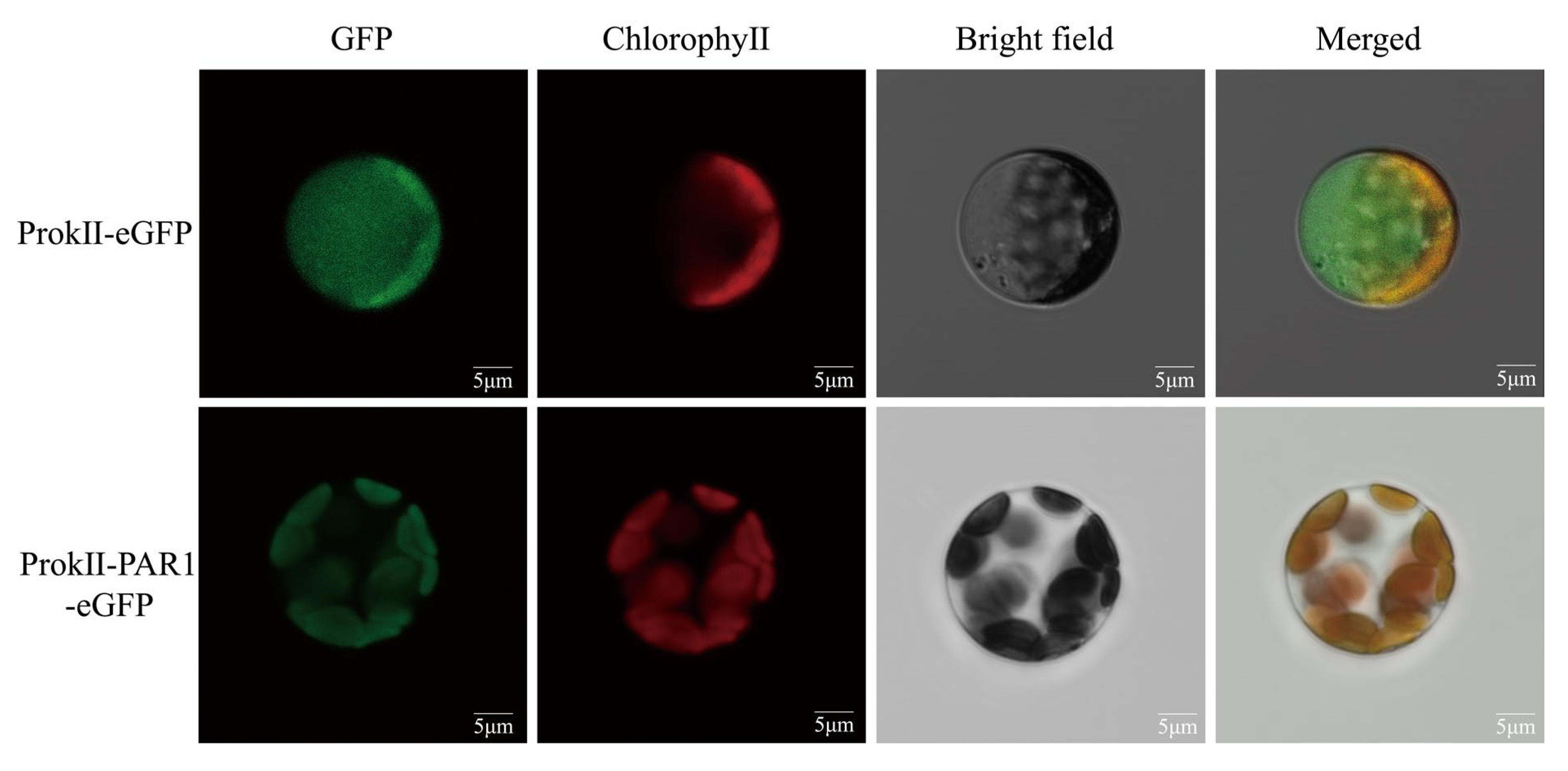
2.8. Subcellular Localization of OePAR1
3. Results
3.1. Identification and Physicochemical Property Analysis of Gene Family Members
3.2. Phylogenetic Analysis
3.3. Conserved Motif and Gene Structure Analysis
3.4. Chromosomal Localization and Duplication Event Analysis
3.5. Promoter cis-Acting Elements Analysis
3.6. Expression Pattern Analysis
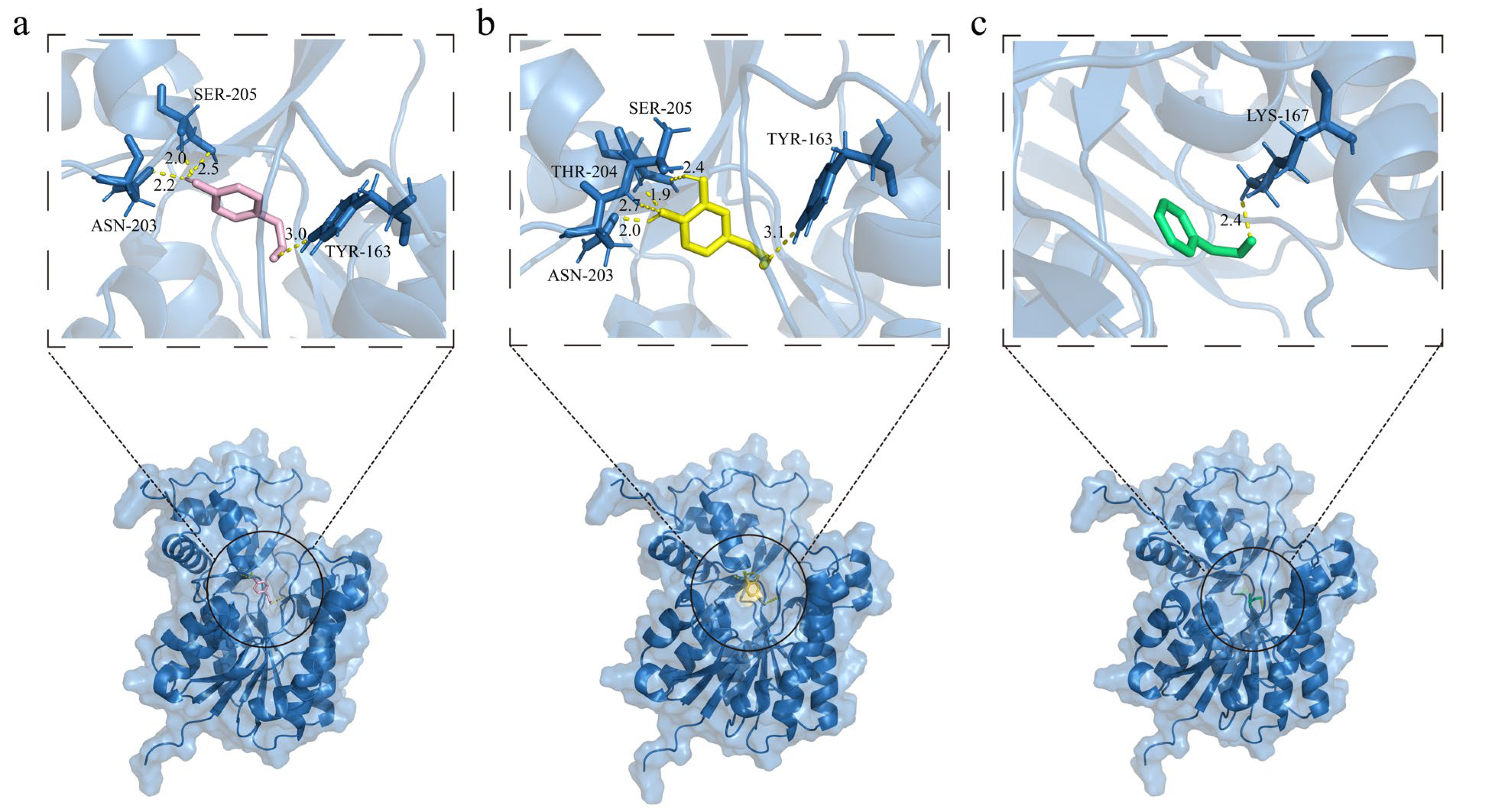
3.7. Three-Dimensional Structure Modeling of OePAR1 and Molecular Docking with Different Substrates
3.8. Subcellular Localization of OePAR1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Unver, T.; Wu, Z.; Sterck, L.; Turktas, M.; Lohaus, R.; Li, Z.; Yang, M.; He, L.; Deng, T.; Escalante, F.J.; et al. Genome of wild olive and the evolution of oil biosynthesis. Proc. Natl. Acad. Sci. USA 2017, 114, E9413–E9422. [Google Scholar] [PubMed]
- Gullón, P.; Gullón, B.; Astray, G.; Carpena, M.; Fraga-Corral, M.; Prieto, M.A.; Simal-Gandara, J. Valorization of by-products from olive oil industry and added-value applications for innovative functional foods. Food Res. Int. 2020, 137, 109683. [Google Scholar] [PubMed]
- Tripoli, E.; Giammanco, M.; Tabacchi, G.; Di Majo, D.; Giammanco, S.; La Guardia, M. The phenolic compounds of olive oil: Structure, biological activity and beneficial effects on human health. Nutr. Res. Rev. 2005, 18, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Alagna, F.; Geu-Flores, F.; Kries, H.; Panara, F.; Baldoni, L.; O’Connor, S.E.; Osbourn, A. Identification and Characterization of the Iridoid Synthase Involved in Oleuropein Biosynthesis in Olive (Olea europaea) Fruits. J. Biol. Chem. 2016, 291, 5542–5554. [Google Scholar] [CrossRef]
- Raederstorff, D. Antioxidant activity of olive polyphenols in humans: A review. Int. J. Vitam. Nutr. Res. 2009, 79, 152–165. [Google Scholar]
- Guodong, R.; Jianguo, Z.; Xiaoxia, L.; Ying, L. Identification of putative genes for polyphenol biosynthesis in olive fruits and leaves using full-length transcriptome sequencing. Food Chem. 2019, 300, 125246. [Google Scholar]
- Sánchez, R.; García-Vico, L.; Sanz, C.; Pérez, A.G. An Aromatic Aldehyde Synthase Controls the Synthesis of Hydroxytyrosol Derivatives Present in Virgin Olive Oil. Antioxidants 2019, 8, 352. [Google Scholar] [CrossRef]
- Rao, G.; Zhang, J.; Liu, X.; Lin, C.; Xin, H.; Xue, L.; Wang, C. De novo assembly of a new Olea europaea genome accession using nanopore sequencing. Hortic. Res. 2021, 8, 64. [Google Scholar]
- Torrens-Spence, M.P.; Pluskal, T.; Li, F.S.; Carballo, V.; Weng, J.K. Complete Pathway Elucidation and Heterologous Reconstitution of Rhodiola Salidroside Biosynthesis. Mol. Plant 2018, 11, 205–217. [Google Scholar]
- Tieman, D.M.; Loucas, H.M.; Kim, J.Y.; Clark, D.G.; Klee, H.J. Tomato phenylacetaldehyde reductases catalyze the last step in the synthesis of the aroma volatile 2-phenylethanol. Phytochemistry 2007, 68, 2660–2669. [Google Scholar]
- Chen, X.M.; Kobayashi, H.; Sakai, M.; Hirata, H.; Asai, T.; Ohnishi, T.; Baldermann, S.; Watanabe, N. Functional characterization of rose phenylacetaldehyde reductase (PAR), an enzyme involved in the biosynthesis of the scent compound 2-phenylethanol. J. Plant Physiol. 2011, 168, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Liu, Q.; Fan, Y.; Wang, C.; Li, Y.; Li, S.; Zhang, J.; Rao, G. Functional differentiation of olive PLP_deC genes: Insights into metabolite biosynthesis and genetic improvement at the whole-genome level. Plant Cell Rep. 2024, 43, 127. [Google Scholar] [PubMed]
- Filling, C.; Filling, C.; Berndt, K.D.; Benach, J.; Knapp, S.; Prozorovski, T.; Nordling, E.; Ladenstein, R.; Jörnvall, H.; Oppermann, U. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J. Biol. Chem. 2002, 277, 25677–25684. [Google Scholar]
- Kavanagh, K.L.; Jörnvall, H.; Persson, B.; Oppermann, U. Medium- and short-chain dehydrogenase/reductase gene and protein families: The SDR superfamily: Functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol. Life Sci. 2008, 65, 3895–3906. [Google Scholar]
- Moummou, H.; Kallberg, Y.; Tonfack, L.B.; Persson, B.; van der Rest, B. The plant short-chain dehydrogenase (SDR) superfamily: Genome-wide inventory and diversification patterns. BMC Plant Biol. 2012, 12, 219. [Google Scholar]
- Ladenstein, R.; Winberg, J.O.; Benach, J. Medium- and short-chain dehydrogenase/reductase gene and protein families: Structure-function relationships in short-chain alcohol dehydrogenases. Cell Mol. Life Sci. 2008, 65, 3918–3935. [Google Scholar]
- Kim, J.S.; Patel, S.K.S.; Tiwari, M.K.; Lai, C.; Kumar, A.; Kim, Y.S.; Kalia, V.C.; Lee, J.K. Phe-140 Determines the Catalytic Efficiency of Arylacetonitrilase from Alcaligenes faecalis. Int. J. Mol. Sci. 2020, 21, 7859. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Wang, C.; Xue, L.; Cui, Q.; Liu, Q.; Zhang, J.; Rao, G. Genome-wide identification of the cytochrome P450 superfamily in Olea europaea helps elucidate the synthesis pathway of oleuropein to improve the quality of olive oil. Sci. Hortic. 2022, 304, 111291. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Sirén, J.; Välimäki, N.; Mäkinen, V. Indexing Graphs for Path Queries with Applications in Genome Research. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 375–388. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Xiaoxia, L.; Zhang, J.; Jinkai, S.; Ying, L.; Guodong, R. The Salix SmSPR1 Involved in Light-Regulated Cell Expansion by Modulating Microtubule Arrangement. Front. Cell Dev. Biol. 2019, 7, 309. [Google Scholar] [CrossRef]
- Moore, R.C.; Purugganan, M.D. The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 15682–15687. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Luo, L.; Zheng, L. Lignins: Biosynthesis and Biological Functions in Plants. Int. J. Mol. Sci. 2018, 19, 335. [Google Scholar] [CrossRef] [PubMed]
- van der Rest, B.; Danoun, S.; Boudet, A.M.; Rochange, S.F. Down-regulation of cinnamoyl-CoA reductase in tomato (Solanum lycopersicum L.) induces dramatic changes in soluble phenolic pools. J. Exp. Bot. 2006, 57, 1399–1411. [Google Scholar] [CrossRef]
- Chao, N.; Li, N.; Qi, Q.; Li, S.; Lv, T.; Jiang, X.N.; Gai, Y. Characterization of the cinnamoyl-CoA reductase (CCR) gene family in Populus tomentosa reveals the enzymatic active sites and evolution of CCR. Planta 2017, 245, 61–75. [Google Scholar] [CrossRef]
- Barakat, A.; Yassin, N.B.; Park, J.S.; Choi, A.; Herr, J.; Carlson, J.E. Comparative and phylogenomic analyses of cinnamoyl-CoA reductase and cinnamoyl-CoA-reductase-like gene family in land plants. Plant Sci. 2011, 181, 249–257. [Google Scholar] [CrossRef]
- Sánchez, R.; Bahamonde, C.; Sanz, C.; Pérez, A.G. Identification and Functional Characterization of Genes Encoding Phenylacetaldehyde Reductases That Catalyze the Last Step in the Biosynthesis of Hydroxytyrosol in Olive. Plants 2021, 10, 1268. [Google Scholar] [CrossRef]
- Luan, Y.; Chen, Z.; Tang, Y.; Sun, J.; Meng, J.; Tao, J.; Zhao, D. Tree peony PsMYB44 negatively regulates petal blotch distribution by inhibiting dihydroflavonol-4-reductase gene expression. Ann. Bot. 2023, 131, 323–334. [Google Scholar] [CrossRef]
- Ruan, H.; Shi, X.; Gao, L.; Rashid, A.; Li, Y.; Lei, T.; Dai, X.; Xia, T.; Wang, Y. Functional analysis of the dihydroflavonol 4-reductase family of Camellia sinensis: Exploiting key amino acids to reconstruct reduction activity. Hortic. Res. 2022, 9, uhac098. [Google Scholar] [CrossRef]
- Yin, G.; Xu, H.; Xiao, S.; Qin, Y.; Li, Y.; Yan, Y.; Hu, Y. The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 2013, 13, 148. [Google Scholar] [CrossRef]
- Liu, L.; Xu, W.; Hu, X.; Liu, H.; Lin, Y. W-box and G-box elements play important roles in early senescence of rice flag leaf. Sci. Rep. 2016, 6, 20881. [Google Scholar]
- Müller, M.; Niesar, M.; Berens, I.; Gailing, O. Genotyping by sequencing reveals lack of local genetic structure between two German Ips typographus L. populations. For. Res. 2022, 2, 1. [Google Scholar]
- Li, J.; Terzaghi, W.; Gong, Y.; Li, C.; Ling, J.J.; Fan, Y.; Qin, N.; Gong, X.; Zhu, D.; Deng, X.W. Modulation of BIN2 kinase activity by HY5 controls hypocotyl elongation in the light. Nat. Commun. 2020, 11, 1592. [Google Scholar] [PubMed]
- Zhang, K.; Lin, C.; Chen, B.; Lin, Y.; Su, H.; Du, Y.; Zhang, H.; Zhou, H.; Ji, R.; Zhang, L. A light responsive transcription factor CsbHLH89 positively regulatesanthocyanidin synthesis in tea (Camellia sinensis). Sci. Hortic. 2023, 327, 112784. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, C.; Cui, Y.; Du, Q.; Tang, W.; Yang, W.; Kou, G.; Tang, W.; Chen, H.; Gong, R. Potential regulatory genes of light induced anthocyanin accumulation in sweet cherry identified by combining transcriptome and metabolome analysis. Front. Plant Sci. 2023, 14, 1238624. [Google Scholar]
- Zagoskina, N.V.; Zubova, M.Y.; Nechaeva, T.L.; Kazantseva, V.V.; Goncharuk, E.A.; Katanskaya, V.M.; Baranova, E.N.; Aksenova, M.A. Polyphenols in Plants: Structure, Biosynthesis, Abiotic Stress Regulation, and Practical Applications (Review). Int. J. Mol. Sci. 2023, 24, 13874. [Google Scholar] [CrossRef]
- Rao, G.; Zhang, J.; Liu, X.; Li, X.; Wang, C. Combined Metabolome and Transcriptome Profiling Reveal Optimal Harvest Strategy Model Based on Different Production Purposes in Olive. Foods 2021, 10, 360. [Google Scholar] [CrossRef]
- Sun, N.; Hu, J.; Li, C.; Wang, X.; Gai, Y.; Jiang, X. Fusion gene 4CL-CCR promotes lignification in tobacco suspension cells. Plant Cell Rep. 2023, 42, 939–952. [Google Scholar]
- Xiao, W.; Liu, A.; Lai, W.; Wang, J.; Li, X.; Zha, Y.; Zhao, B.; Chen, X.; Yu, H. Combined transcriptome and metabolome analysis revealed the molecular mechanisms of fruit skin coloration in pink strawberry. Front. Plant Sci. 2024, 15, 1486892. [Google Scholar] [CrossRef]
- Anterola, A.M.; Lewis, N.G. Trends in lignin modification: A comprehensive analysis of the effects of genetic manipulations/mutations on lignification and vascular integrity. Phytochemistry 2002, 61, 221–294. [Google Scholar]
- Petit, P.; Granier, T.; d’Estaintot, B.L.; Manigand, C.; Bathany, K.; Schmitter, J.M.; Lauvergeat, V.; Hamdi, S.; Gallois, B. Crystal structure of grape dihydroflavonol 4-reductase, a key enzyme in flavonoid biosynthesis. J. Mol. Biol. 2007, 368, 1345–1357. [Google Scholar]
- Jose, S.; Gupta, M.; Sharma, U.; Quintero-Saumeth, J.; Dwivedi, M. Potential of phytocompounds from Brassica oleracea targeting S2-domain of SARS-CoV-2 spike glycoproteins: Structural and molecular insights. J. Mol. Struct. 2022, 1254, 132369. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| seq1 | seq2 | Ka | Ks | Ka/Ks |
|---|---|---|---|---|
| OeDFR1 | OeDFR3 | 5.61 × 10−2 | 5.38 × 10−2 | 1.04 |
| OeCCR3 | OeCCR2 | 1.37 × 10−2 | 1.60 × 10−2 | 0.86 |
| OePAR1 | OePAR3 | 1.33 × 10−3 | 4.35 × 10−3 | 0.31 |
| OePAR5 | OePAR4 | 0.29 | 1.11 | 0.26 |
| OeCCR8 | OeCCR9 | 8.19 × 10−2 | 0.31 | 0.26 |
| OeCCR6 | OePAR4 | 0.50 | 2.07 | 0.24 |
| OeCCR5 | OeCCR6 | 4.73 × 10−2 | 0.25 | 0.19 |
| OeCCR3 | OeCCR1 | 5.35 × 10−2 | 0.31 | 0.17 |
| OeCCR4 | OeCCR5 | 2.47 × 10−2 | 0.15 | 0.17 |
| OePAR1 | OePAR4 | 0.15 | 1.06 | 0.14 |
| OePAR4 | OePAR3 | 0.15 | 1.06 | 0.14 |
| OeCCR3 | OeCCR7 | 9.70 × 10−2 | 0.82 | 0.12 |
| OeCCR7 | OeCCR1 | 8.71 × 10−2 | 0.79 | 0.11 |
| Compound | Structure 2D | Chemical Formula | H Bond Residues | Hydrophobic Interaction Residues | π-Stacking Residues | Binding Affinity (kcal/mol) |
|---|---|---|---|---|---|---|
| 3,4-DHPAA | 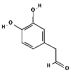 | C8H8O3 | TYR-163 ASN-203 THR-204 SER-205 | ILE-17 PRO-190 VAL-193 | - | −4.98 |
| 4-HPAA | 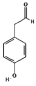 | C8H8O2 | TYR-163 ASN-203 SER-205 | VAL-193 | PHE-88 | −3.59 |
| PAA | 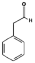 | C8H8O | LYS-167 | ILE-17 THR-126 PRO-190 | - | −3.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Cui, Q.; Li, S.; Li, Y.; Yi, G.; Wang, C.; Liu, Q.; Zhang, J.; Rao, G. Genome-Wide Identification of Phenylacetaldehyde Reductase Genes and Molecular Docking Simulation Study of OePAR1 in Olives. Forests 2025, 16, 630. https://doi.org/10.3390/f16040630
Fan Y, Cui Q, Li S, Li Y, Yi G, Wang C, Liu Q, Zhang J, Rao G. Genome-Wide Identification of Phenylacetaldehyde Reductase Genes and Molecular Docking Simulation Study of OePAR1 in Olives. Forests. 2025; 16(4):630. https://doi.org/10.3390/f16040630
Chicago/Turabian StyleFan, Yutong, Qizhen Cui, Shuyuan Li, Yufei Li, Gang Yi, Chenhe Wang, Qingqing Liu, Jianguo Zhang, and Guodong Rao. 2025. "Genome-Wide Identification of Phenylacetaldehyde Reductase Genes and Molecular Docking Simulation Study of OePAR1 in Olives" Forests 16, no. 4: 630. https://doi.org/10.3390/f16040630
APA StyleFan, Y., Cui, Q., Li, S., Li, Y., Yi, G., Wang, C., Liu, Q., Zhang, J., & Rao, G. (2025). Genome-Wide Identification of Phenylacetaldehyde Reductase Genes and Molecular Docking Simulation Study of OePAR1 in Olives. Forests, 16(4), 630. https://doi.org/10.3390/f16040630