A Divergent Hepatitis D-Like Agent in Birds

 , , , , , and
, , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Selection, RNA Library Construction and Sequencing
2.3. RNA Virus Discovery
2.4. Characterisation of Novel Hepatitis D-Like Virus
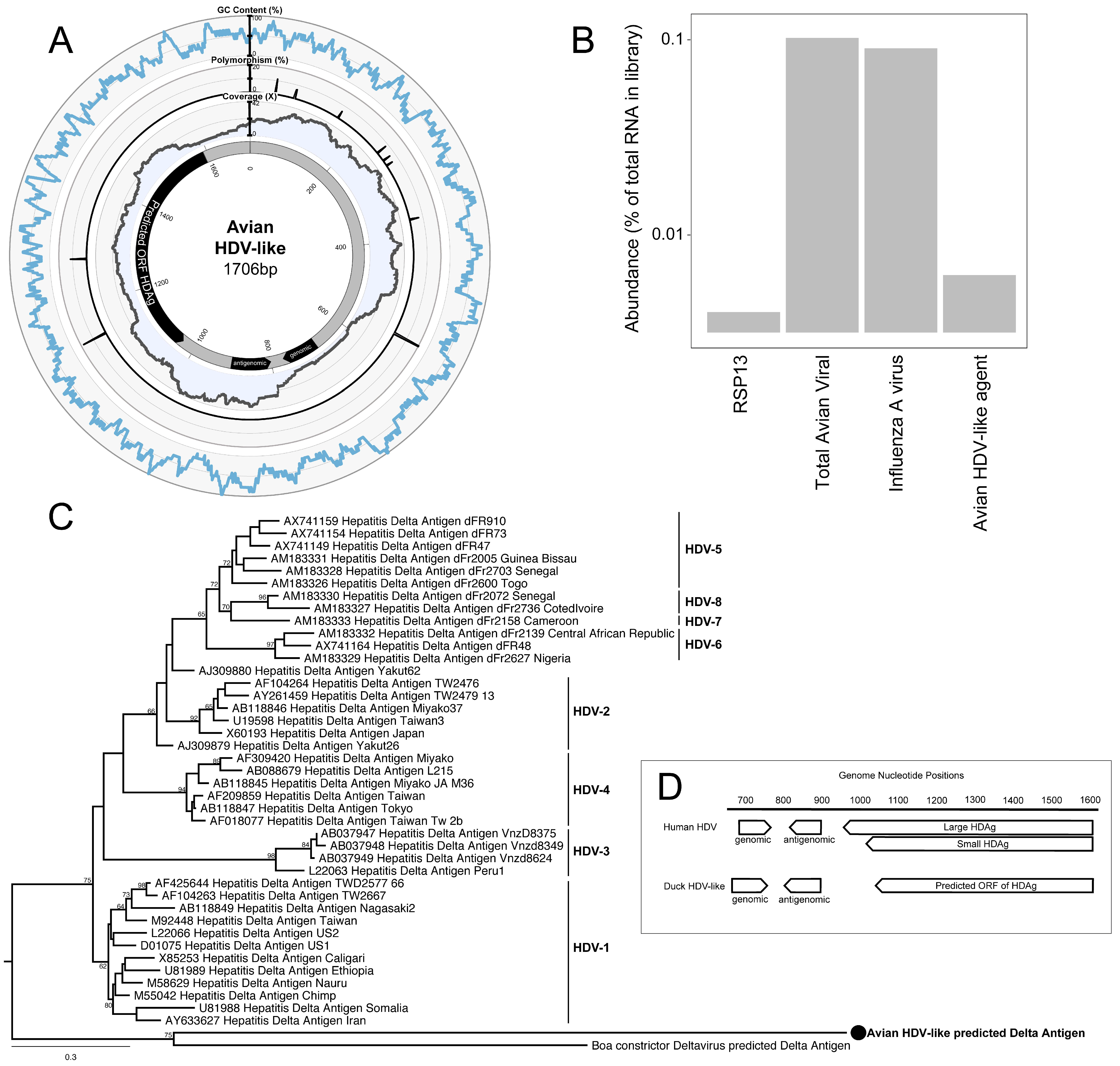
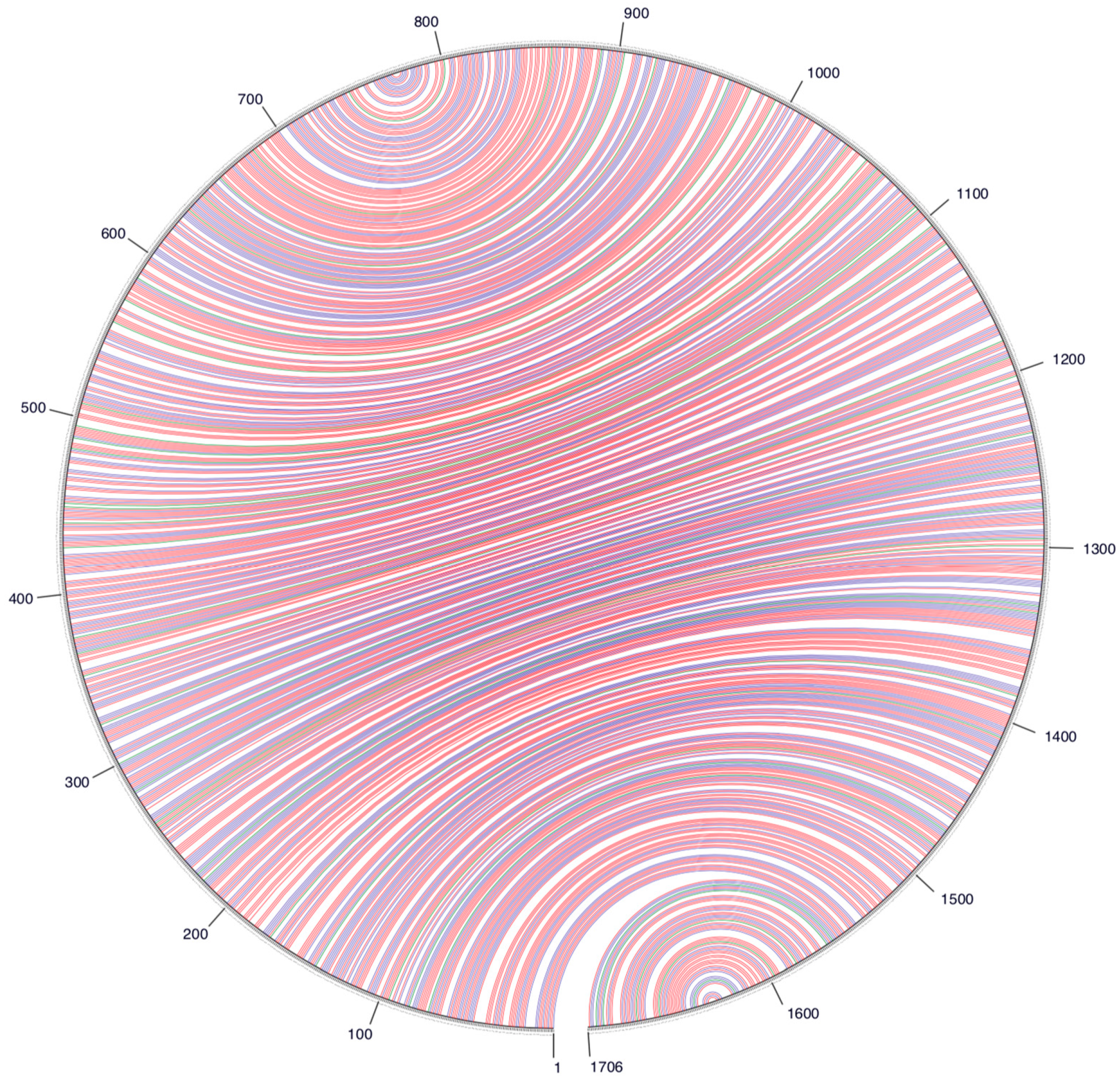
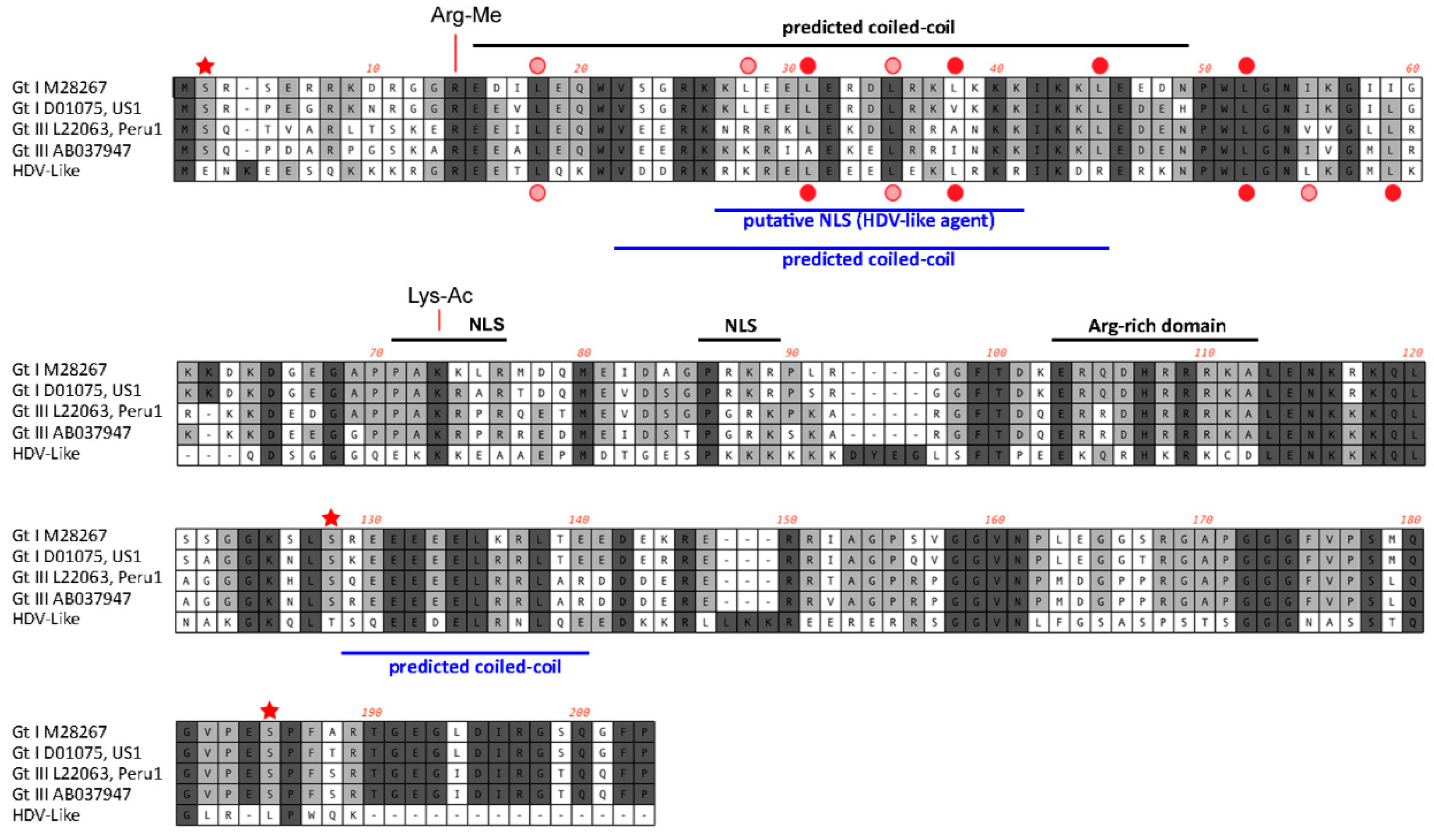
3. Results and Discussion
4. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, J.; Pelchat, M. Origin of hepatitis Delta virus. Future Microbiol. 2010, 5, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Purcell, R.H.; Farci, P. Hepatitis D (D) virus. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2222–2241. [Google Scholar]
- Wedemeyer, H.; Manns, M.P. Epidemiology, pathogenesis and management of hepatitis D: Update and challenges ahead. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- WHO. Hepatitis D Fact Sheet. 2017. Available online: http://www.who.int/mediacentre/factsheets/hepatitis-d/en/ (accessed on 24 February 2018).
- Le Gal, F.; Gault, E.; Ripault, M.P.; Serpaggi, J.; Trinchet, J.C.; Gordien, E.; Deny, P. Eighth major clade for hepatitis Delta virus. Emerg. Infect. Dis. 2006, 12, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, M.; Locarnini, S.; Yuen, L. Origins and evolution of hepatitis B virus and hepatitis D virus. Cold Spring Harb. Perspect. Med. 2016, 6, a021360. [Google Scholar] [CrossRef] [PubMed]
- Brazas, R.; Ganem, D. Delta-interacting protein and the origin of hepatitis Delta antigen-Response. Science 1997, 276, 825. [Google Scholar]
- Long, M.; de Souza, S.J.; Gilbert, W. Delta-interacting protein and the origin of hepatitis Delta antigen. Science 1997, 276, 824–825. [Google Scholar] [CrossRef] [PubMed]
- Brazas, R.; Ganem, D. A cellular homolog of hepatitis Delta antigen: Implications for viral replication and evolution. Science 1996, 274, 90–94. [Google Scholar] [CrossRef]
- Salehi-Ashtiani, K.; Luptak, A.; Litovchick, A.; Szostak, J.W. A genomewide search for ribozymes reveals an HDV-like sequence in the human CPEB3 gene. Science 2006, 313, 1788–1792. [Google Scholar] [CrossRef]
- Taylor, J.M. Host RNA circles and the origin of hepatitis Delta virus. World J. Gastroenterol. 2014, 20, 2971–2978. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-SEQ data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Chapman, J.R.; Helin, A.S.; Wille, M.; Atterby, C.; Jarhult, J.D.; Fridlund, J.S.; Waldenstrom, J. A panel of stably expressed reference genes for real-time qPCR gene expression studies of mallards (Anas platyrhynchos). PLoS ONE 2016, 11, e0149454. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Soding, J. Hhblits: Lightning-fast iterative protein sequence searching by hmm-hmm alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. Trimal: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [Green Version]
- Webb, C.H.T.; Luptak, A. HDV-like self-cleaving ribozymes. RNA Biol. 2011, 8, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Byun, Y.; Han, K. Pseudoviewer3: Generating planar drawings of large-scale RNA structures with pseudoknots. Bioinformatics 2009, 25, 1435–1437. [Google Scholar] [CrossRef]
- Wolf, E.; Kim, P.S.; Berger, B. Multicoil: A program for predicting two- and three-stranded coiled coils. Protein Sci. 1997, 6, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis Delta (delta) viral genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef]
- Hetzel, U.; Szirovicza, L.; Smura, T.; Prähauser, B.; Vapalahti, O.; Kipar, A.; Hepojoki, J. Identification of a novel deltavirus in boa constrictor. BioRxiv Preprint 2018. [Google Scholar] [CrossRef]
- Perrotta, A.T.; Been, M.D. A pseudoknot-like structure required for efficient self-cleavage of hepatitis Delta-virus RNA. Nature 1991, 350, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Determinants of translational fidelity and efficiency in vertebrate mRNAs. Biochimie 1994, 76, 815–821. [Google Scholar] [CrossRef]
- Proudfoot, N.J. Ending the message: Poly(a) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.Y.; Taylor, J. Regulation of polyadenylation of hepatitis Delta virus antigenomic RNA. J. Virol. 1991, 65, 6438–6446. [Google Scholar] [PubMed]
- Casey, J.L.; Bergmann, K.F.; Brown, T.L.; Gerin, J.L. Structural requirements for RNA editing in hepatitis Delta virus: Evidence for a uridine-to-cytidine editing mechanism. Proc. Natl. Acad. Sci. USA 1992, 89, 7149–7153. [Google Scholar] [CrossRef]
- Glenn, J.S.; Watson, J.A.; Havel, C.M.; White, J.M. Identification of a prenylation site in delta-virus large antigen. Science 1992, 256, 1331–1333. [Google Scholar] [CrossRef]
- Xia, Y.P.; Lai, M.M. Oligomerization of hepatitis Delta antigen is required for both the trans-activating and trans-dominant inhibitory activities of the delta antigen. J. Virol. 1992, 66, 6641–6648. [Google Scholar]
- Lazinski, D.W.; Taylor, J.M. Relating structure to function in the hepatitis Delta virus antigen. J. Virol. 1993, 67, 2672–2680. [Google Scholar] [PubMed]
- Huang, W.H.; Chen, C.W.; Wu, H.L.; Chen, P.J. Post-translational modification of delta antigen of hepatitis D virus. Curr. Top. Microbiol. 2006, 307, 91–112. [Google Scholar]
- Lai, M.M.C. RNA replication without RNA-dependent RNA polymerase: Surprises from hepatitis Delta virus. J. Virol. 2005, 79, 7951–7958. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wille, M.; Netter, H.J.; Littlejohn, M.; Yuen, L.; Shi, M.; Eden, J.-S.; Klaassen, M.; Holmes, E.C.; Hurt, A.C. A Divergent Hepatitis D-Like Agent in Birds. Viruses 2018, 10, 720. https://doi.org/10.3390/v10120720
Wille M, Netter HJ, Littlejohn M, Yuen L, Shi M, Eden J-S, Klaassen M, Holmes EC, Hurt AC. A Divergent Hepatitis D-Like Agent in Birds. Viruses. 2018; 10(12):720. https://doi.org/10.3390/v10120720
Chicago/Turabian StyleWille, Michelle, Hans J. Netter, Margaret Littlejohn, Lilly Yuen, Mang Shi, John-Sebastian Eden, Marcel Klaassen, Edward C. Holmes, and Aeron C. Hurt. 2018. "A Divergent Hepatitis D-Like Agent in Birds" Viruses 10, no. 12: 720. https://doi.org/10.3390/v10120720