Genetic Structure and Population Demographic History of a Widespread Mangrove Plant Xylocarpus granatum J. Koenig across the Indo-West Pacific Region

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Chloroplast DNA Sequencing and Data Analysis
2.3. Nuclear SSR Genotyping and Data Analysis of X. Granatum
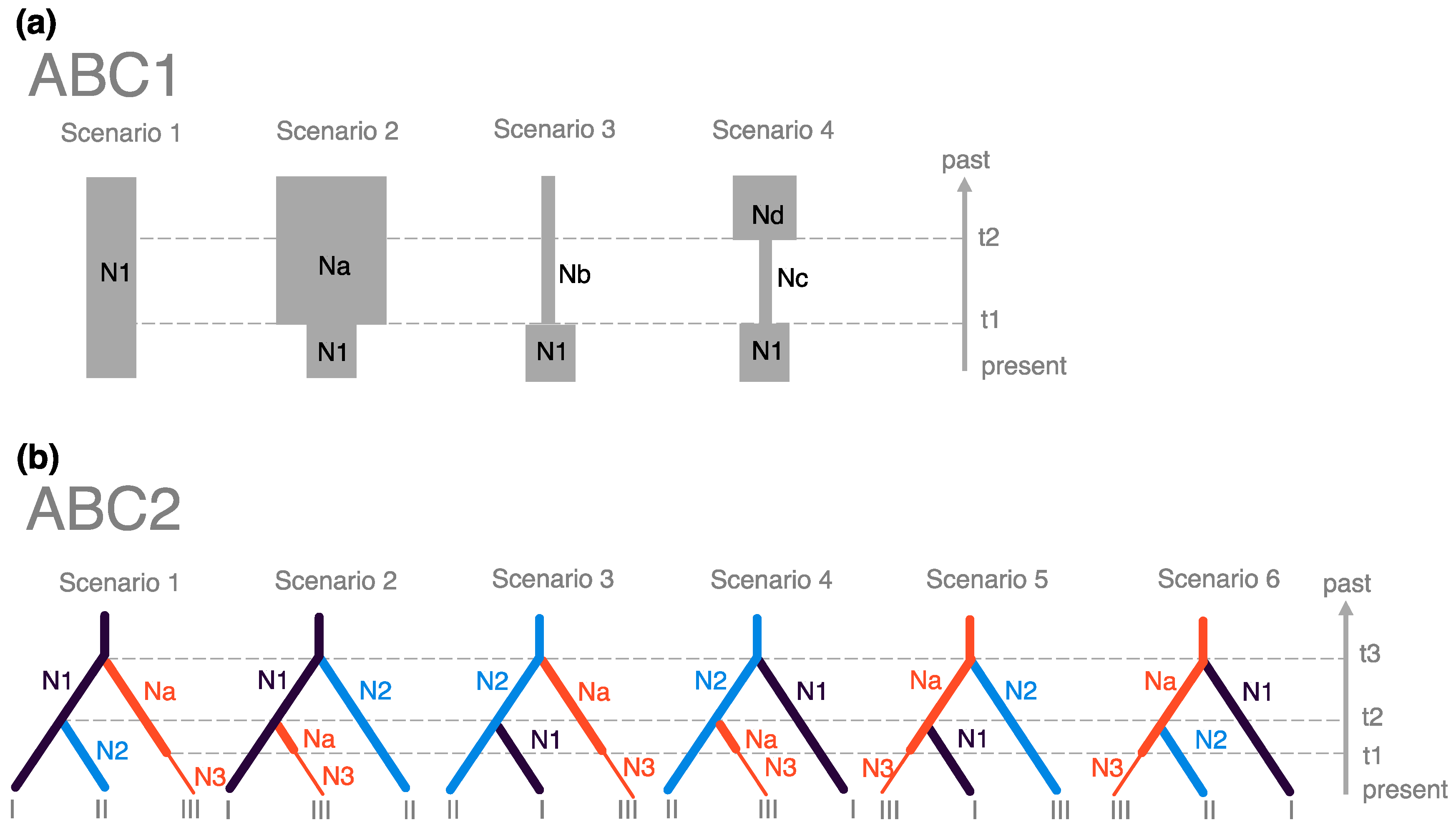
2.4. Inference of Demographic History
- Scenario 1—Constant population size model: The effective population size of Pop# was constant at N1 from the past to the present.
- Scenario 2—Population shrinkage model: The effective population size of Pop# reduced from Na to N1 at t1.
- Scenario 3—Population expansion model: The effective population size of Pop# increased from Nb to N1 at t1.
- Scenario 4—Bottleneck model: The effective population size of Pop# experienced a bottleneck from Nd to Nc at t2 followed by population size recovery from Nc to N1 at t1.
3. Results
3.1. Phylogenetic Relationships of the cpDNA Haplotypes in Xylocarpus
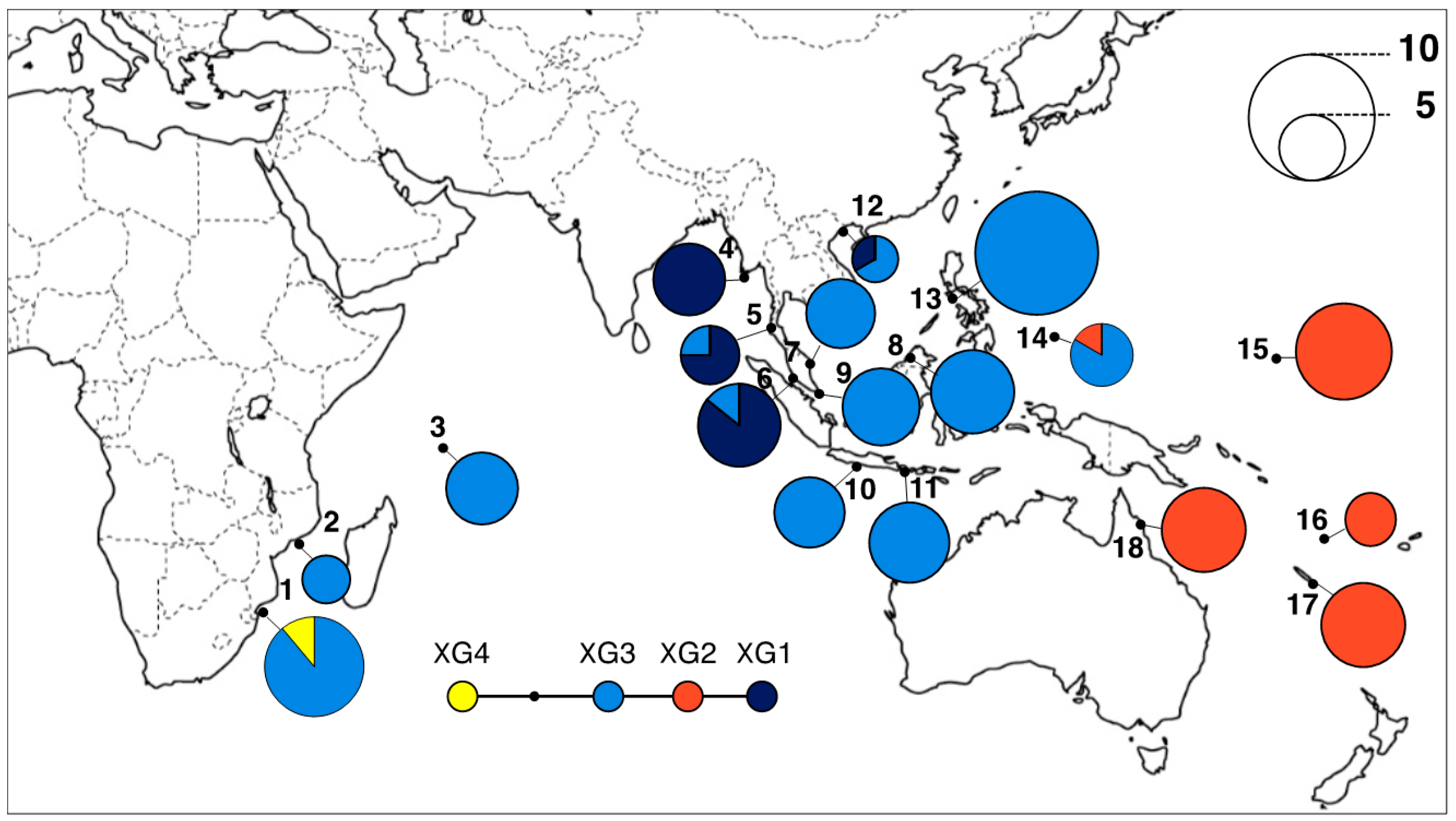
3.2. Geographical Distribution of cpDNA Haplotypes
3.3. Genetic Structure Revealed by SSR Markers
3.4. Inferences of Demographic History
4. Discussion
4.1. Genetic Structure of a Widespread Mangrove Plant, X. granatum, across the IWP Region
4.2. Inferences of the Demographic History of X. granatum
4.3. Malay Peninsula as a Common Barrier to Widespread Mangrove Plants
4.4. Relationships with Other Xylocarpus Species
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Spalding, M. World Atlas of Mangroves; Routledge: Abingdon, UK, 2010. [Google Scholar]
- Duke, N.C.; Meynecke, J.-O.; Dittmann, S.; Ellison, A.M.; Anger, K.; Berger, U.; Cannicci, S.; Diele, K.; Ewel, K.C.; Field, C.D.; et al. A World without Mangroves? Science 2007, 317, 41–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polidoro, B.A.; Carpenter, K.E.; Collins, L.; Duke, N.C.; Ellison, A.M.; Ellison, J.C.; Farnsworth, E.J.; Fernando, E.S.; Kathiresan, K.; Koedam, N.E.; et al. The loss of species: Mangrove extinction risk and geographic areas of global concern. PLoS ONE 2010, 5, e10095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, D.R.; Friess, D.A. Rates and drivers of mangrove deforestation in Southeast Asia, 2000–2012. Proc. Natl. Acad. Sci. USA 2016, 113, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Gilman, E.L.; Ellison, J.; Duke, N.C.; Field, C. Threats to mangroves from climate change and adaptation options: A review. Aquat. Bot. 2008, 89, 237–250. [Google Scholar] [CrossRef]
- Duke, N.C.; Ball, M.C.; Ellison, J.C. Factors influencing biodiversity and distributional gradients in mangroves. Glob. Ecol. Biogeogr. Lett. 1998, 7, 27–47. [Google Scholar] [CrossRef]
- Liao, P.-C.; Havanond, S.; Huang, S. Phylogeography of Ceriops tagal (Rhizophoraceae) in Southeast Asia: The land barrier of the Malay Peninsula has caused population differentiation between the Indian Ocean and South China Sea. Conserv. Genet. 2007, 8, 89–98. [Google Scholar] [CrossRef]
- Minobe, S.; Fukui, S.; Saiki, R.; Kajita, T.; Changtragoon, S.; Ab Shukor, N.A.; Latiff, A.; Ramesh, B.R.; Koizumi, O.; Yamazaki, T. Highly differentiated population structure of a Mangrove species, Bruguiera gymnorhiza (Rhizophoraceae) revealed by one nuclear GapCp and one chloroplast intergenic spacer trnF-trnL. Conserv. Genet. 2010, 11, 301–310. [Google Scholar] [CrossRef]
- Urashi, C.; Teshima, K.M.; Minobe, S.; Koizumi, O.; Inomata, N. Inferences of evolutionary history of a widely distributed mangrove species, Bruguiera gymnorrhiza, in the Indo-West Pacific region. Ecol. Evol. 2013, 3, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Tamura, M.; Tateishi, Y.; Webb, E.L.; Kajita, T. Strong genetic structure over the American continents and transoceanic dispersal in the mangrove genus Rhizophora (Rhizophoraceae) revealed by broad-scale nuclear and chloroplast DNA analysis. Am. J. Bot. 2013, 100, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.L.; Onishi, Y.; Inomata, N.; Teshima, K.M.; Chan, H.T.; Baba, S.; Changtragoon, S.; Siregar, I.Z.; Szmidt, A.E. Closely related and sympatric but not all the same: Genetic variation of Indo-West Pacific Rhizophora mangroves across the Malay Peninsula. Conserv. Genet. 2015, 16, 137–150. [Google Scholar] [CrossRef]
- Wee, A.K.S.; Takayama, K.; Chua, J.L.; Asakawa, T.; Meenakshisundaram, S.H.; Onrizal, B.A.; Ardli, E.R.; Sungkaew, S.; Malekal, N.B.; Tung, N.X.; et al. Genetic differentiation and phylogeography of partially sympatric species complex Rhizophora mucronata Lam. and R. stylosa Griff. using SSR markers. BMC Evol. Biol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Tan, F.; Su, G.; Deng, S.; He, H.; Shi, S. Population genetic structure of three tree species in the mangrove genus Ceriops (Rhizophoraceae) from the Indo West Pacific. Genetica 2008, 133, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Ono, J.; Tsuda, Y.; Wee, A.K.S.; Takayama, K.; Onrizal, B.A.; Ardli, E.R.; Sungkaew, S.; Suleiman, M.; Tung, N.X.; Salmo, S.G., III; et al. Genetic structure of a widespread mangrove species Bruguiera gymnorhiza suggests limited gene flow by sea-dispersal in oceanic regions. Unpublished work. 2017. [Google Scholar]
- Budde, K.B.; González-Martínez, S.C.; Hardy, O.J.; Heuertz, M. The ancient tropical rainforest tree Symphonia globulifera L. f. (Clusiaceae) was not restricted to postulated Pleistocene refugia in Atlantic Equatorial Africa. Heredity 2013, 111, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, Y.; Nakao, K.; Ide, Y.; Tsumura, Y. The population demography of Betula maximowicziana, a cool-temperate tree species in Japan, in relation to the last glacial period: Its admixture-like genetic structure is the result of simple population splitting not admixing. Mol. Ecol. 2015, 24, 1403–1418. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Ortega, J.S.; Yamamoto, T.; Vovides, A.P.; Pérez-Farrera, A.M.; Martínez, J.F.; Molina-Freaner, F.; Watano, Y.; Kajita, T. Aridification as a driver of biodiversity: A case study for the cycad genus Dioon (Zamiaceae). Ann. Bot. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, F.; Tsuda, Y.; Fineschi, S.; Bruschi, P.; Magri, D.; Zhelev, P.; Paule, L.; Simeone, M.C.; González-Martínez, S.C.; Vendramin, G.G. Combining molecular and fossil data to infer demographic history of Quercus cerris: Insights on European eastern glacial refugia. J. Biogeogr. 2016, 43, 679–690. [Google Scholar] [CrossRef]
- Soliani, C.; Tsuda, Y.; Bagnoli, F.; Gallo, L.A.; Vendramin, G.G.; Marchelli, P. Halfway encounters: Meeting points of colonization routes among the southern beeches Nothofagus pumilio and N. antarctica. Mol. Phylogenet. Evol. 2015, 85, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, Y.; Semerikov, V.; Sebastiani, F.; Vendramin, G.G.; Lascoux, M. Multispecies genetic structure and hybridization in the Betula genus across Eurasia. Mol. Ecol. 2017, 26, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Mabberley, D.J.; Pannell, C.M.; Sing, A.M. Flora Malesiana: Series I. Spermatophyta Volume 12, Part 1, 1995. Meliaceae; Rijksherbarium, Foundation Flora Malesiana: Leiden, The Netherlands, 1995; ISBN 9071236269. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Demesure, B.; Sodzi, N.; Petit, R.J. A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol. Ecol. 1995, 4, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 1991, 17, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Small, R.L.; Ryburn, J.A.; Cronn, R.C.; Seelanan, T.; Wendel, J.F. The tortoise and the hare: Choosing between noncoding plastome and nuclear Adh sequences for phylogeny reconstruction in a recently diverged plant group. Am. J. Bot. 1998, 85, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G.; Hedrick, P.W. Assessing population structure: FST and related measures. Mol. Ecol. Resour. 2011, 11, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, Y.; Shinmura, Y.; Wee, A.K.S.; Takayama, K.; Asakawa, T.; Yllano, O.B.; Salmo, S.G., III; Ardli, E.R.; Tung, N.X.; Binti Malekal, N.; et al. Development of 11 polymorphic microsatellite markers for Xylocarpus granatum (Meliaceae) using next-generation sequencing technology. Conserv. Genet. Resour. 2013, 5, 1159–1162. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Tajima, F.; Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Langella, O. Populations 1.2.30: Population Genetic Software (Individuals or Population Distances, Phylogenetic Trees). 2007. Available online: http://bioinformatics.org/~tryphon/populations/ (accessed on 25 July 2017).
- Parks, D.H.; Porter, M.; Churcher, S.; Wang, S.; Blouin, C.; Whalley, J.; Brooks, S.; Beiko, R.G. GenGIS: A geospatial information system for genomic data. Genome Res. 2009, 19, 1896–1904. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.-M.; Santos, F.; Beaumont, M.A.; Robert, C.P.; Marin, J.-M.; Balding, D.J.; Guillemaud, T.; Estoup, A. Inferring population history with DIY ABC: A user-friendly approach to approximate Bayesian computation. Bioinformatics 2008, 24, 2713–2719. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.-M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Leblois, R.; Marin, J.-M.; Estoup, A. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Estoup, A.; Jarne, P.; Cornuet, J.-M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol. Ecol. 2002, 11, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Garza, J.C.; Williamson, E.G. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 2001, 10, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.B.; Ruiz Linares, A.; Cavalli-Sforza, L.L.; Feldman, M.W. Genetic absolute dating based on microsatellites and the origin of modern humans. Proc. Natl. Acad. Sci. USA 1995, 92, 6723–6727. [Google Scholar] [CrossRef] [PubMed]
- Wee, A.K.S.; Teo, J.X.H.; Chua, J.L.; Takayama, K.; Asakawa, T.; Meenakshisundaram, S.H.; Onrizal, B.A.; Ardli, E.R.; Sungkaew, S.; Suleiman, M.; et al. Vicariance, oceanic barriers and geographic distance drive contemporary genetic structure of widespread mangrove species Sonneratia alba in the Indo-West Pacific. Forests 2017, in press. [Google Scholar]
- Yamamoto, T.; Tsuda, Y.; Takayama, K.; Nagashima, R.; Tateishi, Y.; Kajita, T. The presence of a cryptic barrier in the West Pacific Ocean suggests the effect of glacial climate changes on a widespread sea-dispersed plant, Vigna marina (Fabaceae). Ecol. Evol. 2017. under review. [Google Scholar]
- Harwell, M.C.; Orth, R.J. Long-distance dispersal potential in a marine macrophyte. Ecology 2002, 83, 3319–3330. [Google Scholar] [CrossRef]
- Takayama, K.; Kajita, T.; Murata, J.; Tateishi, Y. Phylogeography and genetic structure of Hibiscus tiliaceus—speciation of a pantropical plant with sea-drifted seeds. Mol. Ecol. 2006, 15, 2871–2881. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Tateishi, Y.; Murata, J.; Kajita, T. Gene flow and population subdivision in a pantropical plant with sea-drifted seeds Hibiscus tiliaceus and its allied species: Evidence from microsatellite analyses. Mol. Ecol. 2008, 17, 2730–2742. [Google Scholar] [CrossRef] [PubMed]
- Miryeganeh, M.; Takayama, K.; Tateishi, Y.; Kajita, T. Long-distance dispersal by sea-drifted seeds has maintained the global distribution of Ipomoea pes-caprae subsp. brasiliensis (Convolvulaceae). PLoS ONE 2014, 9, e91836. [Google Scholar] [CrossRef] [PubMed]
- Voris, H.K. Maps of Pleistocene sea levels in Southeast Asia: Shorelines, river systems and time durations. J. Biogeogr. 2000, 27, 1153–1167. [Google Scholar] [CrossRef]
- Wyrtki, K. Physical Oceanography of the Southeast Asian Waters; Scientific Results of Marine Investigations of the South China Sea and the Gulf of Thailand; University of California, Scripps Institution of Oceanography: San Diego, CA, USA, 1961; Volume 2, p. 195. [Google Scholar] [CrossRef]
- Magri, D.; Vendramin, G.G.; Comps, B.; Dupanloup, I.; Geburek, T.; Gömöry, D.; Latałowa, M.; Litt, T.; Paule, L.; Roure, J.M.; et al. A new scenario for the Quaternary history of European beech populations: Palaeobotanical evidence and genetic consequences. New Phytol. 2006, 171, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, Y.; Ide, Y. Chloroplast DNA phylogeography of Betula maximowicziana, a long-lived pioneer tree species and noble hardwood in Japan. J. Plant Res. 2010, 123, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Triest, L. Molecular ecology and biogeography of mangrove trees towards conceptual insights on gene flow and barriers: A review. Aquat. Bot. 2008, 89, 138–154. [Google Scholar] [CrossRef]
- Tomlinson, P.B. The Botany of Mangroves; Cambridge University Press: Cambridge, UK, 1986. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Country | Locality | Coordinate | Chloroplast DNA | h | Nulcear SSRs | ||
|---|---|---|---|---|---|---|---|---|
| n | H | n | Voucher | |||||
| Xylocarpus granatum J. Koenig | ||||||||
| 1 | Mozambique | Maputo | 25.85° S, E32.696° E | 9 | 2 | 0.198 | - | TK10122702 |
| 2 | Mozambique | Quelimane | 17.883° S, 36.861° E | 4 | 1 | 0.000 | 4 | TK10122504 |
| 3 | Seychelles | Mahe | 4.654° S, 55.403° E | 6 | 1 | 0.000 | - | KT12022201 |
| 4 | Myanmar | Ayeyarwady | 15.95° N, 95.357° E | 6 | 1 | 0.000 | 4 | TK11100805 |
| 5 | Thailand | Phuket | 8.204° N, 98.442° E | 4 | 2 | 0.375 | 4 | KT09121701 |
| 6 | Malaysia | Klang | 2.988° N, 101.355° E | 7 | 2 | 0.245 | 4 | TK11121403 |
| 7 | Malaysia | Kemaman | 4.65° N, 103.427° E | 6 | 1 | 0.000 | 4 | TK11121603 |
| 8 | Malaysia | Sabah | 5.945° N, 118.022° E | 6 | 1 | 0.000 | 4 | TK11072202 |
| 9 | Singapore | Sungei Buloh | 1.448° N, 103.73° E | 5 | 1 | 0.000 | - | KT09111202 |
| 10 | Indonesia | Java | 7.709° S, 108.9° E | 6 | 1 | 0.000 | - | KT09111508 |
| 11 | Indonesia | Bali | 8.734° S, 115.197° E | 8 | 1 | 0.000 | 4 | KT09111704 |
| 12 | Vietnam | Dong Rui | 21.226° N, 107.375° E | 3 | 2 | 0.444 | - | TK10050104 |
| 13 | Philippines | Panay | 11.802° N, 122.207° E | 11 | 1 | 0.000 | 4 | TK11062404 |
| 14 | Palau | Airai | 7.368° N, 134.576° E | 5 | 2 | 0.320 | 4 | YT60917 |
| 15 | Micronesia | Kosrae | 5.279° N, 162.973° E | 8 | 1 | 0.000 | 3 | TK11121601 |
| 16 | Vanuatu | Malatie | 17.55° S, 168.341° E | 3 | 1 | 0.000 | 4 | KT10073101 |
| 17 | New Caledonia | Baie de Taare | 22.259° S, 167.013° E | 7 | 1 | 0.000 | 4 | KT13022607 |
| 18 | Australia | Daintree River | 16.258° S, 145.399° E | 7 | 1 | 0.000 | 3 | KT13031902 |
| Total 111 | Total 50 | |||||||
| Xylocarpus moluccensis M.Roem. | ||||||||
| 19 | India | Pichavaram | 11.431° N, 79.794° E | 2 | 1 | 0.000 | - | TK10112308 |
| 20 | Thailand | Trat | 12.165° N, 102.482° E | 2 | 2 | 0.500 | - | KT09121902 |
| 21 | Malaysia | Klang | 3.038° N, 101.264° E | 7 | 1 | 0.000 | - | TK11121404 |
| 22 | Vietnam | Ca Mau | 8.716° N, 104.814° E | 1 | 1 | 0.000 | - | TK10042705 |
| 23 | Philippines | Panay | 11.802° N, 122.2° E | 4 | 1 | 0.000 | - | TK11062405 |
| 24 | Myanmar | Ayeyarwady | 15.95° N, 95.357° E | 1 | 1 | 0.000 | - | KK09122608 |
| Total 17 | ||||||||
| Xylocarpus rumphii (Kostel.) Mabb. | ||||||||
| 25 | Malaysia | Kilim | 6.42° N, 99.417° E | 1 | 1 | 0.000 | - | KLG20130620 |
| trnD–trnT | trnL–trnF | accD–psaI | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype | Accession No. | 4 | 185 | 191 | 196–201 | 202 | 223 | 231 | 232 | 404 | Accession No. | 946 | 950 | 979 | Accession No. | 1222 | 1266 | 1325 | 1525 |
| Xylocarpus granatum J. Koenig | |||||||||||||||||||
| XG1 | LC217845 | C | A | A | - | T | T | A | A | T | LC217851 | C | C | C | LC217857 | G | C | A | - |
| XG2 | LC217846 | C | A | C | - | T | T | A | A | T | LC217851 | C | C | C | LC217857 | G | C | A | - |
| XG3 | LC217846 | C | A | C | - | T | T | A | A | T | LC217851 | C | C | C | LC217858 | G | C | G | - |
| XG4 | LC217846 | C | A | C | - | T | T | A | A | T | LC217852 | T | A | C | LC217858 | G | C | G | - |
| Xylocarpus moluccensis M. Roem. | |||||||||||||||||||
| XM1 | LC217847 | T | C | C | AAAGGA | A | G | A | A | G | LC217853 | T | C | G | LC217859 | G | G | G | - |
| XM2 | LC217847 | T | C | C | AAAGGA | A | G | A | A | G | LC217854 | C | C | C | LC217859 | G | G | G | - |
| XM3 | LC217848 | T | C | C | AAAGGA | A | G | G | A | G | LC217854 | C | C | C | LC217860 | A | G | G | T |
| XM4 | LC217848 | T | C | C | AAAGGA | A | G | G | A | G | LC217855 | T | A | C | LC217860 | A | G | G | T |
| XM5 | LC217849 | T | C | C | AAAGGA | A | G | G | G | G | LC217855 | T | A | C | LC217860 | A | G | G | T |
| Xylocarpus rumphii (Kostel.) Mabb. | |||||||||||||||||||
| XR (XM4) | LC217850 | T | C | C | AAAGGA | A | G | G | A | G | LC217856 | T | A | C | LC217861 | A | G | G | T |
| Source of Variation | Total Variance (%) | F-Statistics | p-Value | F'-Value |
|---|---|---|---|---|
| 13 populations | ||||
| Among populations | 40.4% | FST = 0.404 | 0.001 | F'ST = 0.802 |
| Among individuals within populations | 59.6% | - | - | - |
| K = 2 | ||||
| Among group | 19.8% | FRT = 0.198 | 0.001 | F'RT = 0.748 |
| Among population within groups | 27.2% | FSR = 0.339 | 0.001 | F'SR = 0.678 |
| Among individuals within populations | 53.0% | FST = 0.470 | 0.001 | - |
| K = 3 | ||||
| Among group | 18.3% | FRT = 0.183 | 0.001 | F'RT = 0.648 |
| Among population within groups | 24.9% | FSR = 0.302 | 0.001 | F'SR = 0.610 |
| Among individuals within populations | 56.8% | FST = 0.432 | 0.001 | - |
| Between the Indian Ocean (Pop 2,4,5 and 6) and the South China Sea (Pop 7,8,11,13 and 14) | ||||
| Among group | 10.4% | FRT = 0.104 | 0.001 | F'RT = 0.389 |
| Among population within groups | 23.9% | FSR = 0.266 | 0.001 | F'SR = 0.577 |
| Among individuals within populations | 65.8% | FST = 0.342 | 0.001 | - |
| Between the Indian Ocean (Pop 2,4,5 and 6) and Oceania (Pop 15,16,17 and 18) | ||||
| Among group | 11.6% | FRT = 0.116 | 0.001 | F'RT = 0.416 |
| Among population within groups | 23.5% | FSR = 0.266 | 0.001 | F'SR = 0.567 |
| Among individuals within populations | 64.9% | FST = 0.351 | 0.001 | - |
| Between the South China Sea (Pop 7,8,11,13 and 14) and Oceania (Pop 15,16,17 and 18) | ||||
| Among group | 25% | FRT = 0.246 | 0.001 | F'RT = 0.815 |
| Among population within groups | 21% | FSR = 0.283 | 0.001 | F'SR = 0.572 |
| Among individuals within populations | 54% | FST = 0.459 | 0.001 | - |
| ABC1 | Posterior Probability (95% Confidence Interval (Lower–Upper)) | ||
|---|---|---|---|
| Scenario | Region 1 | Region 2 | Region 3 |
| 1 | 0.2640 (0.2586–0.2693) | 0.2564 (0.2477–0.2651) | 0.2554 (0.2500–0.2607) |
| 2 | 0.2543 (0.2491–0.2596) | 0.2645 (0.2482–0.2660) | 0.3917 (0.3858–0.3977) |
| 3 | 0.2340 (0.2288–0.2392) | 0.2221 (0.2136–0.2305) | 0.1624 (0.1579–0.1669) |
| 4 | 0.2477 (0.2420–0.2533) | 0.2571 (0.2482–0.2660) | 0.1905 (0.1858–0.1952) |
| ABC2 | |
|---|---|
| Scenario | Posterior Probability (95% Confidence Interval: Lower–Upper) |
| 1 | 0.3794 (0.3712–0.3877) |
| 2 | 0.0839 (0.0784–0.0893) |
| 3 | 0.3111 (0.3031–0.3190) |
| 4 | 0.0877 (0.0821–0.0934) |
| 5 | 0.0590 (0.0541–0.0639) |
| 6 | 0.0789 (0.0734–0.0844) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomizawa, Y.; Tsuda, Y.; Saleh, M.N.; Wee, A.K.S.; Takayama, K.; Yamamoto, T.; Yllano, O.B.; Salmo III, S.G.; Sungkaew, S.; Adjie, B.; et al. Genetic Structure and Population Demographic History of a Widespread Mangrove Plant Xylocarpus granatum J. Koenig across the Indo-West Pacific Region. Forests 2017, 8, 480. https://doi.org/10.3390/f8120480
Tomizawa Y, Tsuda Y, Saleh MN, Wee AKS, Takayama K, Yamamoto T, Yllano OB, Salmo III SG, Sungkaew S, Adjie B, et al. Genetic Structure and Population Demographic History of a Widespread Mangrove Plant Xylocarpus granatum J. Koenig across the Indo-West Pacific Region. Forests. 2017; 8(12):480. https://doi.org/10.3390/f8120480
Chicago/Turabian StyleTomizawa, Yuki, Yoshiaki Tsuda, Mohd Nazre Saleh, Alison K. S. Wee, Koji Takayama, Takashi Yamamoto, Orlex Baylen Yllano, Severino G. Salmo III, Sarawood Sungkaew, Bayu Adjie, and et al. 2017. "Genetic Structure and Population Demographic History of a Widespread Mangrove Plant Xylocarpus granatum J. Koenig across the Indo-West Pacific Region" Forests 8, no. 12: 480. https://doi.org/10.3390/f8120480
APA StyleTomizawa, Y., Tsuda, Y., Saleh, M. N., Wee, A. K. S., Takayama, K., Yamamoto, T., Yllano, O. B., Salmo III, S. G., Sungkaew, S., Adjie, B., Ardli, E., Suleiman, M., Tung, N. X., Soe, K. K., Kandasamy, K., Asakawa, T., Watano, Y., Baba, S., & Kajita, T. (2017). Genetic Structure and Population Demographic History of a Widespread Mangrove Plant Xylocarpus granatum J. Koenig across the Indo-West Pacific Region. Forests, 8(12), 480. https://doi.org/10.3390/f8120480